
The Role of Epigenetics in Neuroinflammatory-Driven Diseases

 , , , , , ,
, , , , , ,  ,
,  ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Epigenetic Alterations in Alzheimer’s Disease (AD)
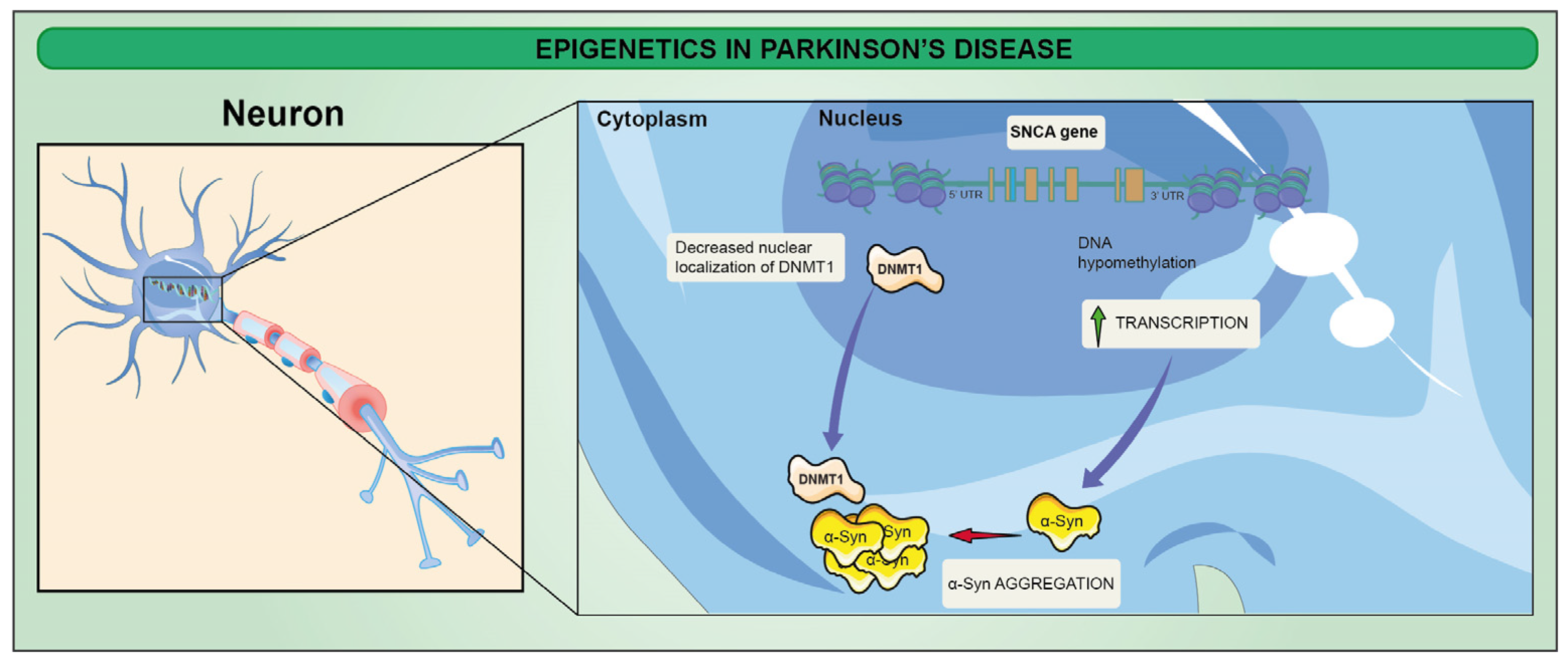
3. Epigenetic Alterations in Parkinson’s Disease (PD)
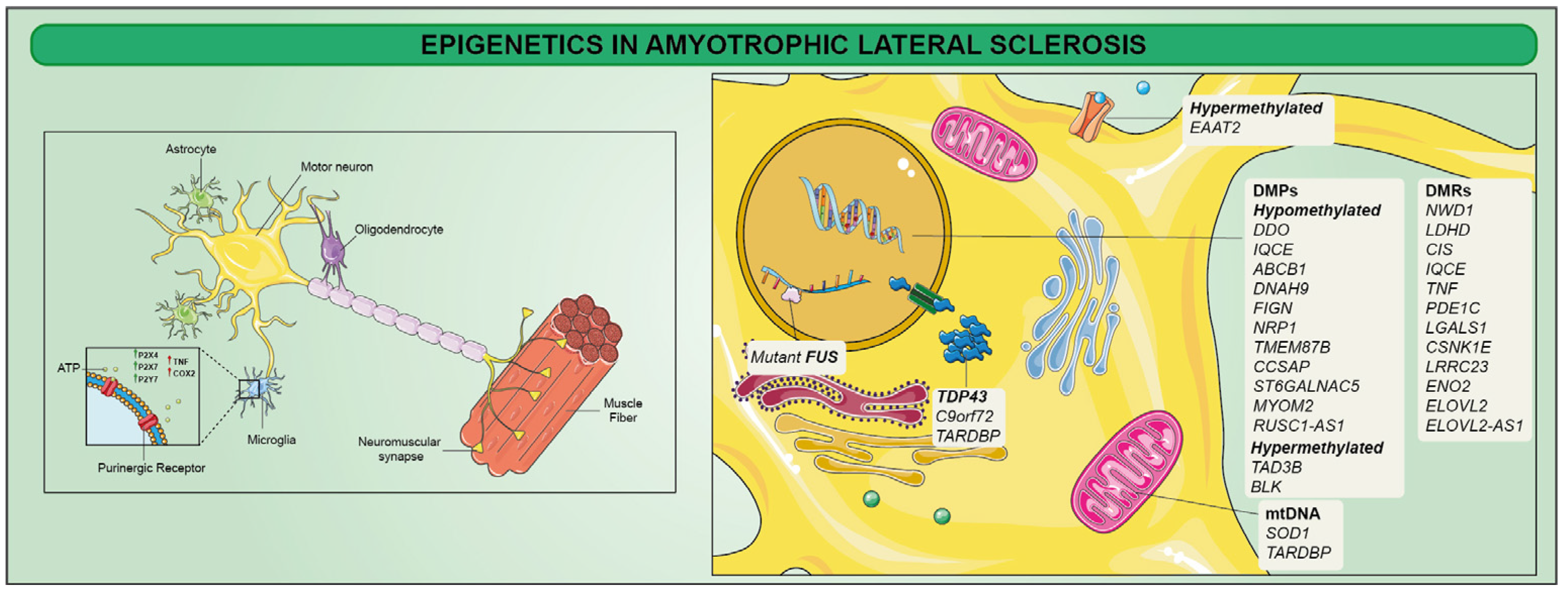
4. Epigenetics in Amyotrophic Lateral Sclerosis (ALS)
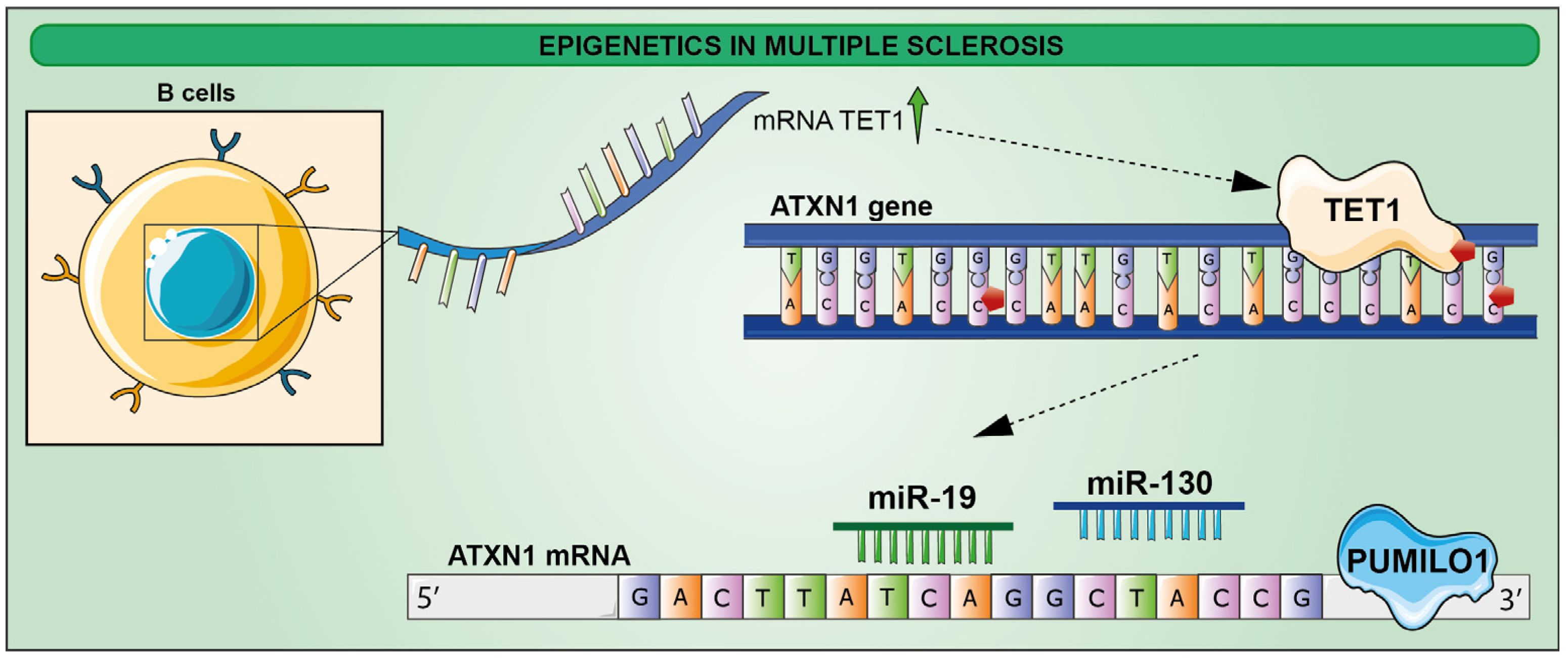
5. Epigenetics in Multiple Sclerosis (MS)
6. Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Dugger, B.N.; Dickson, D.W. Pathology of Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef] [PubMed]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Ghezzi, P.; Bernardini, R.; Giuffrida, R.; Bellomo, M.; Manzoni, C.; Comoletti, D.; Di Santo, E.; Benigni, F.; Mennini, T. Tumor necrosis factor is increased in the spinal cord of an animal model of motor neuron degeneration. Eur. Cytokine Netw. 1998, 9, 139–144. [Google Scholar]
- Vicario, N.; Castrogiovanni, P.; Imbesi, R.; Giallongo, S.; Mannino, G.; Furno, D.L.; Giuffrida, R.; Zappala, A.; Li Volti, G.; Tibullo, D.; et al. GJA1/CX43 High Expression Levels in the Cervical Spinal Cord of ALS Patients Correlate to Microglia-Mediated Neuroinflammatory Profile. Biomedicines 2022, 10, 2246. [Google Scholar] [CrossRef] [PubMed]
- Vicario, N.; Parenti, R. Connexins Signatures of the Neurovascular Unit and Their Physio-Pathological Functions. Int. J. Mol. Sci. 2022, 23, 9510. [Google Scholar] [CrossRef]
- Mannino, G.; Russo, C.; Maugeri, G.; Musumeci, G.; Vicario, N.; Tibullo, D.; Giuffrida, R.; Parenti, R.; Lo Furno, D. Adult stem cell niches for tissue homeostasis. J. Cell Physiol. 2022, 237, 239–257. [Google Scholar] [CrossRef]
- Mannino, G.; Vicario, N.; Parenti, R.; Giuffrida, R.; Lo Furno, D. Connexin expression decreases during adipogenic differentiation of human adipose-derived mesenchymal stem cells. Mol. Biol. Rep. 2020, 47, 9951–9958. [Google Scholar] [CrossRef]
- Spitale, F.M.; Vicario, N.; Rosa, M.D.; Tibullo, D.; Vecchio, M.; Gulino, R.; Parenti, R. Increased expression of connexin 43 in a mouse model of spinal motoneuronal loss. Aging 2020, 12, 12598–12608. [Google Scholar] [CrossRef]
- Kasongo, D.W.; de Leo, G.; Vicario, N.; Leanza, G.; Legname, G. Chronic alpha-Synuclein Accumulation in Rat Hippocampus Induces Lewy Bodies Formation and Specific Cognitive Impairments. eNeuro 2020, 7, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Gulino, R.; Vicario, N.; Giunta, M.A.S.; Spoto, G.; Calabrese, G.; Vecchio, M.; Gulisano, M.; Leanza, G.; Parenti, R. Neuromuscular Plasticity in a Mouse Neurotoxic Model of Spinal Motoneuronal Loss. Int. J. Mol. Sci. 2019, 20, 1500. [Google Scholar] [CrossRef]
- Vicario, N.; Calabrese, G.; Zappala, A.; Parenti, C.; Forte, S.; Graziano, A.C.E.; Vanella, L.; Pellitteri, R.; Cardile, V.; Parenti, R. Inhibition of Cx43 mediates protective effects on hypoxic/reoxygenated human neuroblastoma cells. J. Cell Mol. Med. 2017, 21, 2563–2572. [Google Scholar] [CrossRef] [PubMed]
- Vicario, N.; Turnaturi, R.; Spitale, F.M.; Torrisi, F.; Zappala, A.; Gulino, R.; Pasquinucci, L.; Chiechio, S.; Parenti, C.; Parenti, R. Intercellular communication and ion channels in neuropathic pain chronicization. Inflamm. Res. 2020, 69, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Lo Furno, D.; Mannino, G.; Giuffrida, R.; Gili, E.; Vancheri, C.; Tarico, M.S.; Perrotta, R.E.; Pellitteri, R. Neural differentiation of human adipose-derived mesenchymal stem cells induced by glial cell conditioned media. J. Cell Physiol. 2018, 233, 7091–7100. [Google Scholar] [CrossRef] [PubMed]
- Mannino, G.; Gennuso, F.; Giurdanella, G.; Conti, F.; Drago, F.; Salomone, S.; Furno, D.L.; Bucolo, C.; Giuffrida, R. Pericyte-like differentiation of human adipose-derived mesenchymal stem cells: An in vitro study. World J. Stem Cells 2020, 12, 1152–1170. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, G.; Giuffrida, R.; Lo Furno, D.; Parrinello, N.L.; Forte, S.; Gulino, R.; Colarossi, C.; Schinocca, L.R.; Giuffrida, R.; Cardile, V.; et al. Potential Effect of CD271 on Human Mesenchymal Stromal Cell Proliferation and Differentiation. Int. J. Mol. Sci. 2015, 16, 15609–15624. [Google Scholar] [CrossRef]
- Panto, M.R.; Cicirata, F.; Angaut, P.; Parenti, R.; Serapide, F. The projection from the primary motor and somatic sensory cortex to the basilar pontine nuclei. A detailed electrophysiological and anatomical study in the rat. J. Hirnforsch. 1995, 36, 7–19. [Google Scholar]
- Fagone, E.; Conte, E.; Gili, E.; Fruciano, M.; Pistorio, M.P.; Lo Furno, D.; Giuffrida, R.; Crimi, N.; Vancheri, C. Resveratrol inhibits transforming growth factor-beta-induced proliferation and differentiation of ex vivo human lung fibroblasts into myofibroblasts through ERK/Akt inhibition and PTEN restoration. Exp. Lung Res. 2011, 37, 162–174. [Google Scholar] [CrossRef]
- Gijselinck, I.; Van Mossevelde, S.; van der Zee, J.; Sieben, A.; Engelborghs, S.; De Bleecker, J.; Ivanoiu, A.; Deryck, O.; Edbauer, D.; Zhang, M.; et al. The C9orf72 repeat size correlates with onset age of disease, DNA methylation and transcriptional downregulation of the promoter. Mol. Psychiatry 2016, 21, 1112–1124. [Google Scholar] [CrossRef]
- Hinz, F.I.; Geschwind, D.H. Molecular Genetics of Neurodegenerative Dementias. Cold Spring Harb. Perspect. Biol. 2017, 9, a023705. [Google Scholar] [CrossRef]
- Tcw, J.; Goate, A.M. Genetics of beta-Amyloid Precursor Protein in Alzheimer’s Disease. Cold Spring Harb. Perspect. Med. 2017, 7, a024539. [Google Scholar] [CrossRef]
- Ghasemi, M.; Brown, R.H., Jr. Genetics of Amyotrophic Lateral Sclerosis. Cold Spring Harb. Perspect. Med. 2018, 8, a024125. [Google Scholar] [CrossRef] [PubMed]
- Voglein, J.; Kostova, I.; Arzberger, T.; Roeber, S.; Schmitz, P.; Simons, M.; Ruf, V.; Windl, O.; Herms, J.; Dieterich, M.; et al. First symptom guides diagnosis and prognosis in neurodegenerative diseases-a retrospective study of autopsy proven cases. Eur. J. Neurol. 2021, 28, 1801–1811. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes Dev. 2009, 23, 781–783. [Google Scholar] [CrossRef] [PubMed]
- Giallongo, S.; Lo Re, O.; Vinciguerra, M. Macro Histone Variants: Emerging Rheostats of Gastrointestinal Cancers. Cancers 2019, 11, 676. [Google Scholar] [CrossRef]
- Giallongo, S.; Lo Re, O.; Lochmanova, G.; Parca, L.; Petrizzelli, F.; Zdrahal, Z.; Mazza, T.; Vinciguerra, M. Phosphorylation within Intrinsic Disordered Region Discriminates Histone Variant macroH2A1 Splicing Isoforms-macroH2A1.1 and macroH2A1.2. Biology 2021, 10, 659. [Google Scholar] [CrossRef]
- Giallongo, S.; Rehakova, D.; Biagini, T.; Lo Re, O.; Raina, P.; Lochmanova, G.; Zdrahal, Z.; Resnick, I.; Pata, P.; Pata, I.; et al. Histone Variant macroH2A1.1 Enhances Nonhomologous End Joining-dependent DNA Double-strand-break Repair and Reprogramming Efficiency of Human iPSCs. Stem Cells 2022, 40, 35–48. [Google Scholar] [CrossRef]
- Giallongo, S.; Rehakova, D.; Raffaele, M.; Lo Re, O.; Koutna, I.; Vinciguerra, M. Redox and Epigenetics in Human Pluripotent Stem Cells Differentiation. Antioxid. Redox Signal. 2021, 34, 335–349. [Google Scholar] [CrossRef]
- Hu, B.; Won, H.; Mah, W.; Park, R.B.; Kassim, B.; Spiess, K.; Kozlenkov, A.; Crowley, C.A.; Pochareddy, S.; Psych, E.C.; et al. Neuronal and glial 3D chromatin architecture informs the cellular etiology of brain disorders. Nat. Commun. 2021, 12, 3968. [Google Scholar] [CrossRef]
- Mannino, G.; Cristaldi, M.; Giurdanella, G.; Perrotta, R.E.; Lo Furno, D.; Giuffrida, R.; Rusciano, D. ARPE-19 conditioned medium promotes neural differentiation of adipose-derived mesenchymal stem cells. World J. Stem Cells 2021, 13, 1783–1796. [Google Scholar] [CrossRef]
- Lo Furno, D.; Mannino, G.; Pellitteri, R.; Zappala, A.; Parenti, R.; Gili, E.; Vancheri, C.; Giuffrida, R. Conditioned Media From Glial Cells Promote a Neural-Like Connexin Expression in Human Adipose-Derived Mesenchymal Stem Cells. Front. Physiol. 2018, 9, 1742. [Google Scholar] [CrossRef]
- Lo Furno, D.; Pellitteri, R.; Graziano, A.C.; Giuffrida, R.; Vancheri, C.; Gili, E.; Cardile, V. Differentiation of human adipose stem cells into neural phenotype by neuroblastoma- or olfactory ensheathing cells-conditioned medium. J. Cell Physiol. 2013, 228, 2109–2118. [Google Scholar] [CrossRef] [PubMed]
- Lo Furno, D.; Mannino, G.; Giuffrida, R. Functional role of mesenchymal stem cells in the treatment of chronic neurodegenerative diseases. J. Cell Physiol. 2018, 233, 3982–3999. [Google Scholar] [CrossRef] [PubMed]
- Russo, C.; Mannino, G.; Patane, M.; Parrinello, N.L.; Pellitteri, R.; Stanzani, S.; Giuffrida, R.; Lo Furno, D.; Russo, A. Ghrelin peptide improves glial conditioned medium effects on neuronal differentiation of human adipose mesenchymal stem cells. Histochem. Cell Biol. 2021, 156, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Lo Furno, D.; Mannino, G.; Cardile, V.; Parenti, R.; Giuffrida, R. Potential Therapeutic Applications of Adipose-Derived Mesenchymal Stem Cells. Stem Cells Dev. 2016, 25, 1615–1628. [Google Scholar] [CrossRef] [PubMed]
- Mannino, G.; Russo, C.; Longo, A.; Anfuso, C.D.; Lupo, G.; Lo Furno, D.; Giuffrida, R.; Giurdanella, G. Potential therapeutic applications of mesenchymal stem cells for the treatment of eye diseases. World J. Stem Cells 2021, 13, 632–644. [Google Scholar] [CrossRef]
- Lo Furno, D.; Graziano, A.C.; Caggia, S.; Perrotta, R.E.; Tarico, M.S.; Giuffrida, R.; Cardile, V. Decrease of apoptosis markers during adipogenic differentiation of mesenchymal stem cells from human adipose tissue. Apoptosis 2013, 18, 578–588. [Google Scholar] [CrossRef]
- Hwang, J.Y.; Aromolaran, K.A.; Zukin, R.S. The emerging field of epigenetics in neurodegeneration and neuroprotection. Nat. Rev. Neurosci. 2017, 18, 347–361. [Google Scholar] [CrossRef]
- Miller, C.A.; Sweatt, J.D. Covalent modification of DNA regulates memory formation. Neuron 2007, 53, 857–869. [Google Scholar] [CrossRef]
- Day, J.J.; Sweatt, J.D. DNA methylation and memory formation. Nat. Neurosci. 2010, 13, 1319–1323. [Google Scholar] [CrossRef]
- Feng, J.; Zhou, Y.; Campbell, S.L.; Le, T.; Li, E.; Sweatt, J.D.; Silva, A.J.; Fan, G. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat. Neurosci. 2010, 13, 423–430. [Google Scholar] [CrossRef]
- Hayaishi, O. Enzymic hydroxylation. Annu. Rev. Biochem. 1969, 38, 21–44. [Google Scholar] [CrossRef] [PubMed]
- Ploumakis, A.; Coleman, M.L. OH, the Places You’ll Go! Hydroxylation, Gene Expression, and Cancer. Mol. Cell 2015, 58, 729–741. [Google Scholar] [CrossRef] [PubMed]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Kaas, G.A.; Zhong, C.; Eason, D.E.; Ross, D.L.; Vachhani, R.V.; Ming, G.L.; King, J.R.; Song, H.; Sweatt, J.D. TET1 controls CNS 5-methylcytosine hydroxylation, active DNA demethylation, gene transcription, and memory formation. Neuron 2013, 79, 1086–1093. [Google Scholar] [CrossRef]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.B.; Gao, Y.; et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef]
- Levine, M.E.; Lu, A.T.; Quach, A.; Chen, B.H.; Assimes, T.L.; Bandinelli, S.; Hou, L.; Baccarelli, A.A.; Stewart, J.D.; Li, Y.; et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging 2018, 10, 573–591. [Google Scholar] [CrossRef]
- Marioni, R.E.; Shah, S.; McRae, A.F.; Chen, B.H.; Colicino, E.; Harris, S.E.; Gibson, J.; Henders, A.K.; Redmond, P.; Cox, S.R.; et al. DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol. 2015, 16, 25. [Google Scholar] [CrossRef]
- Grodstein, F.; Lemos, B.; Yu, L.; Iatrou, A.; De Jager, P.L.; Bennett, D.A. Characteristics of Epigenetic Clocks Across Blood and Brain Tissue in Older Women and Men. Front. Neurosci. 2020, 14, 555307. [Google Scholar] [CrossRef]
- Shireby, G.L.; Davies, J.P.; Francis, P.T.; Burrage, J.; Walker, E.M.; Neilson, G.W.A.; Dahir, A.; Thomas, A.J.; Love, S.; Smith, R.G.; et al. Recalibrating the epigenetic clock: Implications for assessing biological age in the human cortex. Brain 2020, 143, 3763–3775. [Google Scholar] [CrossRef]
- Raneros, A.B.; Bernet, C.R.; Florez, A.B.; Suarez-Alvarez, B. An Epigenetic Insight into NLRP3 Inflammasome Activation in Inflammation-Related Processes. Biomedicines 2021, 9, 1614. [Google Scholar] [CrossRef] [PubMed]
- Fang, P.; Chen, C.; Zheng, F.; Jia, J.; Chen, T.; Zhu, J.; Chang, J.; Zhang, Z. NLRP3 inflammasome inhibition by histone acetylation ameliorates sevoflurane-induced cognitive impairment in aged mice by activating the autophagy pathway. Brain Res. Bull. 2021, 172, 79–88. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Wang, L.; Chen, K.; Wan, X.; Wang, F.; Guo, Z.; Mo, Z. NLRP3 inflammasome activation in mesenchymal stem cells inhibits osteogenic differentiation and enhances adipogenic differentiation. Biochem. Biophys. Res. Commun. 2017, 484, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Baardman, J.; Licht, I.; de Winther, M.P.; Van den Bossche, J. Metabolic-epigenetic crosstalk in macrophage activation. Epigenomics 2015, 7, 1155–1164. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Sananbenesi, F.; Wang, X.; Dobbin, M.; Tsai, L.H. Recovery of learning and memory is associated with chromatin remodelling. Nature 2007, 447, 178–182. [Google Scholar] [CrossRef]
- Levenson, J.M.; O’Riordan, K.J.; Brown, K.D.; Trinh, M.A.; Molfese, D.L.; Sweatt, J.D. Regulation of histone acetylation during memory formation in the hippocampus. J. Biol. Chem. 2004, 279, 40545–40559. [Google Scholar] [CrossRef]
- Bourtchouladze, R.; Lidge, R.; Catapano, R.; Stanley, J.; Gossweiler, S.; Romashko, D.; Scott, R.; Tully, T. A mouse model of Rubinstein-Taybi syndrome: Defective long-term memory is ameliorated by inhibitors of phosphodiesterase 4. Proc. Natl. Acad. Sci. USA 2003, 100, 10518–10522. [Google Scholar] [CrossRef]
- Gulino, R.; Forte, S.; Parenti, R.; Memeo, L.; Gulisano, M. MicroRNA and pediatric tumors: Future perspectives. Acta Histochem. 2015, 117, 339–354. [Google Scholar] [CrossRef]
- Stevanovic, M.; Stanisavljevic Ninkovic, D.; Mojsin, M.; Drakulic, D.; Schwirtlich, M. Interplay of SOX transcription factors and microRNAs in the brain under physiological and pathological conditions. Neural Regen. Res. 2022, 17, 2325–2334. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, X.; Yang, K.; Shao, Y.; Gu, R.; Liu, X.; Liu, H.; Liu, Y.; Zhou, Y. Macrophage-derived apoptotic vesicles regulate fate commitment of mesenchymal stem cells via miR155. Stem Cell Res. Ther. 2022, 13, 323. [Google Scholar] [CrossRef] [PubMed]
- Pounders, J.; Hill, E.J.; Hooper, D.; Zhang, X.; Biesiada, J.; Kuhnell, D.; Greenland, H.L.; Esfandiari, L.; Timmerman, E.; Foster, F.; et al. MicroRNA expression within neuronal-derived small extracellular vesicles in frontotemporal degeneration. Medicine 2022, 101, e30854. [Google Scholar] [CrossRef]
- Sufianov, A.; Begliarzade, S.; Ilyasova, T.; Xu, X.; Beylerli, O. MicroRNAs as potential diagnostic markers of glial brain tumors. Noncoding RNA Res. 2022, 7, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Shan, B.Q.; Zhao, H.Y.; He, H.; Tian, M.L.; Cheng, X.; Qin, J.B.; Jin, G.H. MiR-130a-3p regulates neural stem cell differentiation in vitro by targeting Acsl4. J. Cell Mol. Med. 2022, 26, 2717–2727. [Google Scholar] [CrossRef] [PubMed]
- Estrada-Meza, C.; Torres-Copado, A.; Loreti Gonzalez-Melgoza, L.; Ruiz-Manriquez, L.M.; De Donato, M.; Sharma, A.; Pathak, S.; Banerjee, A.; Paul, S. Recent insights into the microRNA and long non-coding RNA-mediated regulation of stem cell populations. 3 Biotech 2022, 12, 270. [Google Scholar] [CrossRef] [PubMed]
- Naseer, S.; Abelleira-Hervas, L.; Savani, D.; de Burgh, R.; Aleksynas, R.; Donat, C.K.; Syed, N.; Sastre, M. Traumatic Brain Injury Leads to Alterations in Contusional Cortical miRNAs Involved in Dementia. Biomolecules 2022, 12, 1457. [Google Scholar] [CrossRef] [PubMed]
- Schratt, G. microRNAs at the synapse. Nat. Rev. Neurosci. 2009, 10, 842–849. [Google Scholar] [CrossRef]
- Woldemichael, B.T.; Mansuy, I.M. Micro-RNAs in cognition and cognitive disorders: Potential for novel biomarkers and therapeutics. Biochem. Pharmacol. 2016, 104, 1–7. [Google Scholar] [CrossRef]
- Packer, A.N.; Xing, Y.; Harper, S.Q.; Jones, L.; Davidson, B.L. The bifunctional microRNA miR-9/miR-9* regulates REST and CoREST and is downregulated in Huntington’s disease. J. Neurosci. 2008, 28, 14341–14346. [Google Scholar] [CrossRef]
- Schiffer, D.; Caldera, V.; Mellai, M.; Conforti, P.; Cattaneo, E.; Zuccato, C. Repressor element-1 silencing transcription factor (REST) is present in human control and Huntington’s disease neurones. Neuropathol. Appl. Neurobiol. 2014, 40, 899–910. [Google Scholar] [CrossRef]
- Tsai, M.C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef] [PubMed]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Murgas, P.; Godoy, B.; von Bernhardi, R. Abeta potentiates inflammatory activation of glial cells induced by scavenger receptor ligands and inflammatory mediators in culture. Neurotox. Res. 2012, 22, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Mandelkow, E.M.; Mandelkow, E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb. Perspect. Med. 2012, 2, a006247. [Google Scholar] [CrossRef]
- Hardy, J.; Duff, K.; Hardy, K.G.; Perez-Tur, J.; Hutton, M. Genetic dissection of Alzheimer’s disease and related dementias: Amyloid and its relationship to tau. Nat. Neurosci. 1998, 1, 355–358. [Google Scholar] [CrossRef]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef]
- Allen, N.J.; Eroglu, C. Cell Biology of Astrocyte-Synapse Interactions. Neuron 2017, 96, 697–708. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef]
- Batiuk, M.Y.; Martirosyan, A.; Wahis, J.; de Vin, F.; Marneffe, C.; Kusserow, C.; Koeppen, J.; Viana, J.F.; Oliveira, J.F.; Voet, T.; et al. Identification of region-specific astrocyte subtypes at single cell resolution. Nat. Commun. 2020, 11, 1220. [Google Scholar] [CrossRef]
- Endo, F.; Kasai, A.; Soto, J.S.; Yu, X.; Qu, Z.; Hashimoto, H.; Gradinaru, V.; Kawaguchi, R.; Khakh, B.S. Molecular basis of astrocyte diversity and morphology across the CNS in health and disease. Science 2022, 378, eadc9020. [Google Scholar] [CrossRef] [PubMed]
- Swardfager, W.; Lanctot, K.; Rothenburg, L.; Wong, A.; Cappell, J.; Herrmann, N. A meta-analysis of cytokines in Alzheimer’s disease. Biol. Psychiatry 2010, 68, 930–941. [Google Scholar] [CrossRef] [PubMed]
- El Kadmiri, N.; Said, N.; Slassi, I.; El Moutawakil, B.; Nadifi, S. Biomarkers for Alzheimer Disease: Classical and Novel Candidates’ Review. Neuroscience 2018, 370, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Edison, P.; Brooks, D.J. Role of Neuroinflammation in the Trajectory of Alzheimer’s Disease and in vivo Quantification Using PET. J. Alzheimers Dis. 2018, 64, S339–S351. [Google Scholar] [CrossRef] [PubMed]
- Beaino, W.; Janssen, B.; Kooij, G.; van der Pol, S.M.A.; van Het Hof, B.; van Horssen, J.; Windhorst, A.D.; de Vries, H.E. Purinergic receptors P2Y12R and P2X7R: Potential targets for PET imaging of microglia phenotypes in multiple sclerosis. J. Neuroinflamm. 2017, 14, 259. [Google Scholar] [CrossRef]
- Narayanaswami, V.; Dahl, K.; Bernard-Gauthier, V.; Josephson, L.; Cumming, P.; Vasdev, N. Emerging PET Radiotracers and Targets for Imaging of Neuroinflammation in Neurodegenerative Diseases: Outlook Beyond TSPO. Mol. Imaging 2018, 17, 1536012118792317. [Google Scholar] [CrossRef]
- Husain, M.A.; Laurent, B.; Plourde, M. APOE and Alzheimer’s Disease: From Lipid Transport to Physiopathology and Therapeutics. Front. Neurosci. 2021, 15, 630502. [Google Scholar] [CrossRef]
- Mise, A.; Yoshino, Y.; Yamazaki, K.; Ozaki, Y.; Sao, T.; Yoshida, T.; Mori, T.; Mori, Y.; Ochi, S.; Iga, J.I.; et al. TOMM40 and APOE Gene Expression and Cognitive Decline in Japanese Alzheimer’s Disease Subjects. J. Alzheimers Dis. 2017, 60, 1107–1117. [Google Scholar] [CrossRef]
- Lee, E.G.; Tulloch, J.; Chen, S.; Leong, L.; Saxton, A.D.; Kraemer, B.; Darvas, M.; Keene, C.D.; Shutes-David, A.; Todd, K.; et al. Redefining transcriptional regulation of the APOE gene and its association with Alzheimer’s disease. PLoS ONE 2020, 15, e0227667. [Google Scholar] [CrossRef]
- Shao, Y.; Shaw, M.; Todd, K.; Khrestian, M.; D’Aleo, G.; Barnard, P.J.; Zahratka, J.; Pillai, J.; Yu, C.E.; Keene, C.D.; et al. DNA methylation of TOMM40-APOE-APOC2 in Alzheimer’s disease. J. Hum. Genet. 2018, 63, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Nagata, T.; Kobayashi, N.; Ishii, J.; Shinagawa, S.; Nakayama, R.; Shibata, N.; Kuerban, B.; Ohnuma, T.; Kondo, K.; Arai, H.; et al. Association between DNA Methylation of the BDNF Promoter Region and Clinical Presentation in Alzheimer’s Disease. Dement. Geriatr. Cogn. Dis. Extra 2015, 5, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Nicolia, V.; Ciraci, V.; Cavallaro, R.A.; Ferrer, I.; Scarpa, S.; Fuso, A. GSK3beta 5′-flanking DNA Methylation and Expression in Alzheimer’s Disease Patients. Curr. Alzheimer. Res. 2017, 14, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, Y.; Yoshino, Y.; Yamazaki, K.; Sao, T.; Mori, Y.; Ochi, S.; Yoshida, T.; Mori, T.; Iga, J.I.; Ueno, S.I. DNA methylation changes at TREM2 intron 1 and TREM2 mRNA expression in patients with Alzheimer’s disease. J. Psychiatr. Res. 2017, 92, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.R.; Smith, R.G.; Burrage, J.; Troakes, C.; Al-Sarraj, S.; Kalaria, R.N.; Sloan, C.; Robinson, A.C.; Mill, J.; Lunnon, K. A cross-brain regions study of ANK1 DNA methylation in different neurodegenerative diseases. Neurobiol. Aging 2019, 74, 70–76. [Google Scholar] [CrossRef]
- Semick, S.A.; Bharadwaj, R.A.; Collado-Torres, L.; Tao, R.; Shin, J.H.; Deep-Soboslay, A.; Weiss, J.R.; Weinberger, D.R.; Hyde, T.M.; Kleinman, J.E.; et al. Integrated DNA methylation and gene expression profiling across multiple brain regions implicate novel genes in Alzheimer’s disease. Acta Neuropathol. 2019, 137, 557–569. [Google Scholar] [CrossRef]
- Villela, D.; Ramalho, R.F.; Silva, A.R.; Brentani, H.; Suemoto, C.K.; Pasqualucci, C.A.; Grinberg, L.T.; Krepischi, A.C.; Rosenberg, C. Differential DNA Methylation of MicroRNA Genes in Temporal Cortex from Alzheimer’s Disease Individuals. Neural Plast. 2016, 2016, 2584940. [Google Scholar] [CrossRef]
- Watson, C.T.; Roussos, P.; Garg, P.; Ho, D.J.; Azam, N.; Katsel, P.L.; Haroutunian, V.; Sharp, A.J. Genome-wide DNA methylation profiling in the superior temporal gyrus reveals epigenetic signatures associated with Alzheimer’s disease. Genome Med. 2016, 8, 5. [Google Scholar] [CrossRef]
- Li, P.; Marshall, L.; Oh, G.; Jakubowski, J.L.; Groot, D.; He, Y.; Wang, T.; Petronis, A.; Labrie, V. Epigenetic dysregulation of enhancers in neurons is associated with Alzheimer’s disease pathology and cognitive symptoms. Nat. Commun. 2019, 10, 2246. [Google Scholar] [CrossRef]
- Coppieters, N.; Dieriks, B.V.; Lill, C.; Faull, R.L.; Curtis, M.A.; Dragunow, M. Global changes in DNA methylation and hydroxymethylation in Alzheimer’s disease human brain. Neurobiol. Aging 2014, 35, 1334–1344. [Google Scholar] [CrossRef]
- Fetahu, I.S.; Ma, D.; Rabidou, K.; Argueta, C.; Smith, M.; Liu, H.; Wu, F.; Shi, Y.G. Epigenetic signatures of methylated DNA cytosine in Alzheimer’s disease. Sci. Adv. 2019, 5, eaaw2880. [Google Scholar] [CrossRef] [PubMed]
- Hroudova, J.; Singh, N.; Fisar, Z. Mitochondrial dysfunctions in neurodegenerative diseases: Relevance to Alzheimer’s disease. Biomed. Res. Int. 2014, 2014, 175062. [Google Scholar] [CrossRef] [PubMed]
- Shock, L.S.; Thakkar, P.V.; Peterson, E.J.; Moran, R.G.; Taylor, S.M. DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc. Natl. Acad. Sci. USA 2011, 108, 3630–3635. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.A.; Hyun, J.W. Oxidative Stress, Nrf2, and Epigenetic Modification Contribute to Anticancer Drug Resistance. Toxicol. Res. 2017, 33, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Blanch, M.; Mosquera, J.L.; Ansoleaga, B.; Ferrer, I.; Barrachina, M. Altered Mitochondrial DNA Methylation Pattern in Alzheimer Disease-Related Pathology and in Parkinson Disease. Am. J. Pathol. 2016, 186, 385–397. [Google Scholar] [CrossRef]
- Stoccoro, A.; Siciliano, G.; Migliore, L.; Coppede, F. Decreased Methylation of the Mitochondrial D-Loop Region in Late-Onset Alzheimer’s Disease. J. Alzheimers Dis. 2017, 59, 559–564. [Google Scholar] [CrossRef]
- Mastroeni, D.; Grover, A.; Delvaux, E.; Whiteside, C.; Coleman, P.D.; Rogers, J. Epigenetic mechanisms in Alzheimer’s disease. Neurobiol. Aging 2011, 32, 1161–1180. [Google Scholar] [CrossRef]
- Frost, B.; Hemberg, M.; Lewis, J.; Feany, M.B. Tau promotes neurodegeneration through global chromatin relaxation. Nat. Neurosci. 2014, 17, 357–366. [Google Scholar] [CrossRef]
- Marzi, S.J.; Leung, S.K.; Ribarska, T.; Hannon, E.; Smith, A.R.; Pishva, E.; Poschmann, J.; Moore, K.; Troakes, C.; Al-Sarraj, S.; et al. A histone acetylome-wide association study of Alzheimer’s disease identifies disease-associated H3K27ac differences in the entorhinal cortex. Nat. Neurosci. 2018, 21, 1618–1627. [Google Scholar] [CrossRef]
- Klein, H.U.; McCabe, C.; Gjoneska, E.; Sullivan, S.E.; Kaskow, B.J.; Tang, A.; Smith, R.V.; Xu, J.; Pfenning, A.R.; Bernstein, B.E.; et al. Epigenome-wide study uncovers large-scale changes in histone acetylation driven by tau pathology in aging and Alzheimer’s human brains. Nat. Neurosci. 2019, 22, 37–46. [Google Scholar] [CrossRef]
- Berson, A.; Nativio, R.; Berger, S.L.; Bonini, N.M. Epigenetic Regulation in Neurodegenerative Diseases. Trends Neurosci. 2018, 41, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Stoccoro, A.; Coppede, F. Role of epigenetics in Alzheimer’s disease pathogenesis. Neurodegener. Dis. Manag. 2018, 8, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jiao, B.; Shen, L. The Epigenetics of Alzheimer’s Disease: Factors and Therapeutic Implications. Front. Genet. 2018, 9, 579. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Sheng, S.; Qin, C. The role of HDAC6 in Alzheimer’s disease. J. Alzheimers Dis. 2013, 33, 283–295. [Google Scholar] [CrossRef]
- Xu, K.; Dai, X.L.; Huang, H.C.; Jiang, Z.F. Targeting HDACs: A promising therapy for Alzheimer’s disease. Oxid. Med. Cell. Longev. 2011, 2011, 143269. [Google Scholar] [CrossRef]
- Lu, X.; Wang, L.; Yu, C.; Yu, D.; Yu, G. Histone Acetylation Modifiers in the Pathogenesis of Alzheimer’s Disease. Front. Cell. Neurosci. 2015, 9, 226. [Google Scholar] [CrossRef]
- Konsoula, Z.; Barile, F.A. Epigenetic histone acetylation and deacetylation mechanisms in experimental models of neurodegenerative disorders. J. Pharmacol. Toxicol. Methods 2012, 66, 215–220. [Google Scholar] [CrossRef]
- Nikolac Perkovic, M.; Videtic Paska, A.; Konjevod, M.; Kouter, K.; Svob Strac, D.; Nedic Erjavec, G.; Pivac, N. Epigenetics of Alzheimer’s Disease. Biomolecules 2021, 11, 195. [Google Scholar] [CrossRef]
- Fischer, A. Targeting histone-modifications in Alzheimer’s disease. What is the evidence that this is a promising therapeutic avenue? Neuropharmacology 2014, 80, 95–102. [Google Scholar] [CrossRef]
- Herrera-Espejo, S.; Santos-Zorrozua, B.; Alvarez-Gonzalez, P.; Lopez-Lopez, E.; Garcia-Orad, A. A Systematic Review of MicroRNA Expression as Biomarker of Late-Onset Alzheimer’s Disease. Mol. Neurobiol. 2019, 56, 8376–8391. [Google Scholar] [CrossRef]
- Holohan, K.N.; Lahiri, D.K.; Schneider, B.P.; Foroud, T.; Saykin, A.J. Functional microRNAs in Alzheimer’s disease and cancer: Differential regulation of common mechanisms and pathways. Front. Genet. 2012, 3, 323. [Google Scholar] [CrossRef]
- Chang, F.; Zhang, L.H.; Xu, W.P.; Jing, P.; Zhan, P.Y. microRNA-9 attenuates amyloidbeta-induced synaptotoxicity by targeting calcium/calmodulin-dependent protein kinase kinase 2. Mol. Med. Rep. 2014, 9, 1917–1922. [Google Scholar] [CrossRef] [PubMed]
- Miya Shaik, M.; Tamargo, I.A.; Abubakar, M.B.; Kamal, M.A.; Greig, N.H.; Gan, S.H. The Role of microRNAs in Alzheimer’s Disease and Their Therapeutic Potentials. Genes 2018, 9, 174. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J.; Zhao, Y.; Cui, J.G. An NF-kappaB-sensitive micro RNA-146a-mediated inflammatory circuit in Alzheimer disease and in stressed human brain cells. J. Biol. Chem. 2008, 283, 31315–31322. [Google Scholar] [CrossRef] [PubMed]
- Shioya, M.; Obayashi, S.; Tabunoki, H.; Arima, K.; Saito, Y.; Ishida, T.; Satoh, J. Aberrant microRNA expression in the brains of neurodegenerative diseases: miR-29a decreased in Alzheimer disease brains targets neurone navigator 3. Neuropathol. Appl. Neurobiol. 2010, 36, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.X.; Rajeev, B.W.; Stromberg, A.J.; Ren, N.; Tang, G.; Huang, Q.; Rigoutsos, I.; Nelson, P.T. The expression of microRNA miR-107 decreases early in Alzheimer’s disease and may accelerate disease progression through regulation of beta-site amyloid precursor protein-cleaving enzyme 1. J. Neurosci. 2008, 28, 1213–1223. [Google Scholar] [CrossRef]
- Kole, A.J.; Swahari, V.; Hammond, S.M.; Deshmukh, M. miR-29b is activated during neuronal maturation and targets BH3-only genes to restrict apoptosis. Genes Dev. 2011, 25, 125–130. [Google Scholar] [CrossRef]
- Rohn, T.T.; Vyas, V.; Hernandez-Estrada, T.; Nichol, K.E.; Christie, L.A.; Head, E. Lack of pathology in a triple transgenic mouse model of Alzheimer’s disease after overexpression of the anti-apoptotic protein Bcl-2. J. Neurosci. 2008, 28, 3051–3059. [Google Scholar] [CrossRef]
- Banzhaf-Strathmann, J.; Benito, E.; May, S.; Arzberger, T.; Tahirovic, S.; Kretzschmar, H.; Fischer, A.; Edbauer, D. MicroRNA-125b induces tau hyperphosphorylation and cognitive deficits in Alzheimer’s disease. EMBO J. 2014, 33, 1667–1680. [Google Scholar] [CrossRef]
- Smith, P.Y.; Hernandez-Rapp, J.; Jolivette, F.; Lecours, C.; Bisht, K.; Goupil, C.; Dorval, V.; Parsi, S.; Morin, F.; Planel, E.; et al. miR-132/212 deficiency impairs tau metabolism and promotes pathological aggregation in vivo. Hum. Mol. Genet. 2015, 24, 6721–6735. [Google Scholar] [CrossRef]
- Geekiyanage, H.; Chan, C. MicroRNA-137/181c regulates serine palmitoyltransferase and in turn amyloid beta, novel targets in sporadic Alzheimer’s disease. J. Neurosci. 2011, 31, 14820–14830. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Wang, B.; Kodali, M.C.; Chen, C.; Kim, E.; Patters, B.J.; Lan, L.; Kumar, S.; Wang, X.; Yue, J.; et al. In vivo evidence for the contribution of peripheral circulating inflammatory exosomes to neuroinflammation. J. Neuroinflamm. 2018, 15, 8. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J.; Tan, E.K. Parkinson’s disease: Etiopathogenesis and treatment. J. Neurol. Neurosurg. Psychiatry 2020, 91, 795–808. [Google Scholar] [CrossRef] [PubMed]
- Ascherio, A.; Schwarzschild, M.A. The epidemiology of Parkinson’s disease: Risk factors and prevention. Lancet Neurol. 2016, 15, 1257–1272. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, C.H.; Del Tredici, K.; Braak, H. Parkinson’s disease: A dual-hit hypothesis. Neuropathol. Appl. Neurobiol. 2007, 33, 599–614. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Mashima, K. Neuroprotection and Disease Modification by Astrocytes and Microglia in Parkinson Disease. Antioxidants 2022, 11, 170. [Google Scholar] [CrossRef]
- Jewell, S.; Herath, A.M.; Gordon, R. Inflammasome Activation in Parkinson’s Disease. J. Parkinsons Dis. 2022, 12, S113–S128. [Google Scholar] [CrossRef]
- Tang, Y.; Li, T.; Li, J.; Yang, J.; Liu, H.; Zhang, X.J.; Le, W. Jmjd3 is essential for the epigenetic modulation of microglia phenotypes in the immune pathogenesis of Parkinson’s disease. Cell Death Differ. 2014, 21, 369–380. [Google Scholar] [CrossRef]
- Long-Smith, C.M.; Collins, L.; Toulouse, A.; Sullivan, A.M.; Nolan, Y.M. Interleukin-1beta contributes to dopaminergic neuronal death induced by lipopolysaccharide-stimulated rat glia in vitro. J. Neuroimmunol. 2010, 226, 20–26. [Google Scholar] [CrossRef]
- Na, S.J.; DiLella, A.G.; Lis, E.V.; Jones, K.; Levine, D.M.; Stone, D.J.; Hess, J.F. Molecular profiling of a 6-hydroxydopamine model of Parkinson’s disease. Neurochem. Res. 2010, 35, 761–772. [Google Scholar] [CrossRef]
- Tieu, K. A guide to neurotoxic animal models of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2011, 1, a009316. [Google Scholar] [CrossRef]
- Tapias, V.; Hu, X.; Luk, K.C.; Sanders, L.H.; Lee, V.M.; Greenamyre, J.T. Synthetic alpha-synuclein fibrils cause mitochondrial impairment and selective dopamine neurodegeneration in part via iNOS-mediated nitric oxide production. Cell Mol. Life Sci. 2017, 74, 2851–2874. [Google Scholar] [CrossRef] [PubMed]
- Theodore, S.; Cao, S.; McLean, P.J.; Standaert, D.G. Targeted overexpression of human alpha-synuclein triggers microglial activation and an adaptive immune response in a mouse model of Parkinson disease. J. Neuropathol. Exp. Neurol. 2008, 67, 1149–1158. [Google Scholar] [CrossRef] [PubMed]
- Booth, H.D.E.; Hirst, W.D.; Wade-Martins, R. The Role of Astrocyte Dysfunction in Parkinson’s Disease Pathogenesis. Trends Neurosci. 2017, 40, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Suk, J.E.; Patrick, C.; Bae, E.J.; Cho, J.H.; Rho, S.; Hwang, D.; Masliah, E.; Lee, S.J. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J. Biol. Chem. 2010, 285, 9262–9272. [Google Scholar] [CrossRef]
- Pajares, M.I.; Rojo, A.; Manda, G.; Bosca, L.; Cuadrado, A. Inflammation in Parkinson’s Disease: Mechanisms and Therapeutic Implications. Cells 2020, 9, 1687. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.C.; Breidert, T.; Rousselet, E.; Hunot, S.; Hartmann, A.; Michel, P.P. The role of glial reaction and inflammation in Parkinson’s disease. Ann. N. Y. Acad. Sci. 2003, 991, 214–228. [Google Scholar] [CrossRef]
- Guhathakurta, S.; Bok, E.; Evangelista, B.A.; Kim, Y.S. Deregulation of alpha-synuclein in Parkinson’s disease: Insight from epigenetic structure and transcriptional regulation of SNCA. Prog. Neurobiol. 2017, 154, 21–36. [Google Scholar] [CrossRef]
- Desplats, P.; Spencer, B.; Coffee, E.; Patel, P.; Michael, S.; Patrick, C.; Adame, A.; Rockenstein, E.; Masliah, E. Alpha-synuclein sequesters Dnmt1 from the nucleus: A novel mechanism for epigenetic alterations in Lewy body diseases. J. Biol. Chem. 2011, 286, 9031–9037. [Google Scholar] [CrossRef]
- Chen, S.; Bellew, C.; Yao, X.; Stefkova, J.; Dipp, S.; Saifudeen, Z.; Bachvarov, D.; El-Dahr, S.S. Histone deacetylase (HDAC) activity is critical for embryonic kidney gene expression, growth, and differentiation. J. Biol. Chem. 2011, 286, 32775–32789. [Google Scholar] [CrossRef]
- van Heesbeen, H.J.; Smidt, M.P. Entanglement of Genetics and Epigenetics in Parkinson’s Disease. Front. Neurosci. 2019, 13, 277. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; Bjornevik, K.; Im, D.S.; Flierl, A.; Dong, X.; Locascio, J.J.; Abo, K.M.; Long, E.; Jin, M.; Xu, B.; et al. beta2-Adrenoreceptor is a regulator of the alpha-synuclein gene driving risk of Parkinson’s disease. Science 2017, 357, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Toker, L.; Tran, G.T.; Sundaresan, J.; Tysnes, O.B.; Alves, G.; Haugarvoll, K.; Nido, G.S.; Dolle, C.; Tzoulis, C. Genome-wide histone acetylation analysis reveals altered transcriptional regulation in the Parkinson’s disease brain. Mol. Neurodegener. 2021, 16, 31. [Google Scholar] [CrossRef]
- Jing, H.; Lin, H. Sirtuins in epigenetic regulation. Chem. Rev. 2015, 115, 2350–2375. [Google Scholar] [CrossRef] [PubMed]
- Chio, A.; Logroscino, G.; Hardiman, O.; Swingler, R.; Mitchell, D.; Beghi, E.; Traynor, B.G.; Eurals, C. Prognostic factors in ALS: A critical review. Amyotroph. Lateral Scler. 2009, 10, 310–323. [Google Scholar] [CrossRef]
- Lomen-Hoerth, C.; Anderson, T.; Miller, B. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology 2002, 59, 1077–1079. [Google Scholar] [CrossRef]
- Ince, P.G.; Highley, J.R.; Kirby, J.; Wharton, S.B.; Takahashi, H.; Strong, M.J.; Shaw, P.J. Molecular pathology and genetic advances in amyotrophic lateral sclerosis: An emerging molecular pathway and the significance of glial pathology. Acta Neuropathol. 2011, 122, 657–671. [Google Scholar] [CrossRef]
- Tarr, I.S.; McCann, E.P.; Benyamin, B.; Peters, T.J.; Twine, N.A.; Zhang, K.Y.; Zhao, Q.; Zhang, Z.H.; Rowe, D.B.; Nicholson, G.A.; et al. Monozygotic twins and triplets discordant for amyotrophic lateral sclerosis display differential methylation and gene expression. Sci. Rep. 2019, 9, 8254. [Google Scholar] [CrossRef]
- Vicario, N.; Spitale, F.M.; Tibullo, D.; Giallongo, C.; Amorini, A.M.; Scandura, G.; Spoto, G.; Saab, M.W.; D’Aprile, S.; Alberghina, C.; et al. Clobetasol promotes neuromuscular plasticity in mice after motoneuronal loss via sonic hedgehog signaling, immunomodulation and metabolic rebalancing. Cell Death Dis. 2021, 12, 625. [Google Scholar] [CrossRef]
- Liu, E.; Karpf, L.; Bohl, D. Neuroinflammation in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia and the Interest of Induced Pluripotent Stem Cells to Study Immune Cells Interactions With Neurons. Front. Mol. Neurosci. 2021, 14, 767041. [Google Scholar] [CrossRef]
- Gandelman, M.; Peluffo, H.; Beckman, J.S.; Cassina, P.; Barbeito, L. Extracellular ATP and the P2X7 receptor in astrocyte-mediated motor neuron death: Implications for amyotrophic lateral sclerosis. J. Neuroinflamm. 2010, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Apolloni, S.; Amadio, S.; Parisi, C.; Matteucci, A.; Potenza, R.L.; Armida, M.; Popoli, P.; D’Ambrosi, N.; Volonte, C. Spinal cord pathology is ameliorated by P2X7 antagonism in a SOD1-mutant mouse model of amyotrophic lateral sclerosis. Dis. Model Mech. 2014, 7, 1101–1109. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, R.; Sluyter, V.; Watson, D.; Sluyter, R.; Yerbury, J.J. P2X7 antagonism using Brilliant Blue G reduces body weight loss and prolongs survival in female SOD1(G93A) amyotrophic lateral sclerosis mice. PeerJ 2017, 5, e3064. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, K.; Komine, O. The multi-dimensional roles of astrocytes in ALS. Neurosci. Res. 2018, 126, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Coppede, F.; Stoccoro, A. Mitoepigenetics and Neurodegenerative Diseases. Front. Endocrinol. 2019, 10, 86. [Google Scholar] [CrossRef]
- Stoccoro, A.; Mosca, L.; Carnicelli, V.; Cavallari, U.; Lunetta, C.; Marocchi, A.; Migliore, L.; Coppede, F. Mitochondrial DNA copy number and D-loop region methylation in carriers of amyotrophic lateral sclerosis gene mutations. Epigenomics 2018, 10, 1431–1443. [Google Scholar] [CrossRef]
- Chevin, M.; Sebire, G. Necroptosis in ALS: A hot topic in-progress. Cell Death Discov. 2021, 7, 79. [Google Scholar] [CrossRef]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17085. [Google Scholar] [CrossRef]
- Lian, L.; Liu, M.; Cui, L.; Guan, Y.; Liu, T.; Cui, B.; Zhang, K.; Tai, H.; Shen, D. Environmental risk factors and amyotrophic lateral sclerosis (ALS): A case-control study of ALS in China. J. Clin. Neurosci. 2019, 66, 12–18. [Google Scholar] [CrossRef]
- Dickerson, A.S.; Hansen, J.; Thompson, S.; Gredal, O.; Weisskopf, M.G. A mixtures approach to solvent exposures and amyotrophic lateral sclerosis: A population-based study in Denmark. Eur. J. Epidemiol. 2020, 35, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Bellavia, A.; Dickerson, A.S.; Rotem, R.S.; Hansen, J.; Gredal, O.; Weisskopf, M.G. Joint and interactive effects between health comorbidities and environmental exposures in predicting amyotrophic lateral sclerosis. Int. J. Hyg. Environ. Health 2021, 231, 113655. [Google Scholar] [CrossRef] [PubMed]
- Figueroa-Romero, C.; Hur, J.; Bender, D.E.; Delaney, C.E.; Cataldo, M.D.; Smith, A.L.; Yung, R.; Ruden, D.M.; Callaghan, B.C.; Feldman, E.L. Identification of epigenetically altered genes in sporadic amyotrophic lateral sclerosis. PLoS ONE 2012, 7, e52672. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Jia, X.; Liu, M.; Yang, X.; Cui, L. Epigenome-wide DNA methylation study of whole blood in patients with sporadic amyotrophic lateral sclerosis. Chin. Med. J. 2022, 135, 1466–1473. [Google Scholar] [CrossRef]
- Oates, N.; Pamphlett, R. An epigenetic analysis of SOD1 and VEGF in ALS. Amyotroph. Lateral Scler. 2007, 8, 83–86. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Van Kammen, M.; Levey, A.I.; Martin, L.J.; Kuncl, R.W. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann. Neurol. 1995, 38, 73–84. [Google Scholar] [CrossRef]
- Stoccoro, A.; Smith, A.R.; Mosca, L.; Marocchi, A.; Gerardi, F.; Lunetta, C.; Cereda, C.; Gagliardi, S.; Lunnon, K.; Migliore, L.; et al. Reduced mitochondrial D-loop methylation levels in sporadic amyotrophic lateral sclerosis. Clin. Epigenetics 2020, 12, 137. [Google Scholar] [CrossRef]
- Orton, S.M.; Herrera, B.M.; Yee, I.M.; Valdar, W.; Ramagopalan, S.V.; Sadovnick, A.D.; Ebers, G.C.; Canadian Collaborative Study Group. Sex ratio of multiple sclerosis in Canada: A longitudinal study. Lancet Neurol. 2006, 5, 932–936. [Google Scholar] [CrossRef]
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2008, 372, 1502–1517. [Google Scholar] [CrossRef]
- Gale, C.R.; Martyn, C.N. Migrant studies in multiple sclerosis. Prog. Neurobiol. 1995, 47, 425–448. [Google Scholar] [CrossRef]
- Hedstrom, A.K.; Sundqvist, E.; Baarnhielm, M.; Nordin, N.; Hillert, J.; Kockum, I.; Olsson, T.; Alfredsson, L. Smoking and two human leukocyte antigen genes interact to increase the risk for multiple sclerosis. Brain 2011, 134, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, B.; Matyszak, M.K.; Esiri, M.M.; Perry, V.H. Axonal damage in acute multiple sclerosis lesions. Brain 1997, 120 Pt 3, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Trapp, B.D.; Peterson, J.; Ransohoff, R.M.; Rudick, R.; Mork, S.; Bo, L. Axonal transection in the lesions of multiple sclerosis. N. Engl. J. Med. 1998, 338, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Chandran, P.K.; Kuttan, R. Effect of Calendula officinalis Flower Extract on Acute Phase Proteins, Antioxidant Defense Mechanism and Granuloma Formation During Thermal Burns. J. Clin. Biochem. Nutr. 2008, 43, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Cossburn, M.; Tackley, G.; Baker, K.; Ingram, G.; Burtonwood, M.; Malik, G.; Pickersgill, T.; te Water Naude, J.; Robertson, N. The prevalence of neuromyelitis optica in South East Wales. Eur. J. Neurol. 2012, 19, 655–659. [Google Scholar] [CrossRef] [PubMed]
- Kalincik, T.; Buzzard, K.; Jokubaitis, V.; Trojano, M.; Duquette, P.; Izquierdo, G.; Girard, M.; Lugaresi, A.; Grammond, P.; Grand’Maison, F.; et al. Risk of relapse phenotype recurrence in multiple sclerosis. Mult. Scler. 2014, 20, 1511–1522. [Google Scholar] [CrossRef]
- Confavreux, C.; Vukusic, S. Natural history of multiple sclerosis: A unifying concept. Brain 2006, 129, 606–616. [Google Scholar] [CrossRef]
- Tutuncu, M.; Tang, J.; Zeid, N.A.; Kale, N.; Crusan, D.J.; Atkinson, E.J.; Siva, A.; Pittock, S.J.; Pirko, I.; Keegan, B.M.; et al. Onset of progressive phase is an age-dependent clinical milestone in multiple sclerosis. Mult. Scler. 2013, 19, 188–198. [Google Scholar] [CrossRef]
- Penna, G.; Roncari, A.; Amuchastegui, S.; Daniel, K.C.; Berti, E.; Colonna, M.; Adorini, L. Expression of the inhibitory receptor ILT3 on dendritic cells is dispensable for induction of CD4+Foxp3+ regulatory T cells by 1,25-dihydroxyvitamin D3. Blood 2005, 106, 3490–3497. [Google Scholar] [CrossRef]
- Ma, Q.; Oksenberg, J.R.; Didonna, A. Epigenetic control of ataxin-1 in multiple sclerosis. Ann. Clin. Transl. Neurol. 2022, 9, 1186–1194. [Google Scholar] [CrossRef]
- Didonna, A.; Canto Puig, E.; Ma, Q.; Matsunaga, A.; Ho, B.; Caillier, S.J.; Shams, H.; Lee, N.; Hauser, S.L.; Tan, Q.; et al. Ataxin-1 regulates B cell function and the severity of autoimmune experimental encephalomyelitis. Proc. Natl. Acad. Sci. USA 2020, 117, 23742–23750. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Samaco, R.C.; Gatchel, J.R.; Thaller, C.; Orr, H.T.; Zoghbi, H.Y. miR-19, miR-101 and miR-130 co-regulate ATXN1 levels to potentially modulate SCA1 pathogenesis. Nat. Neurosci. 2008, 11, 1137–1139. [Google Scholar] [CrossRef] [PubMed]
- Gennarino, V.A.; Singh, R.K.; White, J.J.; De Maio, A.; Han, K.; Kim, J.Y.; Jafar-Nejad, P.; di Ronza, A.; Kang, H.; Sayegh, L.S.; et al. Pumilio1 haploinsufficiency leads to SCA1-like neurodegeneration by increasing wild-type Ataxin1 levels. Cell 2015, 160, 1087–1098. [Google Scholar] [CrossRef]
- Lowe, K.; Wu, E.; Wang, N.; Hoerster, G.; Hastings, C.; Cho, M.J.; Scelonge, C.; Lenderts, B.; Chamberlin, M.; Cushatt, J.; et al. Morphogenic Regulators Baby boom and Wuschel Improve Monocot Transformation. Plant Cell 2016, 28, 1998–2015. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Merrill, J.E.; Scolding, N.J. Mechanisms of damage to myelin and oligodendrocytes and their relevance to disease. Neuropathol. Appl. Neurobiol. 1999, 25, 435–458. [Google Scholar] [CrossRef]
- Miyachi, H.; Konishi, T.; Kumazawa, R.; Matsui, H.; Shimizu, S.; Fushimi, K.; Matsue, H.; Yasunaga, H. Treatments and outcomes of generalized pustular psoriasis: A cohort of 1516 patients in a nationwide inpatient database in Japan. J. Am. Acad. Dermatol. 2022, 86, 1266–1274. [Google Scholar] [CrossRef]
- Luo, C.; Keown, C.L.; Kurihara, L.; Zhou, J.; He, Y.; Li, J.; Castanon, R.; Lucero, J.; Nery, J.R.; Sandoval, J.P.; et al. Single-cell methylomes identify neuronal subtypes and regulatory elements in mammalian cortex. Science 2017, 357, 600–604. [Google Scholar] [CrossRef]
- Ponath, G.; Park, C.; Pitt, D. The Role of Astrocytes in Multiple Sclerosis. Front. Immunol. 2018, 9, 217. [Google Scholar] [CrossRef]
- Kular, L.; Klose, D.; Urdanoz-Casado, A.; Ewing, E.; Planell, N.; Gomez-Cabrero, D.; Needhamsen, M.; Jagodic, M. Epigenetic clock indicates accelerated aging in glial cells of progressive multiple sclerosis patients. Front. Aging Neurosci. 2022, 14, 926468. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giallongo, S.; Longhitano, L.; Denaro, S.; D’Aprile, S.; Torrisi, F.; La Spina, E.; Giallongo, C.; Mannino, G.; Lo Furno, D.; Zappalà, A.; et al. The Role of Epigenetics in Neuroinflammatory-Driven Diseases. Int. J. Mol. Sci. 2022, 23, 15218. https://doi.org/10.3390/ijms232315218
Giallongo S, Longhitano L, Denaro S, D’Aprile S, Torrisi F, La Spina E, Giallongo C, Mannino G, Lo Furno D, Zappalà A, et al. The Role of Epigenetics in Neuroinflammatory-Driven Diseases. International Journal of Molecular Sciences. 2022; 23(23):15218. https://doi.org/10.3390/ijms232315218
Chicago/Turabian StyleGiallongo, Sebastiano, Lucia Longhitano, Simona Denaro, Simona D’Aprile, Filippo Torrisi, Enrico La Spina, Cesarina Giallongo, Giuliana Mannino, Debora Lo Furno, Agata Zappalà, and et al. 2022. "The Role of Epigenetics in Neuroinflammatory-Driven Diseases" International Journal of Molecular Sciences 23, no. 23: 15218. https://doi.org/10.3390/ijms232315218
APA StyleGiallongo, S., Longhitano, L., Denaro, S., D’Aprile, S., Torrisi, F., La Spina, E., Giallongo, C., Mannino, G., Lo Furno, D., Zappalà, A., Giuffrida, R., Parenti, R., Li Volti, G., Tibullo, D., & Vicario, N. (2022). The Role of Epigenetics in Neuroinflammatory-Driven Diseases. International Journal of Molecular Sciences, 23(23), 15218. https://doi.org/10.3390/ijms232315218