
Gut Microbiota and Immunotherapy for Alzheimer’s Disease


Abstract

1. Introduction
2. Gut Microbiota and the Microbiota–Gut–Brain Axis
3. Gut Microbiota and AD
4. Immunotherapy for AD
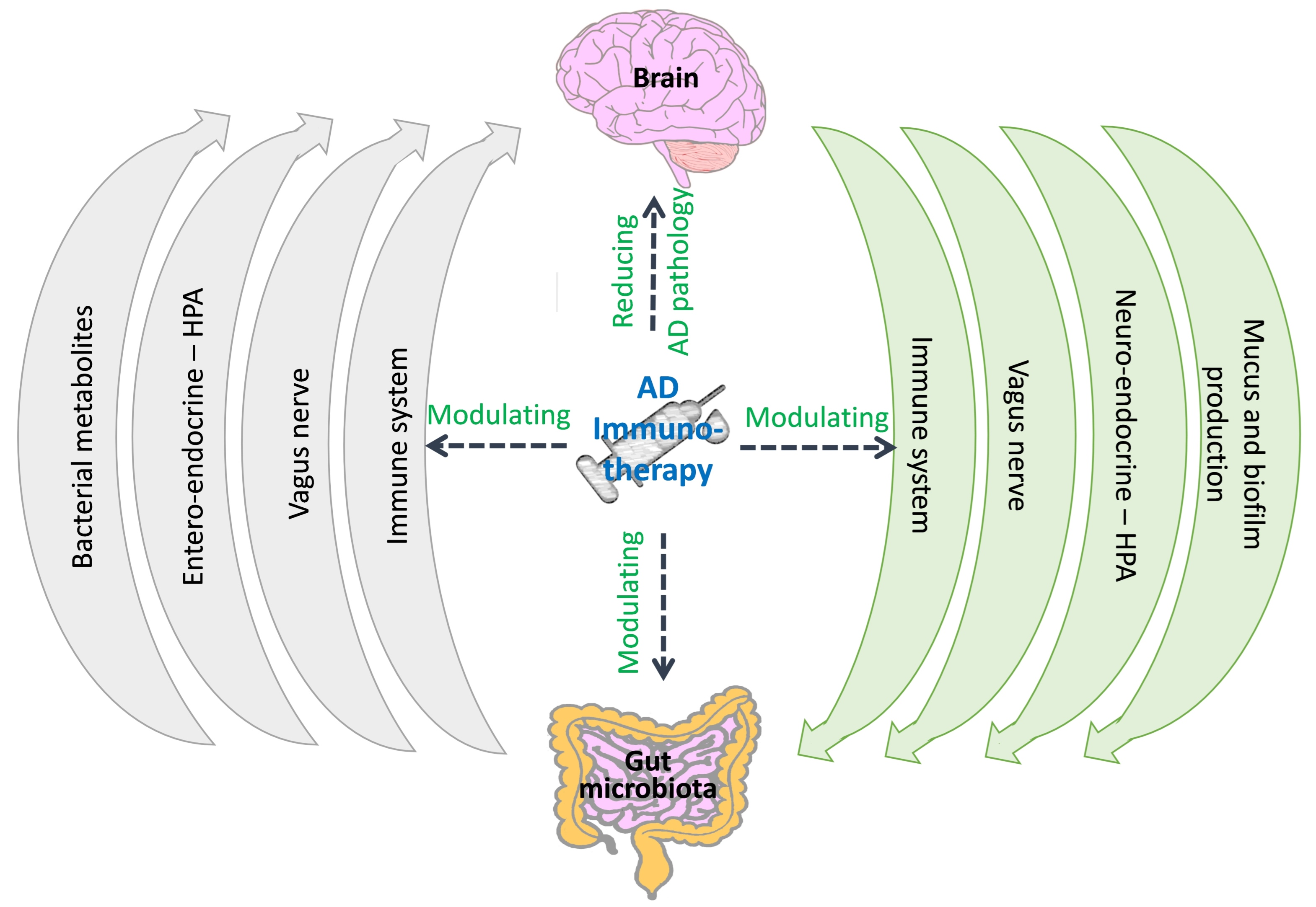
5. Can AD Immunotherapy Act through Modulation of the Gut Microbiota?
6. Does the Gut Microbiota Affect the Therapeutic Effects of AD Immunotherapy?
7. Conclusions and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef] [PubMed]
- 2022 Alzheimer’s disease facts and figures. Alzheimers Dement. 2022, 18, 700–789. [CrossRef] [PubMed]
- Gong, C.X.; Liu, F.; Iqbal, K. Multifactorial Hypothesis and Multi-Targets for Alzheimer’s Disease. J. Alzheimers Dis. 2018, 64, S107–S117. [Google Scholar] [CrossRef]
- Mumtaz, S.; Rana, J.N.; Choi, E.H.; Han, I. Microwave Radiation and the Brain: Mechanisms, Current Status, and Future Prospects. Int. J. Mol. Sci. 2022, 23, 9288. [Google Scholar] [CrossRef] [PubMed]
- Kau, A.L.; Ahern, P.P.; Griffin, N.W.; Goodman, A.L.; Gordon, J.I. Human nutrition, the gut microbiome and the immune system. Nature 2011, 474, 327–336. [Google Scholar] [CrossRef]
- Guard, B.C.; Mila, H.; Steiner, J.M.; Mariani, C.; Suchodolski, J.S.; Chastant-Maillard, S. Characterization of the fecal microbiome during neonatal and early pediatric development in puppies. PLoS ONE 2017, 12, e0175718. [Google Scholar] [CrossRef]
- McKenzie, C.; Tan, J.; Macia, L.; Mackay, C.R. The nutrition-gut microbiome-physiology axis and allergic diseases. Immunol. Rev. 2017, 278, 277–295. [Google Scholar]
- Morais, L.H.; Schreiber, H.L.; Mazmanian, S.K. The gut microbiota-brain axis in behaviour and brain disorders. Nat. Rev. Microbiol. 2021, 19, 241–255. [Google Scholar] [CrossRef]
- Cryan, J.F.; O’Riordan, K.J.; Cowan, C.S.M.; Sandhu, K.V.; Bastiaanssen, T.F.S.; Boehme, M.; Codagnone, M.G.; Cussotto, S.; Fulling, C.; Golubeva, A.V.; et al. The Microbiota-Gut-Brain Axis. Physiol. Rev. 2019, 99, 1877–2013. [Google Scholar]
- Bulgart, H.R.; Neczypor, E.W.; Wold, L.E.; Mackos, A.R. Microbial involvement in Alzheimer disease development and progression. Mol. Neurodegener. 2020, 15, 42. [Google Scholar] [CrossRef]
- Agagunduz, D.; Kocaadam-Bozkurt, B.; Bozkurt, O.; Sharma, H.; Esposito, R.; Ozogul, F.; Capasso, R. Microbiota alteration and modulation in Alzheimer’s disease by gerobiotics: The gut-health axis for a good mind. Biomed. Pharmacother. 2022, 153, 113430. [Google Scholar] [CrossRef]
- Agagunduz, D.; Gencer Bingol, F.; Celik, E.; Cemali, O.; Ozenir, C.; Ozogul, F.; Capasso, R. Recent developments in the probiotics as live biotherapeutic products (LBPs) as modulators of gut brain axis related neurological conditions. J. Transl. Med. 2022, 20, 460. [Google Scholar] [CrossRef]
- Varesi, A.; Pierella, E.; Romeo, M.; Piccini, G.B.; Alfano, C.; Bjorklund, G.; Oppong, A.; Ricevuti, G.; Esposito, C.; Chirumbolo, S.; et al. The Potential Role of Gut Microbiota in Alzheimer’s Disease: From Diagnosis to Treatment. Nutrients 2022, 14, 668. [Google Scholar] [CrossRef]
- Szablewski, L. Human Gut Microbiota in Health and Alzheimer’s Disease. J. Alzheimers Dis. 2018, 62, 549–560. [Google Scholar] [CrossRef]
- Kowalski, K.; Mulak, A. Brain-Gut-Microbiota Axis in Alzheimer’s Disease. J. Neurogastroenterol. Motil. 2019, 25, 48–60. [Google Scholar] [CrossRef]
- Alzheimer’s Association. 2020 Alzheimer’s disease facts and figures. Alzheimers Dement 2020, 16, 391–460. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Nahed, P.; Kambar, M.; Zhong, K.; Fonseca, J.; Taghva, K. Alzheimer’s disease drug development pipeline: 2022. Alzheimers Dement. 2022, 8, e12295. [Google Scholar] [CrossRef]
- Abdalqadir, N.; Adeli, K. GLP-1 and GLP-2 Orchestrate Intestine Integrity, Gut Microbiota, and Immune System Crosstalk. Microorganisms 2022, 10, 2061. [Google Scholar]
- Lazar, V.; Ditu, L.M.; Pircalabioru, G.G.; Gheorghe, I.; Curutiu, C.; Holban, A.M.; Picu, A.; Petcu, L.; Chifiriuc, M.C. Aspects of Gut Microbiota and Immune System Interactions in Infectious Diseases, Immunopathology, and Cancer. Front. Immunol. 2018, 9, 1830. [Google Scholar]
- Ciabattini, A.; Olivieri, R.; Lazzeri, E.; Medaglini, D. Role of the Microbiota in the Modulation of Vaccine Immune Responses. Front. Microbiol. 2019, 10, 1305. [Google Scholar] [CrossRef]
- Yoo, J.Y.; Groer, M.; Dutra, S.V.O.; Sarkar, A.; McSkimming, D.I. Gut Microbiota and Immune System Interactions. Microorganisms 2020, 8, 1587. [Google Scholar] [CrossRef]
- Chaput, N.; Lepage, P.; Coutzac, C.; Soularue, E.; Le Roux, K.; Monot, C.; Boselli, L.; Routier, E.; Cassard, L.; Collins, M.; et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann. Oncol. 2017, 28, 1368–1379. [Google Scholar] [CrossRef]
- Frankel, A.E.; Coughlin, L.A.; Kim, J.; Froehlich, T.W.; Xie, Y.; Frenkel, E.P.; Koh, A.Y. Metagenomic Shotgun Sequencing and Unbiased Metabolomic Profiling Identify Specific Human Gut Microbiota and Metabolites Associated with Immune Checkpoint Therapy Efficacy in Melanoma Patients. Neoplasia 2017, 19, 848–855. [Google Scholar] [CrossRef]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillere, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef]
- Guo, Z.; Peng, X.; Li, H.Y.; Wang, Y.; Qian, Y.; Wang, Z.; Ye, D.; Ji, X.; Wang, Z.; Wang, Y.; et al. Evaluation of Peripheral Immune Dysregulation in Alzheimer’s Disease and Vascular Dementia. J. Alzheimers Dis. 2019, 71, 1175–1186. [Google Scholar] [CrossRef]
- Zhuang, Z.Q.; Shen, L.L.; Li, W.W.; Fu, X.; Zeng, F.; Gui, L.; Lu, Y.; Cai, M.; Zhu, C.; Tan, Y.L.; et al. Gut Microbiota is Altered in Patients with Alzheimer’s Disease. J. Alzheimers Dis. 2018, 63, 1337–1346. [Google Scholar] [CrossRef]
- Vogt, N.M.; Kerby, R.L.; Dill-McFarland, K.A.; Harding, S.J.; Merluzzi, A.P.; Johnson, S.C.; Carlsson, C.M.; Asthana, S.; Zetterberg, H.; Blennow, K.; et al. Gut microbiome alterations in Alzheimer’s disease. Sci. Rep. 2017, 7, 13537. [Google Scholar] [CrossRef]
- Liu, P.; Wu, L.; Peng, G.; Han, Y.; Tang, R.; Ge, J.; Zhang, L.; Jia, L.; Yue, S.; Zhou, K.; et al. Altered microbiomes distinguish Alzheimer’s disease from amnestic mild cognitive impairment and health in a Chinese cohort. Brain Behav. Immun. 2019, 80, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Cox, L.M.; Schafer, M.J.; Sohn, J.; Vincentini, J.; Weiner, H.L.; Ginsberg, S.D.; Blaser, M.J. Calorie restriction slows age-related microbiota changes in an Alzheimer’s disease model in female mice. Sci. Rep. 2019, 9, 17904. [Google Scholar] [CrossRef] [PubMed]
- Bauerl, C.; Collado, M.C.; Diaz Cuevas, A.; Vina, J.; Perez Martinez, G. Shifts in gut microbiota composition in an APP/PSS1 transgenic mouse model of Alzheimer’s disease during lifespan. Lett. Appl. Microbiol. 2018, 66, 464–471. [Google Scholar] [CrossRef]
- Harach, T.; Marungruang, N.; Duthilleul, N.; Cheatham, V.; Mc Coy, K.D.; Frisoni, G.; Neher, J.J.; Fak, F.; Jucker, M.; Lasser, T.; et al. Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci. Rep. 2017, 7, 41802. [Google Scholar] [CrossRef]
- Bonfili, L.; Cecarini, V.; Berardi, S.; Scarpona, S.; Suchodolski, J.S.; Nasuti, C.; Fiorini, D.; Boarelli, M.C.; Rossi, G.; Eleuteri, A.M. Microbiota modulation counteracts Alzheimer’s disease progression influencing neuronal proteolysis and gut hormones plasma levels. Sci. Rep. 2017, 7, 2426. [Google Scholar] [CrossRef]
- Unger, M.M.; Spiegel, J.; Dillmann, K.U.; Grundmann, D.; Philippeit, H.; Burmann, J.; Fassbender, K.; Schwiertz, A.; Schafer, K.H. Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Park. Relat. Disord. 2016, 32, 66–72. [Google Scholar] [CrossRef]
- Pellegrini, C.; Antonioli, L.; Colucci, R.; Blandizzi, C.; Fornai, M. Interplay among gut microbiota, intestinal mucosal barrier and enteric neuro-immune system: A common path to neurodegenerative diseases? Acta Neuropathol. 2018, 136, 345–361. [Google Scholar] [CrossRef]
- Ghaisas, S.; Maher, J.; Kanthasamy, A. Gut microbiome in health and disease: Linking the microbiome-gut-brain axis and environmental factors in the pathogenesis of systemic and neurodegenerative diseases. Pharmacol. Ther. 2016, 158, 52–62. [Google Scholar] [CrossRef]
- Costello, E.K.; Stagaman, K.; Dethlefsen, L.; Bohannan, B.J.; Relman, D.A. The application of ecological theory toward an understanding of the human microbiome. Science 2012, 336, 1255–1262. [Google Scholar] [CrossRef]
- Li, W.; Deng, Y.; Chu, Q.; Zhang, P. Gut microbiome and cancer immunotherapy. Cancer Lett. 2019, 447, 41–47. [Google Scholar] [CrossRef]
- de Vos, W.M.; de Vos, E.A. Role of the intestinal microbiome in health and disease: From correlation to causation. Nutr. Rev. 2012, 70 (Suppl. 1), S45–S56. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.X.; Wang, Y.P. Gut Microbiota-brain Axis. Chin. Med. J. 2016, 129, 2373–2380. [Google Scholar] [CrossRef] [PubMed]
- Dave, M.; Higgins, P.D.; Middha, S.; Rioux, K.P. The human gut microbiome: Current knowledge, challenges, and future directions. Transl. Res. 2012, 160, 246–257. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, P.W. Changes in the intestinal microbiota from adulthood through to old age. Clin. Microbiol. Infect. 2012, 18 (Suppl. 4), 44–46. [Google Scholar] [CrossRef] [PubMed]
- Grenham, S.; Clarke, G.; Cryan, J.F.; Dinan, T.G. Brain-gut-microbe communication in health and disease. Front. Physiol. 2011, 2, 94. [Google Scholar] [CrossRef] [PubMed]
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Diversity of the human intestinal microbial flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef] [PubMed]
- Penney, N.; Barton, W.; Posma, J.M.; Darzi, A.; Frost, G.; Cotter, P.D.; Holmes, E.; Shanahan, F.; O’Sullivan, O.; Garcia-Perez, I. Investigating the Role of Diet and Exercise in Gut Microbe-Host Cometabolism. mSystems 2020, 5, e00677-20. [Google Scholar] [CrossRef]
- Clarke, S.F.; Murphy, E.F.; O’Sullivan, O.; Lucey, A.J.; Humphreys, M.; Hogan, A.; Hayes, P.; O’Reilly, M.; Jeffery, I.B.; Wood-Martin, R.; et al. Exercise and associated dietary extremes impact on gut microbial diversity. Gut 2014, 63, 1913–1920. [Google Scholar] [CrossRef]
- Burger-van Paassen, N.; Vincent, A.; Puiman, P.J.; van der Sluis, M.; Bouma, J.; Boehm, G.; van Goudoever, J.B.; van Seuningen, I.; Renes, I.B. The regulation of intestinal mucin MUC2 expression by short-chain fatty acids: Implications for epithelial protection. Biochem. J. 2009, 420, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Sound, R. The gut microbiota-brain axis and role of probiotics. In Nutraceuticabs in Brain and Health, 1st ed.; Ghosh D. Elsevier Inc.: Amsterdam, The Netherlands, 2020; Chapter 13; pp. 175–191. [Google Scholar]
- Nell, S.; Suerbaum, S.; Josenhans, C. The impact of the microbiota on the pathogenesis of IBD: Lessons from mouse infection models. Nat. Rev. Microbiol. 2010, 8, 564–577. [Google Scholar] [PubMed]
- Rakoff-Nahoum, S.; Paglino, J.; Eslami-Varzaneh, F.; Edberg, S.; Medzhitov, R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 2004, 118, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Carabotti, M.; Scirocco, A.; Maselli, M.A.; Severi, C. The gut-brain axis: Interactions between enteric microbiota, central and enteric nervous systems. Ann. Gastroenterol. 2015, 28, 203–209. [Google Scholar] [PubMed]
- Appleton, J. The Gut-Brain Axis: Influence of Microbiota on Mood and Mental Health. Integr. Med. 2018, 17, 28–32. [Google Scholar]
- Bonaz, B.; Bazin, T.; Pellissier, S. The Vagus Nerve at the Interface of the Microbiota-Gut-Brain Axis. Front. Neurosci. 2018, 12, 49. [Google Scholar] [CrossRef] [PubMed]
- Bravo, J.A.; Forsythe, P.; Chew, M.V.; Escaravage, E.; Savignac, H.M.; Dinan, T.G.; Bienenstock, J.; Cryan, J.F. Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc. Natl. Acad. Sci. USA 2011, 108, 16050–16055. [Google Scholar] [CrossRef]
- Pellissier, S.; Dantzer, C.; Canini, F.; Mathieu, N.; Bonaz, B. Psychological adjustment and autonomic disturbances in inflammatory bowel diseases and irritable bowel syndrome. Psychoneuroendocrinology 2010, 35, 653–662. [Google Scholar] [CrossRef]
- Bonaz, B.; Sinniger, V.; Pellissier, S. Vagal tone: Effects on sensitivity, motility, and inflammation. Neurogastroenterol. Motil. 2016, 28, 455–462. [Google Scholar] [CrossRef]
- Silva, Y.P.; Bernardi, A.; Frozza, R.L. The Role of Short-Chain Fatty Acids From Gut Microbiota in Gut-Brain Communication. Front. Endocrinol. 2020, 11, 25. [Google Scholar] [CrossRef]
- Diaz Heijtz, R.; Wang, S.; Anuar, F.; Qian, Y.; Bjorkholm, B.; Samuelsson, A.; Hibberd, M.L.; Forssberg, H.; Pettersson, S. Normal gut microbiota modulates brain development and behavior. Proc. Natl. Acad. Sci. USA 2011, 108, 3047–3052. [Google Scholar] [CrossRef]
- Daulatzai, M.A. Chronic functional bowel syndrome enhances gut-brain axis dysfunction, neuroinflammation, cognitive impairment, and vulnerability to dementia. Neurochem. Res. 2014, 39, 624–644. [Google Scholar] [PubMed]
- Fekete, T.; Bencze, D.; Szabo, A.; Csoma, E.; Biro, T.; Bacsi, A.; Pazmandi, K. Regulatory NLRs Control the RLR-Mediated Type I Interferon and Inflammatory Responses in Human Dendritic Cells. Front. Immunol. 2018, 9, 2314. [Google Scholar] [CrossRef] [PubMed]
- Sharon, G.; Sampson, T.R.; Geschwind, D.H.; Mazmanian, S.K. The Central Nervous System and the Gut Microbiome. Cell 2016, 167, 915–932. [Google Scholar] [CrossRef] [PubMed]
- Ojeda, J.; Avila, A.; Vidal, P.M. Gut Microbiota Interaction with the Central Nervous System throughout Life. J. Clin. Med. 2021, 10, 1299. [Google Scholar] [CrossRef] [PubMed]
- Nagu, P.; Parashar, A.; Behl, T.; Mehta, V. Gut Microbiota Composition and Epigenetic Molecular Changes Connected to the Pathogenesis of Alzheimer’s Disease. J. Mol. Neurosci. 2021, 71, 1436–1455. [Google Scholar] [PubMed]
- Zhao, Y.; Jaber, V.; Lukiw, W.J. Gastrointestinal Tract Microbiome-Derived Pro-inflammatory Neurotoxins in Alzheimer’s Disease. J. Aging Sci. 2021, 9 (Suppl. 5), 002. [Google Scholar]
- Zhao, Y.; Jaber, V.; Lukiw, W.J. Secretory Products of the Human GI Tract Microbiome and Their Potential Impact on Alzheimer’s Disease (AD): Detection of Lipopolysaccharide (LPS) in AD Hippocampus. Front. Cell Infect. Microbiol. 2017, 7, 318. [Google Scholar] [CrossRef]
- Zhan, X.; Stamova, B.; Sharp, F.R. Lipopolysaccharide Associates with Amyloid Plaques, Neurons and Oligodendrocytes in Alzheimer’s Disease Brain: A Review. Front. Aging Neurosci. 2018, 10, 42. [Google Scholar] [CrossRef]
- Kim, H.S.; Kim, S.; Shin, S.J.; Park, Y.H.; Nam, Y.; Kim, C.W.; Lee, K.W.; Kim, S.M.; Jung, I.D.; Yang, H.D.; et al. Gram-negative bacteria and their lipopolysaccharides in Alzheimer’s disease: Pathologic roles and therapeutic implications. Transl. Neurodegener. 2021, 10, 49. [Google Scholar]
- Wang, X.; Sun, G.; Feng, T.; Zhang, J.; Huang, X.; Wang, T.; Xie, Z.; Chu, X.; Yang, J.; Wang, H.; et al. Sodium oligomannate therapeutically remodels gut microbiota and suppresses gut bacterial amino acids-shaped neuroinflammation to inhibit Alzheimer’s disease progression. Cell Res. 2019, 29, 787–803. [Google Scholar] [CrossRef]
- Syed, Y.Y. Sodium Oligomannate: First Approval. Drugs 2020, 80, 441–444. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Xu, J.; Ling, Y.; Wang, F.; Gong, T.; Yang, C.; Ye, S.; Ye, K.; Wei, D.; Song, Z.; et al. Fecal microbiota transplantation alleviated Alzheimer’s disease-like pathogenesis in APP/PS1 transgenic mice. Transl. Psychiatry 2019, 9, 189. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Kim, Y.; Choi, H.; Kim, W.; Park, S.; Lee, D.; Kim, D.K.; Kim, H.J.; Choi, H.; Hyun, D.W.; et al. Transfer of a healthy microbiota reduces amyloid and tau pathology in an Alzheimer’s disease animal model. Gut 2020, 69, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Lee, J.H.; Shin, J.; Kim, J.S.; Cha, B.; Lee, S.; Kwon, K.S.; Shin, Y.W.; Choi, S.H. Cognitive function improvement after fecal microbiota transplantation in Alzheimer’s dementia patient: A case report. Curr. Med. Res. Opin. 2021, 37, 1739–1744. [Google Scholar] [CrossRef] [PubMed]
- Hazan, S. Rapid improvement in Alzheimer’s disease symptoms following fecal microbiota transplantation: A case report. J. Int. Med. Res. 2020, 48, 300060520925930. [Google Scholar] [CrossRef] [PubMed]
- Wieckowska-Gacek, A.; Mietelska-Porowska, A.; Wydrych, M.; Wojda, U. Western diet as a trigger of Alzheimer’s disease: From metabolic syndrome and systemic inflammation to neuroinflammation and neurodegeneration. Ageing Res. Rev. 2021, 70, 101397. [Google Scholar] [PubMed]
- Garcia-Mantrana, I.; Selma-Royo, M.; Alcantara, C.; Collado, M.C. Shifts on Gut Microbiota Associated to Mediterranean Diet Adherence and Specific Dietary Intakes on General Adult Population. Front. Microbiol. 2018, 9, 890. [Google Scholar] [CrossRef]
- Casas, R.; Sacanella, E.; Estruch, R. The immune protective effect of the Mediterranean diet against chronic low-grade inflammatory diseases. Endocr. Metab. Immune Disord. Drug Targets 2014, 14, 245–254. [Google Scholar] [CrossRef]
- Nayak, D.; Roth, T.L.; McGavern, D.B. Microglia development and function. Annu. Rev. Immunol. 2014, 32, 367–402. [Google Scholar] [CrossRef]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System during Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Perry, V.H.; Nicoll, J.A.; Holmes, C. Microglia in neurodegenerative disease. Nat. Rev. Neurol. 2010, 6, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Lemloh, M.L.; Fromont, J.; Brummer, F.; Usher, K.M. Diversity and abundance of photosynthetic sponges in temperate Western Australia. BMC Ecol. 2009, 9, 4. [Google Scholar] [CrossRef] [PubMed]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Frost, J.L.; Schafer, D.P. Microglia: Architects of the Developing Nervous System. Trends Cell Biol. 2016, 26, 587–597. [Google Scholar] [CrossRef]
- Thomas, J.O.; Sowa, J.K.; Limburg, B.; Bian, X.; Evangeli, C.; Swett, J.L.; Tewari, S.; Baugh, J.; Schatz, G.C.; Briggs, G.A.D.; et al. Charge transport through extended molecular wires with strongly correlated electrons. Chem. Sci. 2021, 12, 11121–11129. [Google Scholar] [CrossRef]
- Griciuc, A.; Tanzi, R.E. The role of innate immune genes in Alzheimer’s disease. Curr. Opin. Neurol. 2021, 34, 228–236. [Google Scholar] [CrossRef]
- Rivest, S. Regulation of innate immune responses in the brain. Nat. Rev. Immunol. 2009, 9, 429–439. [Google Scholar] [CrossRef]
- Ahmed, M.M.; Johnson, N.R.; Boyd, T.D.; Coughlan, C.; Chial, H.J.; Potter, H. Innate Immune System Activation and Neuroinflammation in down Syndrome and Neurodegeneration: Therapeutic Targets or Partners? Front. Aging Neurosci. 2021, 13, 718426. [Google Scholar] [CrossRef]
- Burgaletto, C.; Munafo, A.; Di Benedetto, G.; De Francisci, C.; Caraci, F.; Di Mauro, R.; Bucolo, C.; Bernardini, R.; Cantarella, G. The immune system on the TRAIL of Alzheimer’s disease. J. Neuroinflammation 2020, 17, 298. [Google Scholar] [CrossRef]
- Nadeau, S.; Rivest, S. Role of microglial-derived tumor necrosis factor in mediating CD14 transcription and nuclear factor kappa B activity in the brain during endotoxemia. J. Neurosci. 2000, 20, 3456–3468. [Google Scholar] [CrossRef]
- Nguyen, M.D.; Julien, J.P.; Rivest, S. Innate immunity: The missing link in neuroprotection and neurodegeneration? Nat. Rev. Neurosci. 2002, 3, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Le Page, A.; Dupuis, G.; Frost, E.H.; Larbi, A.; Pawelec, G.; Witkowski, J.M.; Fulop, T. Role of the peripheral innate immune system in the development of Alzheimer’s disease. Exp. Gerontol. 2018, 107, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Li, G.; Huang, P.; Liu, Z.; Zhao, B. The Gut Microbiota and Alzheimer’s Disease. J. Alzheimers Dis. 2017, 58, 1–15. [Google Scholar] [CrossRef]
- Boutajangout, A.; Wisniewski, T. The innate immune system in Alzheimer’s disease. Int. J. Cell Biol. 2013, 2013, 576383. [Google Scholar] [CrossRef] [PubMed]
- Lannfelt, L.; Relkin, N.R.; Siemers, E.R. Amyloid-ss-directed immunotherapy for Alzheimer’s disease. J. Intern. Med. 2014, 275, 284–295. [Google Scholar] [CrossRef] [PubMed]
- DeMattos, R.B.; Bales, K.R.; Cummins, D.J.; Dodart, J.C.; Paul, S.M.; Holtzman, D.M. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2001, 98, 8850–8855. [Google Scholar] [CrossRef]
- Gelinas, D.S.; DaSilva, K.; Fenili, D.; St George-Hyslop, P.; McLaurin, J. Immunotherapy for Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101 (Suppl. 2), 14657–14662. [Google Scholar] [CrossRef]
- Ghersi-Egea, J.F.; Gorevic, P.D.; Ghiso, J.; Frangione, B.; Patlak, C.S.; Fenstermacher, J.D. Fate of cerebrospinal fluid-borne amyloid beta-peptide: Rapid clearance into blood and appreciable accumulation by cerebral arteries. J. Neurochem. 1996, 67, 880–883. [Google Scholar] [CrossRef]
- Poduslo, J.F.; Curran, G.L.; Sanyal, B.; Selkoe, D.J. Receptor-mediated transport of human amyloid beta-protein 1–40 and 1–42 at the blood-brain barrier. Neurobiol. Dis. 1999, 6, 190–199. [Google Scholar] [CrossRef]
- Shibata, M.; Yamada, S.; Kumar, S.R.; Calero, M.; Bading, J.; Frangione, B.; Holtzman, D.M.; Miller, C.A.; Strickland, D.K.; Ghiso, J.; et al. Clearance of Alzheimer’s amyloid-ss(1–40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J. Clin. Investig. 2000, 106, 1489–1499. [Google Scholar]
- Bard, F.; Cannon, C.; Barbour, R.; Burke, R.L.; Games, D.; Grajeda, H.; Guido, T.; Hu, K.; Huang, J.; Johnson-Wood, K.; et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat. Med. 2000, 6, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Bacskai, B.J.; Kajdasz, S.T.; McLellan, M.E.; Games, D.; Seubert, P.; Schenk, D.; Hyman, B.T. Non-Fc-mediated mechanisms are involved in clearance of amyloid-beta in vivo by immunotherapy. J. Neurosci. 2002, 22, 7873–7878. [Google Scholar] [CrossRef] [PubMed]
- Das, P.; Howard, V.; Loosbrock, N.; Dickson, D.; Murphy, M.P.; Golde, T.E. Amyloid-beta immunization effectively reduces amyloid deposition in FcRgamma-/- knock-out mice. J. Neurosci. 2003, 23, 8532–8538. [Google Scholar] [CrossRef] [PubMed]
- Frenkel, D.; Katz, O.; Solomon, B. Immunization against Alzheimer’s beta-amyloid plaques via EFRH phage administration. Proc. Natl. Acad. Sci. USA 2000, 97, 11455–11459. [Google Scholar] [CrossRef] [PubMed]
- McLaurin, J.; Cecal, R.; Kierstead, M.E.; Tian, X.; Phinney, A.L.; Manea, M.; French, J.E.; Lambermon, M.H.; Darabie, A.A.; Brown, M.E.; et al. Therapeutically effective antibodies against amyloid-beta peptide target amyloid-beta residues 4–10 and inhibit cytotoxicity and fibrillogenesis. Nat. Med. 2002, 8, 1263–1269. [Google Scholar] [CrossRef]
- Solomon, B.; Koppel, R.; Hanan, E.; Katzav, T. Monoclonal antibodies inhibit in vitro fibrillar aggregation of the Alzheimer beta-amyloid peptide. Proc. Natl. Acad. Sci. USA 1996, 93, 452–455. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.A. Antiviral medications. Orthop. Nurs. 1996, 15, 82–91. [Google Scholar] [CrossRef]
- Arriagada, P.V.; Growdon, J.H.; Hedley-Whyte, E.T.; Hyman, B.T. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 1992, 42, 63163–63169. [Google Scholar] [CrossRef]
- Giannakopoulos, P.; Herrmann, F.R.; Bussiere, T.; Bouras, C.; Kovari, E.; Perl, D.P.; Morrison, J.H.; Gold, G.; Hof, P.R. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology 2003, 60, 1495–1500. [Google Scholar] [CrossRef]
- Sebastian-Serrano, A.; de Diego-Garcia, L.; Diaz-Hernandez, M. The Neurotoxic Role of Extracellular Tau Protein. Int. J. Mol. Sci. 2018, 19, 998. [Google Scholar] [CrossRef]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 5–21. [Google Scholar] [PubMed]
- Katsinelos, T.; Zeitler, M.; Dimou, E.; Karakatsani, A.; Muller, H.M.; Nachman, E.; Steringer, J.P.; Ruiz de Almodovar, C.; Nickel, W.; Jahn, T.R. Unconventional Secretion Mediates the Trans-cellular Spreading of Tau. Cell Rep. 2018, 23, 2039–2055. [Google Scholar] [CrossRef] [PubMed]
- Bright, J.; Hussain, S.; Dang, V.; Wright, S.; Cooper, B.; Byun, T.; Ramos, C.; Singh, A.; Parry, G.; Stagliano, N.; et al. Human secreted tau increases amyloid-beta production. Neurobiol. Aging 2015, 36, 693–709. [Google Scholar] [CrossRef] [PubMed]
- Pooler, A.M.; Phillips, E.C.; Lau, D.H.; Noble, W.; Hanger, D.P. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 2013, 14, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.W.; Hussaini, S.A.; Bastille, I.M.; Rodriguez, G.A.; Mrejeru, A.; Rilett, K.; Sanders, D.W.; Cook, C.; Fu, H.; Boonen, R.A.; et al. Neuronal activity enhances tau propagation and tau pathology in vivo. Nat. Neurosci. 2016, 19, 1085–1092. [Google Scholar] [CrossRef]
- Yamada, K.; Holth, J.K.; Liao, F.; Stewart, F.R.; Mahan, T.E.; Jiang, H.; Cirrito, J.R.; Patel, T.K.; Hochgrafe, K.; Mandelkow, E.M.; et al. Neuronal activity regulates extracellular tau in vivo. J. Exp. Med. 2014, 211, 387–393. [Google Scholar] [CrossRef]
- Blomberg, M.; Jensen, M.; Basun, H.; Lannfelt, L.; Wahlund, L.O. Cerebrospinal fluid tau levels increase with age in healthy individuals. Dement. Geriatr. Cogn. Disord. 2001, 12, 127–132. [Google Scholar] [CrossRef]
- Hampel, H.; Blennow, K. CSF tau and beta-amyloid as biomarkers for mild cognitive impairment. Dialogues Clin. Neurosci. 2004, 6, 379–390. [Google Scholar] [CrossRef]
- Okonkwo, O.C.; Alosco, M.L.; Griffith, H.R.; Mielke, M.M.; Shaw, L.M.; Trojanowski, J.Q.; Tremont, G.; Alzheimer’s Disease Neuroimaging Initiative. Cerebrospinal fluid abnormalities and rate of decline in everyday function across the dementia spectrum: Normal aging, mild cognitive impairment, and Alzheimer disease. Arch. Neurol. 2010, 67, 688–696. [Google Scholar] [CrossRef]
- Sato, C.; Barthelemy, N.R.; Mawuenyega, K.G.; Patterson, B.W.; Gordon, B.A.; Jockel-Balsarotti, J.; Sullivan, M.; Crisp, M.J.; Kasten, T.; Kirmess, K.M.; et al. Tau Kinetics in Neurons and the Human Central Nervous System. Neuron 2018, 97, 1284–1298.e7. [Google Scholar] [CrossRef]
- Sjogren, M.; Davidsson, P.; Tullberg, M.; Minthon, L.; Wallin, A.; Wikkelso, C.; Granerus, A.K.; Vanderstichele, H.; Vanmechelen, E.; Blennow, K. Both total and phosphorylated tau are increased in Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2001, 70, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Henriques, A.D.; Benedet, A.L.; Camargos, E.F.; Rosa-Neto, P.; Nobrega, O.T. Fluid and imaging biomarkers for Alzheimer’s disease: Where we stand and where to head to. Exp. Gerontol. 2018, 107, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.L.; Tung, Y.C.; Liu, F.; Gong, C.X.; Iqbal, K. Tau passive immunization inhibits not only tau but also Abeta pathology. Alzheimers Res. Ther. 2017, 9, 1. [Google Scholar] [CrossRef]
- Asuni, A.A.; Boutajangout, A.; Quartermain, D.; Sigurdsson, E.M. Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J. Neurosci. 2007, 27, 9115–9129. [Google Scholar] [CrossRef]
- Gu, J.; Congdon, E.E.; Sigurdsson, E.M. Two novel Tau antibodies targeting the 396/404 region are primarily taken up by neurons and reduce Tau protein pathology. J. Biol. Chem. 2013, 288, 33081–33095. [Google Scholar] [CrossRef] [PubMed]
- Collin, L.; Bohrmann, B.; Gopfert, U.; Oroszlan-Szovik, K.; Ozmen, L.; Gruninger, F. Neuronal uptake of tau/pS422 antibody and reduced progression of tau pathology in a mouse model of Alzheimer’s disease. Brain 2014, 137, 2834–2846. [Google Scholar] [CrossRef] [PubMed]
- Congdon, E.E.; Gu, J.; Sait, H.B.; Sigurdsson, E.M. Antibody uptake into neurons occurs primarily via clathrin-dependent Fcgamma receptor endocytosis and is a prerequisite for acute tau protein clearance. J. Biol. Chem. 2013, 288, 35452–35465. [Google Scholar] [CrossRef]
- Krishnamurthy, P.K.; Deng, Y.; Sigurdsson, E.M. Mechanistic Studies of Antibody-Mediated Clearance of Tau Aggregates Using an ex vivo Brain Slice Model. Front. Psychiatry 2011, 2, 59. [Google Scholar] [CrossRef]
- Watkinson, R.E.; McEwan, W.A.; James, L.C. Intracellular antibody immunity. J. Clin. Immunol. 2014, 34 (Suppl. 1), S30–S34. [Google Scholar] [CrossRef]
- Shamir, D.B.; Rosenqvist, N.; Rasool, S.; Pedersen, J.T.; Sigurdsson, E.M. Internalization of tau antibody and pathological tau protein detected with a flow cytometry multiplexing approach. Alzheimers Dement. 2016, 12, 1098–1107. [Google Scholar] [CrossRef]
- Pedersen, J.T.; Sigurdsson, E.M. Tau immunotherapy for Alzheimer’s disease. Trends Mol. Med. 2015, 21, 394–402. [Google Scholar] [PubMed]
- Mohamed, N.V.; Herrou, T.; Plouffe, V.; Piperno, N.; Leclerc, N. Spreading of tau pathology in Alzheimer’s disease by cell-to-cell transmission. Eur. J. Neurosci. 2013, 37, 1939–1948. [Google Scholar] [CrossRef]
- Avila, J.; Simon, D.; Diaz-Hernandez, M.; Pintor, J.; Hernandez, F. Sources of extracellular tau and its signaling. J. Alzheimers Dis. 2014, 40 (Suppl. 1), S7–S15. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K. The pathological process underlying Alzheimer’s disease in individuals under thirty. Acta Neuropathol. 2011, 121, 171–181. [Google Scholar]
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M.; et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 2009, 11, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Clavaguera, F.; Akatsu, H.; Fraser, G.; Crowther, R.A.; Frank, S.; Hench, J.; Probst, A.; Winkler, D.T.; Reichwald, J.; Staufenbiel, M.; et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc. Natl. Acad. Sci. USA 2013, 110, 9535–9540. [Google Scholar] [CrossRef] [PubMed]
- Iba, M.; Guo, J.L.; McBride, J.D.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M. Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J. Neurosci. 2013, 33, 1024–1037. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Drouet, V.; Wu, J.W.; Witter, M.P.; Small, S.A.; Clelland, C.; Duff, K. Trans-synaptic spread of tau pathology in vivo. PLoS ONE 2012, 7, e31302. [Google Scholar] [CrossRef]
- de Calignon, A.; Polydoro, M.; Suarez-Calvet, M.; William, C.; Adamowicz, D.H.; Kopeikina, K.J.; Pitstick, R.; Sahara, N.; Ashe, K.H.; Carlson, G.A.; et al. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron 2012, 73, 685–697. [Google Scholar] [CrossRef]
- Frost, B.; Jacks, R.L.; Diamond, M.I. Propagation of tau misfolding from the outside to the inside of a cell. J. Biol. Chem. 2009, 284, 12845–12852. [Google Scholar] [CrossRef] [PubMed]
- Sankaranarayanan, S.; Barten, D.M.; Vana, L.; Devidze, N.; Yang, L.; Cadelina, G.; Hoque, N.; DeCarr, L.; Keenan, S.; Lin, A.; et al. Passive immunization with phospho-tau antibodies reduces tau pathology and functional deficits in two distinct mouse tauopathy models. PLoS ONE 2015, 10, e0125614. [Google Scholar] [CrossRef] [PubMed]
- Yanamandra, K.; Kfoury, N.; Jiang, H.; Mahan, T.E.; Ma, S.; Maloney, S.E.; Wozniak, D.F.; Diamond, M.I.; Holtzman, D.M. Anti-tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron 2013, 80, 402–414. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Miao, J.; Chu, D.; Jin, N.; Tung, Y.C.; Dai, C.L.; Hu, W.; Gong, C.X.; Iqbal, K.; Liu, F. Tau antibody 77G7 targeting microtubule binding domain suppresses proteopathic tau to seed tau aggregation. CNS. Neurosci. Ther. 2022, 28, 2245–2259. [Google Scholar] [CrossRef]
- Dai, C.L.; Hu, W.; Tung, Y.C.; Liu, F.; Gong, C.X.; Iqbal, K. Tau passive immunization blocks seeding and spread of Alzheimer hyperphosphorylated Tau-induced pathology in 3 × Tg-AD mice. Alzheimers Res. Ther. 2018, 10, 13. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussiere, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Vander Zanden, C.M.; Chi, E.Y. Passive Immunotherapies Targeting Amyloid Beta and Tau Oligomers in Alzheimer’s Disease. J. Pharm. Sci. 2020, 109, 68–73. [Google Scholar] [CrossRef]
- Swanson, C.J.; Zhang, Y.; Dhadda, S.; Wang, J.; Kaplow, J.; Lai, R.Y.K.; Lannfelt, L.; Bradley, H.; Rabe, M.; Koyama, A.; et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Abeta protofibril antibody. Alzheimers Res. Ther. 2021, 13, 80. [Google Scholar] [CrossRef]
- Guthrie, H.; Honig, L.S.; Lin, H.; Sink, K.M.; Blondeau, K.; Quartino, A.; Dolton, M.; Carrasco-Triguero, M.; Lian, Q.; Bittner, T.; et al. Safety, Tolerability, and Pharmacokinetics of Crenezumab in Patients with Mild-to-Moderate Alzheimer’s Disease Treated with Escalating Doses for up to 133 Weeks. J. Alzheimers Dis. 2020, 76, 967–979. [Google Scholar] [CrossRef]
- Manolopoulos, A.; Andreadis, P.; Malandris, K.; Avgerinos, I.; Karagiannis, T.; Kapogiannis, D.; Tsolaki, M.; Tsapas, A.; Bekiari, E. Intravenous Immunoglobulin for Patients with Alzheimer’s Disease: A Systematic Review and Meta-Analysis. Am. J. Alzheimers Dis. Other Dement. 2019, 34, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Pistollato, F.; Sumalla Cano, S.; Elio, I.; Masias Vergara, M.; Giampieri, F.; Battino, M. Role of gut microbiota and nutrients in amyloid formation and pathogenesis of Alzheimer disease. Nutr. Rev. 2016, 74, 624–634. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Liao, J.; Xia, Y.; Liu, X.; Jones, R.; Haran, J.; McCormick, B.; Sampson, T.R.; Alam, A.; Ye, K. Gut microbiota regulate Alzheimer’s disease pathologies and cognitive disorders via PUFA-associated neuroinflammation. Gut 2022, 71, 2233–2252. [Google Scholar] [CrossRef] [PubMed]
- Minter, M.R.; Hinterleitner, R.; Meisel, M.; Zhang, C.; Leone, V.; Zhang, X.; Oyler-Castrillo, P.; Zhang, X.; Musch, M.W.; Shen, X.; et al. Antibiotic-induced perturbations in microbial diversity during post-natal development alters amyloid pathology in an aged APPSWE/PS1DeltaE9 murine model of Alzheimer’s disease. Sci. Rep. 2017, 7, 10411. [Google Scholar] [CrossRef] [PubMed]
- Rajamohamedsait, H.; Rasool, S.; Rajamohamedsait, W.; Lin, Y.; Sigurdsson, E.M. Prophylactic Active Tau Immunization Leads to Sustained Reduction in Both Tau and Amyloid-beta Pathologies in 3 × Tg Mice. Sci. Rep. 2017, 7, 17034. [Google Scholar] [CrossRef] [PubMed]
- Counts, S.E.; Perez, S.E.; He, B.; Mufson, E.J. Intravenous immunoglobulin reduces tau pathology and preserves neuroplastic gene expression in the 3 × Tg mouse model of Alzheimer’s disease. Curr. Alzheimer Res. 2014, 11, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Sudduth, T.L.; Greenstein, A.; Wilcock, D.M. Intracranial injection of Gammagard, a human IVIg, modulates the inflammatory response of the brain and lowers Abeta in APP/PS1 mice along a different time course than anti-Abeta antibodies. J. Neurosci. 2013, 33, 9684–9692. [Google Scholar] [CrossRef] [PubMed]
- Counts, S.E.; Ray, B.; Mufson, E.J.; Perez, S.E.; He, B.; Lahiri, D.K. Intravenous immunoglobulin (IVIG) treatment exerts antioxidant and neuropreservatory effects in preclinical models of Alzheimer’s disease. J. Clin. Immunol. 2014, 34 (Suppl. 1), S80–S85. [Google Scholar] [CrossRef]
- Barbara Kubickova, P.B.; Klára, H.; Lenka, Š. Effects of cyanobacterial toxins on the human gastrointestinal tract and the mucosal innate immune system. Environ. Sci. Eur. 2019, 31, 31. [Google Scholar] [CrossRef]
- Shin, N.R.; Whon, T.W.; Bae, J.W. Proteobacteria: Microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef]
- Guo, S.; Nighot, M.; Al-Sadi, R.; Alhmoud, T.; Nighot, P.; Ma, T.Y. Lipopolysaccharide Regulation of Intestinal Tight Junction Permeability Is Mediated by TLR4 Signal Transduction Pathway Activation of FAK and MyD88. J. Immunol. 2015, 195, 4999–5010. [Google Scholar] [CrossRef]
- Nighot, M.; Al-Sadi, R.; Guo, S.; Rawat, M.; Nighot, P.; Watterson, M.D.; Ma, T.Y. Lipopolysaccharide-Induced Increase in Intestinal Epithelial Tight Permeability Is Mediated by Toll-Like Receptor 4/Myeloid Differentiation Primary Response 88 (MyD88) Activation of Myosin Light Chain Kinase Expression. Am. J. Pathol. 2017, 187, 2698–2710. [Google Scholar] [CrossRef] [PubMed]
- Andrews, M.C.; Vasanthakumar, A. Gut microbiota—A double-edged sword in cancer immunotherapy. Trends Cancer 2022. [Google Scholar] [CrossRef]
- Davar, D.; Dzutsev, A.K.; McCulloch, J.A.; Rodrigues, R.R.; Chauvin, J.M.; Morrison, R.M.; Deblasio, R.N.; Menna, C.; Ding, Q.; Pagliano, O.; et al. Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science 2021, 371, 595–602. [Google Scholar] [CrossRef]
- Olejnik, B.; Koziol, A.; Brzozowska, E.; Ferens-Sieczkowska, M. Application of selected biosensor techniques in clinical diagnostics. Expert. Rev. Mol. Diagn 2021, 21, 925–937. [Google Scholar] [CrossRef] [PubMed]
- Leeming, E.R.; Johnson, A.J.; Spector, T.D.; Le Roy, C.I. Effect of Diet on the Gut Microbiota: Rethinking Intervention Duration. Nutrients 2019, 11, 2862. [Google Scholar] [CrossRef]
- Iqbal, K. Thinking beyond the Aducanumab Controversy. Ann. Neurol. 2021, 90, 1003–1004. [Google Scholar] [CrossRef] [PubMed]
- Knorz, A.L.; Quante, A. Alzheimer’s Disease: Efficacy of Mono- and Combination Therapy. A Systematic Review. J. Geriatr. Psychiatry Neurol. 2022, 35, 475–486. [Google Scholar] [CrossRef]
- Salloway, S.P.; Sevingy, J.; Budur, K.; Pederson, J.T.; DeMattos, R.B.; Von Rosenstiel, P.; Paez, A.; Evans, R.; Weber, C.J.; Hendrix, J.A.; et al. Advancing combination therapy for Alzheimer’s disease. Alzheimers Dement. 2020, 6, e12073. [Google Scholar] [CrossRef]
- Gauthier, S.; Alam, J.; Fillit, H.; Iwatsubo, T.; Liu-Seifert, H.; Sabbagh, M.; Salloway, S.; Sampaio, C.; Sims, J.R.; Sperling, B.; et al. Combination Therapy for Alzheimer’s Disease: Perspectives of the EU/US CTAD Task Force. J. Prev. Alzheimers Dis. 2019, 6, 164–168. [Google Scholar] [CrossRef]
- Gong, C.X.; Dai, C.L.; Liu, F.; Iqbal, K. Multi-Targets: An Unconventional Drug Development Strategy for Alzheimer’s Disease. Front. Aging Neurosci. 2022, 14, 837649. [Google Scholar] [CrossRef]

{kind=link}
| Agent | Immunization | Target | Phase | Study Population | Clinical Trial # | Status | Sponsor(s) |
|---|---|---|---|---|---|---|---|
| ABvac40 | Active | Aβ (33–40) | Phase II | Amnestic mild cognitive impairment or very mild AD | NCT03461276 | Active, not recruiting | Araclon Biotech S.L. |
| Aducanumab (BIIB037) | Passive | Aβ (3–7) | Phase III | Early AD | NCT04241068 | Active, not recruiting | Biogen |
| Phase IV | Early AD | NCT05310071 | Recruiting | Biogen | |||
| Phase I | Mild cognitive impairment or mild AD | NCT05469009 | Recruiting | Ali Rezai, InSightec | |||
| Gantenerumab (RO4909832) | Passive | Aβ (2–11 and 18–27) | Phase II | Early AD | NCT04592341 | Active, not recruiting | Roche |
| Phase III | Early AD | NCT03443973 | Active, not recruiting | Roche | |||
| Phase III | Early AD | NCT03444870 | Recruiting | Roche | |||
| Phase III | Early AD | NCT04339413 | Active, not recruiting | Roche | |||
| Phase III | With risk for AD or early AD | NCT05256134 | Recruiting | Roche | |||
| Phase III | Prodromal to mild AD | NCT04374253 | Recruiting | Roche | |||
| Phase III | With risk for or with early AD caused by a genetic mutation | NCT05552157 | Not yet recruiting | WUSM, Roche, AA, NIA Genentech, Inc. | |||
| Donanemab (LY3002813) | Passive | Pyrogluta-mate Aβ (p3–7) | Phase I | Healthy | NCT05567159 | Not yet recruiting | Eli Lilly |
| Phase I | Healthy Chinese participants | NCT05533411 | Not yet recruiting | Eli Lilly | |||
| Phase III | Early symptomatic AD | NCT04437511 | Active, not recruiting | Eli Lilly | |||
| Phase III | Preclinical AD | NCT05026866 | Recruiting | Eli Lilly | |||
| Phase II | Symptomatic AD | NCT04640077 | Active, not recruiting | Eli Lilly | |||
| Phase III | Early symptomatic AD | NCT05508789 | Not yet recruiting | Eli Lilly | |||
| Lecanemab (BAN2401) | Passive | Aβ protofibrils (1–16) | Phase I | Healthy | NCT05533801 | Not yet recruiting | Eisai. |
| Phase III | Preclinical AD | NCT04468659 | Recruiting | Eisai. ACTC, Biogen, NIA | |||
| Phase III | Early AD | NCT03887455 | Active, not recruiting | Eisai. Biogen | |||
| Phase II | Early AD | NCT01767311 | Active, not recruiting | Eisai. Biogen | |||
| Solanezumab (LY2062430) | Passive | Aβ protofibrils (1–16) | Phase III | With risk for memory loss | NCT02008357 | Active, not recruiting | Eli Lilly, ATRI |
| RO7126209 * | Passive | Aβ fibrils | Phase I/II | Prodromal or mild to moderate AD | NCT04639050 | Recruiting | Roche |
| Crenezumab (MABT5102A) | Passive | Soluble Aβ oligomers (13–24) | Phase II | Preclinical autosomal dominant AD with PSEN1 E280A mutation | NCT01998841 | Active, not recruiting | Genentech, Inc. NIA, Banner Alzheimer’s Institute |
| SHR−1707 | Passive | Aβ | Phase I | Healthy young adult and elderly | NCT04973189 | Recruiting | Shanghai Hengrui Pharmaceutical Co. |
| LY3372993 | Passive | Pyrogluta-mated form of Aβ | Phase I | Healthy and AD | NCT04451408 | Recruiting | Eli Lilly |
| Phase III | Early AD | NCT05463731 | Recruiting | Eli Lilly | |||
| ACU193 | Passive | Soluble Aβ oligomers | Phase I | MCI or mild AD | NCT04931459 | Recruiting | Acumen Pharmaceuticals, NIA |
| Agent | Immunization | Target | Phase | Study Population | Clinical Trial # | Status | Sponsor(s) |
|---|---|---|---|---|---|---|---|
| ACI−35 | Active | Tau 393–408 (pS396/S404) | Phase II | Early AD | NCT04445831 | Active, not recruiting | AC Immune; Janssen |
| Bepranemab (UCB0107) | Passive | Tau (235–250) | Phase II | Mild cognitive impairment or mild AD | NCT04867616 | Active, not recruiting | UCB Biopharma SRL |
| E2814 | Passive | Tau (273–291, 296–314) | Phase II | Mild to moderate AD | NCT04971733 | Active, not recruiting | Eisai. |
| Phase I | Healthy | NCT04231513 | Recruiting | Eisai. | |||
| JNJ−63733657 | Passive | Tau204–225 (pTau212/217) | Phase I | Healthy Chinese participants | NCT05407818 | Recruiting | Janssen |
| Semorinemab (RO7105705) | Passive | Tau (2–24) | Phase II | Moderate AD | NCT03828747 | Active, not recruiting | Genentech, Inc. |
| Lu AF87908 | Passive | Tau386–408 (pS396/S404) | Phase IPhase II | Healthy and AD Early AD | NCT04149860 NCT04619420 | Recruiting Recruiting | H. Lundbeck A/S Janssen |
| Agents | Immunization | Target | Phase | Study Population | Clinical Trial # | Status | Sponsor(s) |
|---|---|---|---|---|---|---|---|
| Gantenerumab & Solanezumab | Passive | Aβ (2–11, 18–27, and 16–26) | Phase III | With risk for or with early onset AD caused by genetic mutation | NCT01760005 | Recruiting | WUSM; Eli Lilly, Roche; AA; NIA; Avid Radiopharmaceuticals, AMP |
| Donanemab & Aducanumab | Passive | p3–7 and Aβ (3–7) | Phase III | Early symptomatic AD | NCT05108922 | Active, not recruiting | Eli Lilly |
| Lecanemab & E2814 | Passive | Aβ protofibrils (1–16) and Tau (273–291, 296–314) | Phase III | Early onset AD caused by genetic mutation | NCT05269394 | Recruiting | WUSM, NIA, AMP, Eisai, AA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dai, C.-L.; Liu, F.; Iqbal, K.; Gong, C.-X. Gut Microbiota and Immunotherapy for Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 15230. https://doi.org/10.3390/ijms232315230
Dai C-L, Liu F, Iqbal K, Gong C-X. Gut Microbiota and Immunotherapy for Alzheimer’s Disease. International Journal of Molecular Sciences. 2022; 23(23):15230. https://doi.org/10.3390/ijms232315230
Chicago/Turabian StyleDai, Chun-Ling, Fei Liu, Khalid Iqbal, and Cheng-Xin Gong. 2022. "Gut Microbiota and Immunotherapy for Alzheimer’s Disease" International Journal of Molecular Sciences 23, no. 23: 15230. https://doi.org/10.3390/ijms232315230
APA StyleDai, C.-L., Liu, F., Iqbal, K., & Gong, C.-X. (2022). Gut Microbiota and Immunotherapy for Alzheimer’s Disease. International Journal of Molecular Sciences, 23(23), 15230. https://doi.org/10.3390/ijms232315230