

Role of Monovalent Ions in the NKCC1 Inhibition Mechanism Revealed through Molecular Simulations

Abstract
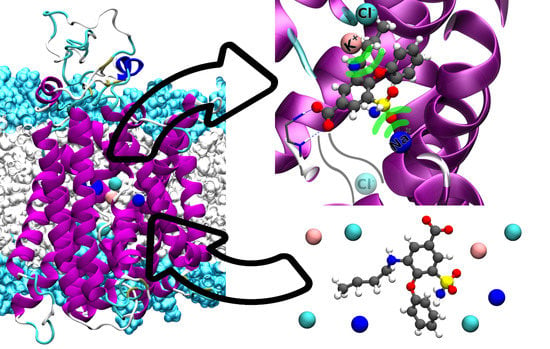
1. Introduction
2. Results
2.1. Bumetanide Binding Pose Refinement
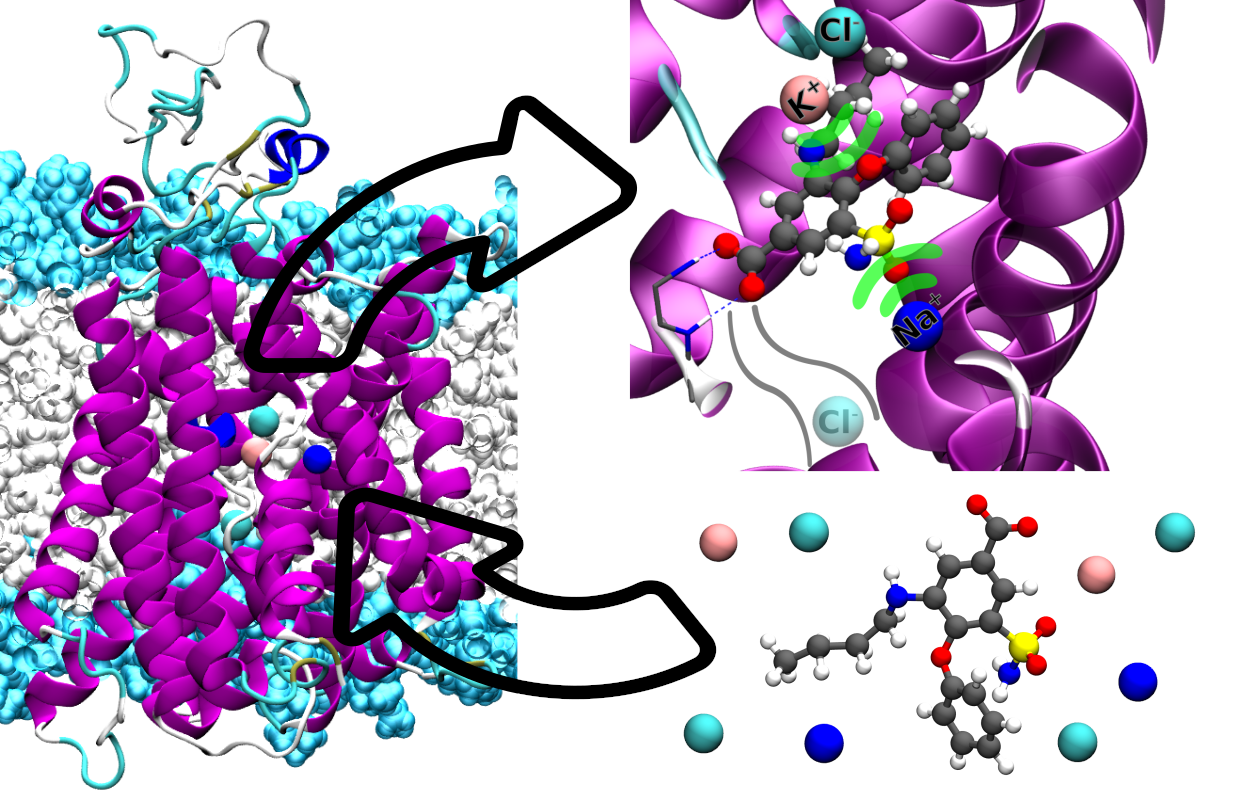
2.2. Bumetanide Binding to Human NKCC1
2.3. Binding Mode of Other Loop Diuretic Drugs
2.3.1. Azosemide Binding Mode
2.3.2. Furosemide Binding Mode
2.3.3. Ethacrynic Acid Binding Mode
3. Discussion
4. Materials and Methods
4.1. Binding Site Location and Docking
4.2. Model Building
4.3. Molecular Dynamics Simulation Details
4.4. Metadynamics Simulation Details
4.5. MM/PBSA Binding Free Energy Calculations and Alanine Scanning Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Markadieu, N.; Delpire, E. Physiology and pathophysiology of SLC12A1/2 transporters. Pflüg. Arch.-Eur. J. Physiol. 2014, 466, 91–105. [Google Scholar] [CrossRef]
- Lytle, C.; Forbush, B., 3rd. Regulatory Phosphorylation of the Secretory Na-K-Cl Cotransporter: Modulation by Cytoplasmic Cl. Am. J. Physiol.-Cell Physiol. 1996, 270, C437–C448. [Google Scholar] [CrossRef]
- Russell, J.M. Sodium-Potassium-Chloride Cotransport. Physiol. Rev. 2000, 80, 211–276. [Google Scholar] [CrossRef] [PubMed]
- Kaila, K.; Price, T.J.; Payne, J.A.; Puskarjov, M.; Voipio, J. Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat. Rev. Neurosci. 2014, 15, 637–654. [Google Scholar] [CrossRef]
- Haas, M.; Forbush III, B. The Na-K-Cl Cotransporter of Secretory Epithelia. Annu. Rev. Physiol. 2000, 62, 515–534. [Google Scholar] [CrossRef]
- Garneau, A.P.; Slimani, S.; Fiola, M.J.; Tremblay, L.E.; Isenring, P. Multiple Facets and Roles of Na+-K+-Cl− Cotransport: Mechanisms and Therapeutic Implications. Physiology 2020, 35, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Schulte, J.T.; Wierenga, C.J.; Bruining, H. Chloride transporters and GABA polarity in developmental, neurological and psychiatric conditions. Neurosci. Biobehav. Rev. 2018, 90, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Contestabile, A.; Magara, S.; Cancedda, L. The GABAergic Hypothesis for Cognitive Disabilities in Down Syndrome. Front. Cell. Neurosci. 2017, 11, 54. [Google Scholar] [CrossRef]
- Cellot, G.; Cherubini, E. GABAergic Signaling as Therapeutic Target for Autism Spectrum Disorders. Front. Pediatr. 2014, 2, 70. [Google Scholar] [CrossRef]
- Gamba, G. Molecular Physiology and Pathophysiology of Electroneutral Cation-Chloride Cotransporters. Physiol. Rev. 2005, 85, 423–493. [Google Scholar] [CrossRef]
- Gagnon, K.B.; Delpire, E. Physiology of SLC12 transporters: Lessons from inherited human genetic mutations and genetically engineered mouse knockouts. Am. J. Physiol.-Cell Physiol. 2013, 304, C693–C714. [Google Scholar] [CrossRef]
- Luo, L.; Wang, J.; Ding, D.; Hasan, N.; Yang, S.-S.; Lin, S.-H.; Schreppel, P.; Sun, B.; Yin, Y.; Erker, T.; et al. Role of NKCC1 Activity in Glioma K+ Homeostasis and Cell Growth: New Insights with the Bumetanide-Derivative STS66. Front. Physiol. 2020, 11, 911. [Google Scholar] [CrossRef]
- Schrier, R.W. Use of Diuretics in Heart Failure and Cirrhosis. Semin. Nephrol. 2011, 31, 503–512. [Google Scholar] [CrossRef]
- Edwards, D.; Shah, H.P.; Cao, W.; Gravenstein, N.; Seubert, C.N.; Martynyuk, A.E. Bumetanide Alleviates Epileptogenic and Neurotoxic Effects of Sevoflurane in Neonatal Rat Brain. Anesthesiol. J. Am. Soc. Anesthesiol. 2010, 112, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Auer, T.; Schreppel, P.; Erker, T.; Schwarzer, C. Functional characterization of novel bumetanide derivatives for epilepsy treatment. Neuropharmacology 2020, 162, 107754. [Google Scholar] [CrossRef] [PubMed]
- Damier, P.; Hammond, C.; Ben-Ari, Y. Bumetanide to Treat Parkinson Disease: A Report of 4 Cases. Clin. Neuropharmacol. 2016, 39, 57–59. [Google Scholar] [CrossRef]
- Forbush, B.; Palfrey, H.C. [3H]bumetanide binding to membranes isolated from dog kidney outer medulla. Relationship to the Na,K,Cl co-transport system. J. Biol. Chem. 1983, 258, 11787–11792. [Google Scholar] [CrossRef] [PubMed]
- Haas, M.; McManus, T.J. Bumetanide inhibits (Na + K + 2Cl) co-transport at a chloride site. Am. J. Physiol.-Cell Physiol. 1983, 245, C235–C240. [Google Scholar] [CrossRef]
- Feig, P.U. Cellular mechanism of action of loop diuretics: Implications for drug effectiveness and adverse effects. Am. J. Cardiol. 1986, 57, A14–A19. [Google Scholar] [CrossRef]
- Nappi, J.M. A retrospective evaluation of the efficacy of intravenous bumetanide and comparison of potency with furosemide. Pharm. Pract. Internet 2013, 11, 44–50. [Google Scholar] [CrossRef][Green Version]
- Lykke, K.; Töllner, K.; Feit, P.W.; Erker, T.; MacAulay, N.; Löscher, W. The search for NKCC1-selective drugs for the treatment of epilepsy: Structure–function relationship of bumetanide and various bumetanide derivatives in inhibiting the human cation-chloride cotransporter NKCC1A. Epilepsy Behav. 2016, 59, 42–49. [Google Scholar] [CrossRef]
- Hannaert, P.; Alvarez-Guerra, M.; Pirot, D.; Nazaret, C.; Garay, R. Rat NKCC2/NKCC1 cotransporter selectivity for loop diuretic drugs. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2002, 365, 193–199. [Google Scholar] [CrossRef]
- Hampel, P.; Römermann, K.; Macaulay, N.; Löscher, W. Azosemide is more potent than bumetanide and various other loop diuretics to inhibit the sodium-potassium-chloride-cotransporter human variants hNKCC1A and hNKCC1B. Sci. Rep. 2018, 8, 9877. [Google Scholar] [CrossRef]
- Blaesse, P.; Airaksinen, M.S.; Rivera, C.; Kaila, K. Cation-Chloride Cotransporters and Neuronal Function. Neuron 2009, 61, 820–838. [Google Scholar] [CrossRef] [PubMed]
- Somasekharan, S.; Tanis, J.; Forbush, B. Loop Diuretic and Ion-binding Residues Revealed by Scanning Mutagenesis of Transmembrane Helix 3 (TM3) of Na-K-Cl Cotransporter (NKCC1). J. Biol. Chem. 2012, 287, 17308–17317. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Kohyama, N.; Kobayashi, Y.; Ohbayashi, M.; Ohtani, H.; Sawada, Y.; Yamamoto, T. Functional characterization of human monocarboxylate transporter 6 (SLC16A5). Drug Metab. Dispos. 2005, 33, 1845–1851. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.S.; Ruszaj, D.; Parker, M.D.; Morris, M.E. Contribution of Monocarboxylate Transporter 6 to the Pharmacokinetics and Pharmacodynamics of Bumetanide in Mice. Drug Metab. Dispos. 2020, 48, 788–795. [Google Scholar] [CrossRef]
- Römermann, K.; Fedrowitz, M.; Hampel, P.; Kaczmarek, E.; Töllner, K.; Erker, T.; Sweet, D.H.; Löscher, W. Multiple blood-brain barrier transport mechanisms limit bumetanide accumulation, and therapeutic potential, in the mammalian brain. Neuropharmacology 2017, 117, 182–194. [Google Scholar] [CrossRef] [PubMed]
- Töllner, K.; Brandt, C.; Römermann, K.; Löscher, W. The organic anion transport inhibitor probenecid increases brain concentrations of the NKCC1 inhibitor bumetanide. Eur. J. Pharmacol. 2015, 746, 167–173. [Google Scholar] [CrossRef]
- Kharod, S.C.; Kang, S.K.; Kadam, S.D. Off-Label Use of Bumetanide for Brain Disorders: An Overview. Front. Neurosci. 2019, 13, 310. [Google Scholar] [CrossRef]
- Löscher, W.; Puskarjov, M.; Kaila, K. Cation-chloride cotransporters NKCC1 and KCC2 as potential targets for novel antiepileptic and antiepileptogenic treatments. Neuropharmacology 2013, 69, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Puskarjov, M.; Kahle, K.T.; Ruusuvuori, E.; Kaila, K. Pharmacotherapeutic targeting of cation-chloride cotransporters in neonatal seizures. Epilepsia 2014, 55, 806–818. [Google Scholar] [CrossRef] [PubMed]
- Wall, G.C.; Bigner, D.; Craig, S. Ethacrynic acid and the sulfa-sensitive patient. Arch. Intern. Med. 2003, 163, 116. [Google Scholar] [CrossRef]
- Ding, D.; Liu, H.; Qi, W.; Jiang, H.; Li, Y.; Wu, X.; Sun, H.; Gross, K.; Salvi, R. Ototoxic effects and mechanisms of loop diuretics. J. Otol. 2016, 11, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Chew, T.A.; Orlando, B.J.; Zhang, J.; Latorraca, N.R.; Wang, A.; Hollingsworth, S.A.; Chen, D.-H.; Dror, R.O.; Liao, M.; Feng, L. Structure and mechanism of the cation–chloride cotransporter NKCC1. Nature 2019, 572, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhou, J.; Zhang, Y.; Liu, T.; Friedel, P.; Zhuo, W.; Somasekharan, S.; Roy, K.; Zhang, L.; Liu, Y.; et al. The structural basis of function and regulation of neuronal cotransporters NKCC1 and KCC2. Commun. Biol. 2021, 4, 226. [Google Scholar] [CrossRef] [PubMed]
- Bisha, I.; Magistrato, A. The molecular mechanism of secondary sodium symporters elucidated through the lens of the computational microscope. RSC Adv. 2016, 6, 9522–9540. [Google Scholar] [CrossRef]
- Delpire, E.; Gagnon, K.B. Kinetics of hyperosmotically stimulated Na-K-2Cl cotransporter in Xenopus laevis oocytes. Am. J. Physiol.-Cell Physiol. 2011, 301, C1074–C1085. [Google Scholar] [CrossRef]
- Bisha, I.; Rodriguez, A.; Laio, A.; Magistrato, A. Metadynamics Simulations Reveal a Na+ Independent Exiting Path of Galactose for the Inward-Facing Conformation of vSGLT. PLOS Comput. Biol. 2014, 10, e1004017. [Google Scholar] [CrossRef] [PubMed]
- Janoš, P.; Magistrato, A. All-Atom Simulations Uncover the Molecular Terms of the NKCC1 Transport Mechanism. J. Chem. Inf. Model. 2021, 61, 3649–3658. [Google Scholar] [CrossRef]
- Neumann, C.; Rosenbæk, L.L.; Flygaard, R.K.; Habeck, M.; Karlsen, J.L.; Wang, Y.; Lindorff-Larsen, K.; Gad, H.H.; Hartmann, R.; Lyons, J.; et al. Cryo-EM Structure of the Human NKCC1 Transporter Reveals Mechanisms of Ion Coupling and Specificity. EMBO J. 2022, 41, e110169. [Google Scholar] [CrossRef]
- Zhao, Y.; Roy, K.; Vidossich, P.; Cancedda, L.; De Vivo, M.; Forbush, B.; Cao, E. Structural basis for inhibition of the Cation-chloride cotransporter NKCC1 by the diuretic drug bumetanide. Nat. Commun. 2022, 13, 2747. [Google Scholar] [CrossRef]
- Moseng, M.A.; Su, C.-C.; Rios, K.; Cui, M.; Lyu, M.; Glaza, P.; Klenotic, P.A.; Delpire, E.; Yu, E.W. Inhibition mechanism of NKCC1 involves the carboxyl terminus and long-range conformational coupling. Sci. Adv. 2022, 8, eabq0952. [Google Scholar] [CrossRef]
- Kozakov, D.; Grove, L.E.; Hall, D.R.; Bohnuud, T.; Mottarella, S.E.; Luo, L.; Xia, B.; Beglov, D.; Vajda, S. The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat. Protoc. 2015, 10, 733–755. [Google Scholar] [CrossRef]
- Grosdidier, A.; Zoete, V.; Michielin, O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011, 39, W270–W277. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2022-3: Glide; Schrödinger LLC: New York, NY, USA, 2021.
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Wu, E.L.; Cheng, X.; Jo, S.; Rui, H.; Song, K.C.; Dávila-Contreras, E.M.; Qi, Y.; Lee, J.; Monje-Galvan, V.; Venable, R.M.; et al. CHARMM-GUI Membrane Builder toward Realistic Biological Membrane Simulations. J. Comput. Chem. 2014, 35, 1997–2004. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Dickson, C.J.; Madej, B.D.; Skjevik, Å.A.; Betz, R.M.; Teigen, K.; Gould, I.R.; Walker, R.C. Lipid14: The Amber Lipid Force Field. J. Chem. Theory Comput. 2014, 10, 865–879. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926. [Google Scholar] [CrossRef]
- Joung, I.S.; Cheatham, T.E. Determination of Alkali and Halide Monovalent Ion Parameters for Use in Explicitly Solvated Biomolecular Simulations. J. Phys. Chem. B 2008, 112, 9020–9041. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Vanquelef, E.; Simon, S.; Marquant, G.; Garcia, E.; Klimerak, G.; Delepine, J.C.; Cieplak, P.; Dupradeau, F.-Y. R.E.D. Server: A web service for deriving RESP and ESP charges and building force field libraries for new molecules and molecular fragments. Nucleic Acids Res. 2011, 39, W511–W517. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Lindahl, L.; Abraham, M.; Hess, B.; Spoel, V.D. GROMACS 2020.4 Manual. 2020. Available online: https://doi.org/10.5281/ZENODO.4054996 (accessed on 13 March 2022).
- Case, D.; Ben-Shalom, I.; Brozell, S.; Cerutti, D.; Cheatham, T., III; Cruzeiro, V.; Darden, T.; Duke, R.; Ghoreishi, D.; Gilson, M.; et al. AMBER 22; University of California: San Francisco, CA, USA, 2022. [Google Scholar]
- Tribello, G.A.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G. PLUMED 2: New feathers for an old bird. Comput. Phys. Commun. 2014, 185, 604–613. [Google Scholar] [CrossRef]
- Capelli, R.; Carloni, P.; Parrinello, M. Exhaustive Search of Ligand Binding Pathways via Volume-Based Metadynamics. J. Phys. Chem. Lett. 2019, 10, 3495–3499. [Google Scholar] [CrossRef]
- Trapl, D.; Spiwok, V. Analysis of the Results of Metadynamics Simulations by Metadynminer and Metadynminer3d. arXiv 2020, arXiv:2009.02241. [Google Scholar]
- Miller, B.R., III; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA. Py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Greene, D.; Qi, R.; Nguyen, R.; Qiu, T.; Luo, R. Heterogeneous Dielectric Implicit Membrane Model for the Calculation of MMPBSA Binding Free Energies. J. Chem. Inf. Model. 2019, 59, 3041–3056. [Google Scholar] [CrossRef]
- Duan, L.; Liu, X.; Zhang, J.Z. Interaction Entropy: A New Paradigm for Highly Efficient and Reliable Computation of Protein–Ligand Binding Free Energy. J. Am. Chem. Soc. 2016, 138, 5722–5728. [Google Scholar] [CrossRef] [PubMed]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI Search and Sequence Analysis Tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview Version 2—A Multiple Sequence Alignment Editor and Analysis Workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ΔHb [kcal/mol] | −TΔS [kcal/mol] | ΔGb [kcal/mol] | |
|---|---|---|---|
| Bumetanide zNKCC1 | −27.8 ± 3.1 | 6.1 ± 2.3 | −21.7 ± 3.9 |
| Bumetanide hNKCC1 | −32.6 ± 3.4 | 6.2 ± 3.3 | −26.4 ± 4.7 |
| Azosemide | −27.6 ± 3.3 | 7.0 ± 3.2 | −20.6 ± 4.6 |
| Furosemide | −24.8 ± 4.3 | 7.9 ± 4.0 | −16.9 ± 5.9 |
| Ethacrynic acid | −22.1 ± 3.6 | 9.0 ± 3.4 | −13.1 ± 5.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janoš, P.; Magistrato, A. Role of Monovalent Ions in the NKCC1 Inhibition Mechanism Revealed through Molecular Simulations. Int. J. Mol. Sci. 2022, 23, 15439. https://doi.org/10.3390/ijms232315439
Janoš P, Magistrato A. Role of Monovalent Ions in the NKCC1 Inhibition Mechanism Revealed through Molecular Simulations. International Journal of Molecular Sciences. 2022; 23(23):15439. https://doi.org/10.3390/ijms232315439
Chicago/Turabian StyleJanoš, Pavel, and Alessandra Magistrato. 2022. "Role of Monovalent Ions in the NKCC1 Inhibition Mechanism Revealed through Molecular Simulations" International Journal of Molecular Sciences 23, no. 23: 15439. https://doi.org/10.3390/ijms232315439
APA StyleJanoš, P., & Magistrato, A. (2022). Role of Monovalent Ions in the NKCC1 Inhibition Mechanism Revealed through Molecular Simulations. International Journal of Molecular Sciences, 23(23), 15439. https://doi.org/10.3390/ijms232315439