
Sensing Magnetic Field and Intermolecular Interactions in Diamagnetic Solution Using Residual Dipolar Couplings of Zephycandidine

Abstract
:1. Introduction
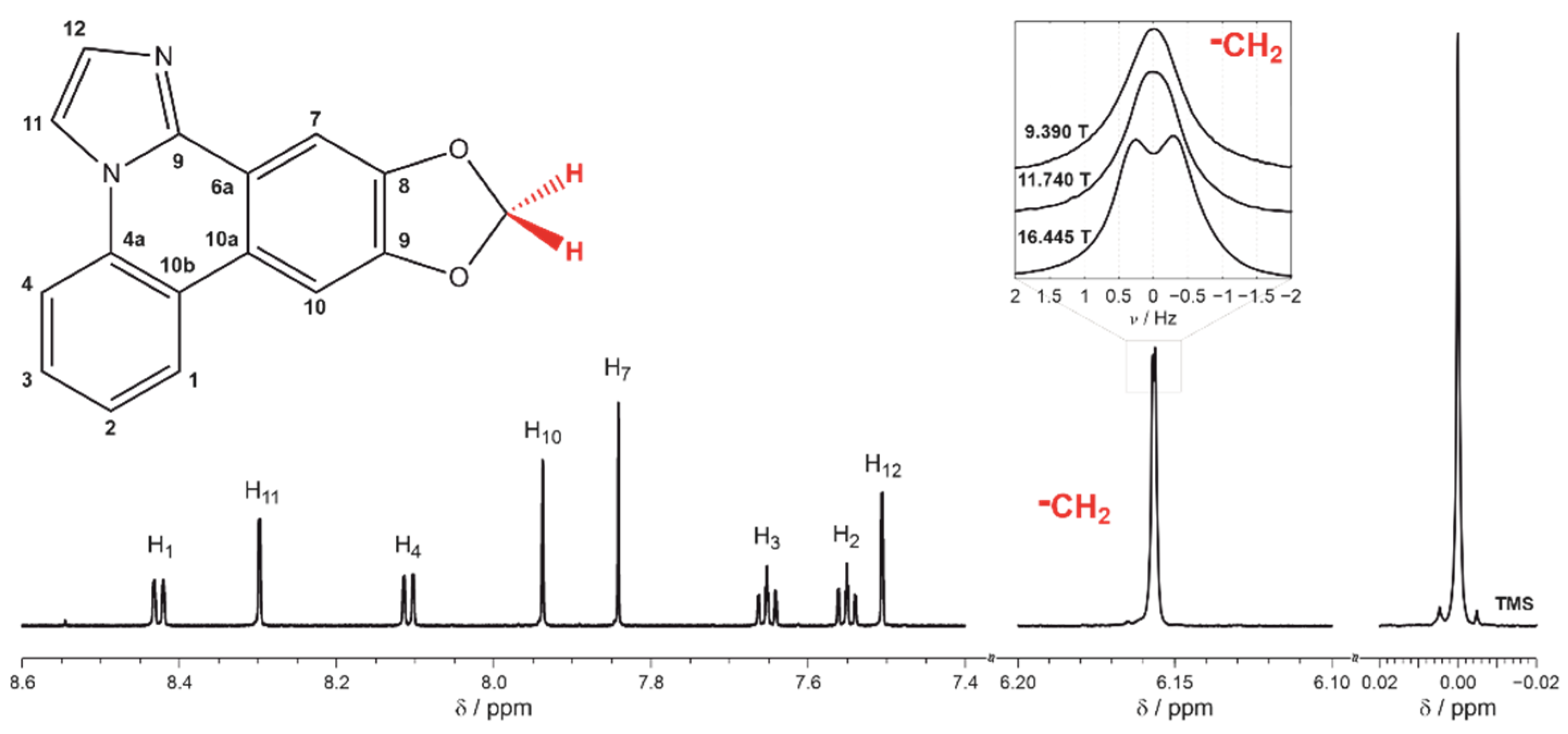
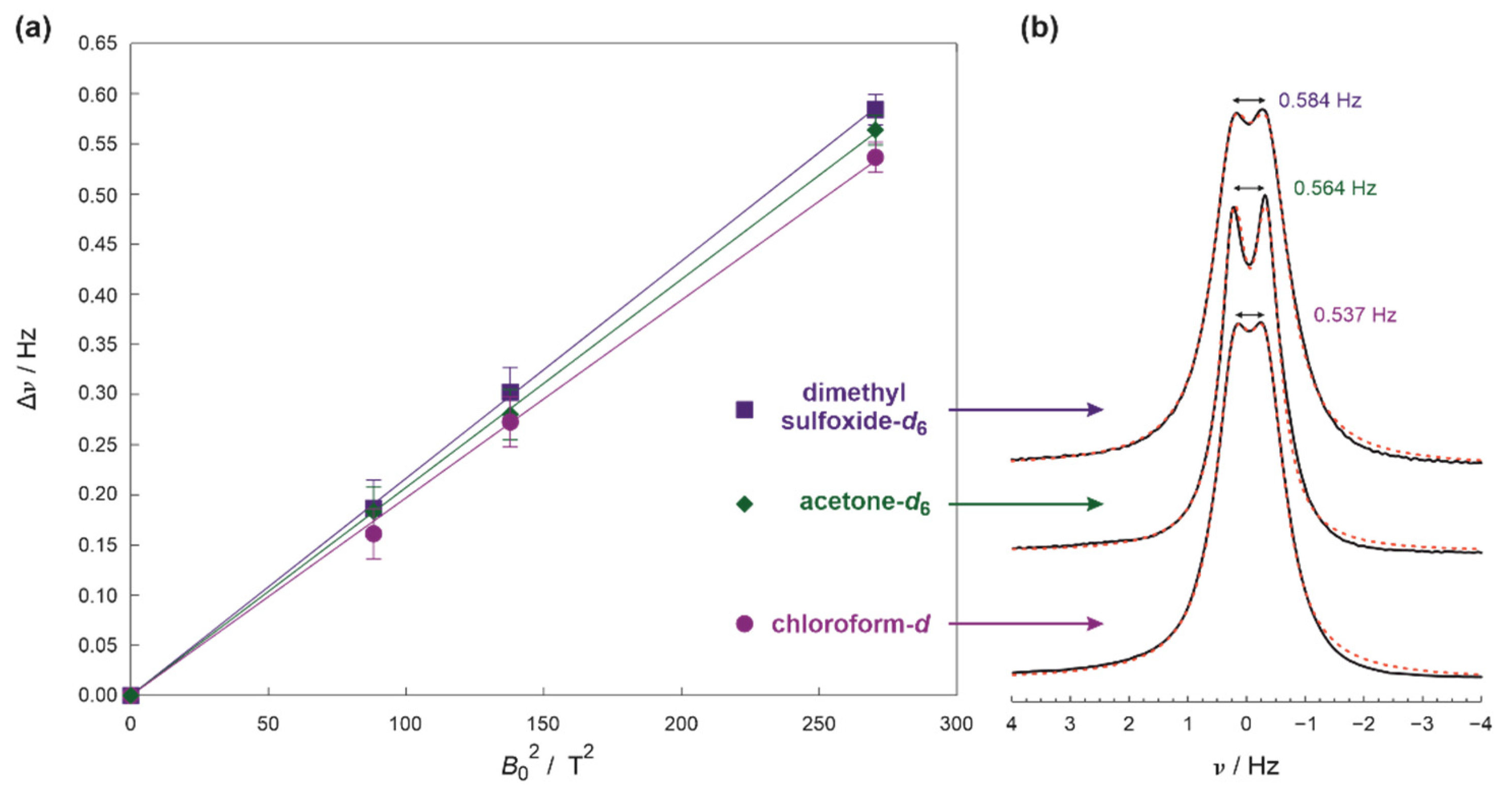
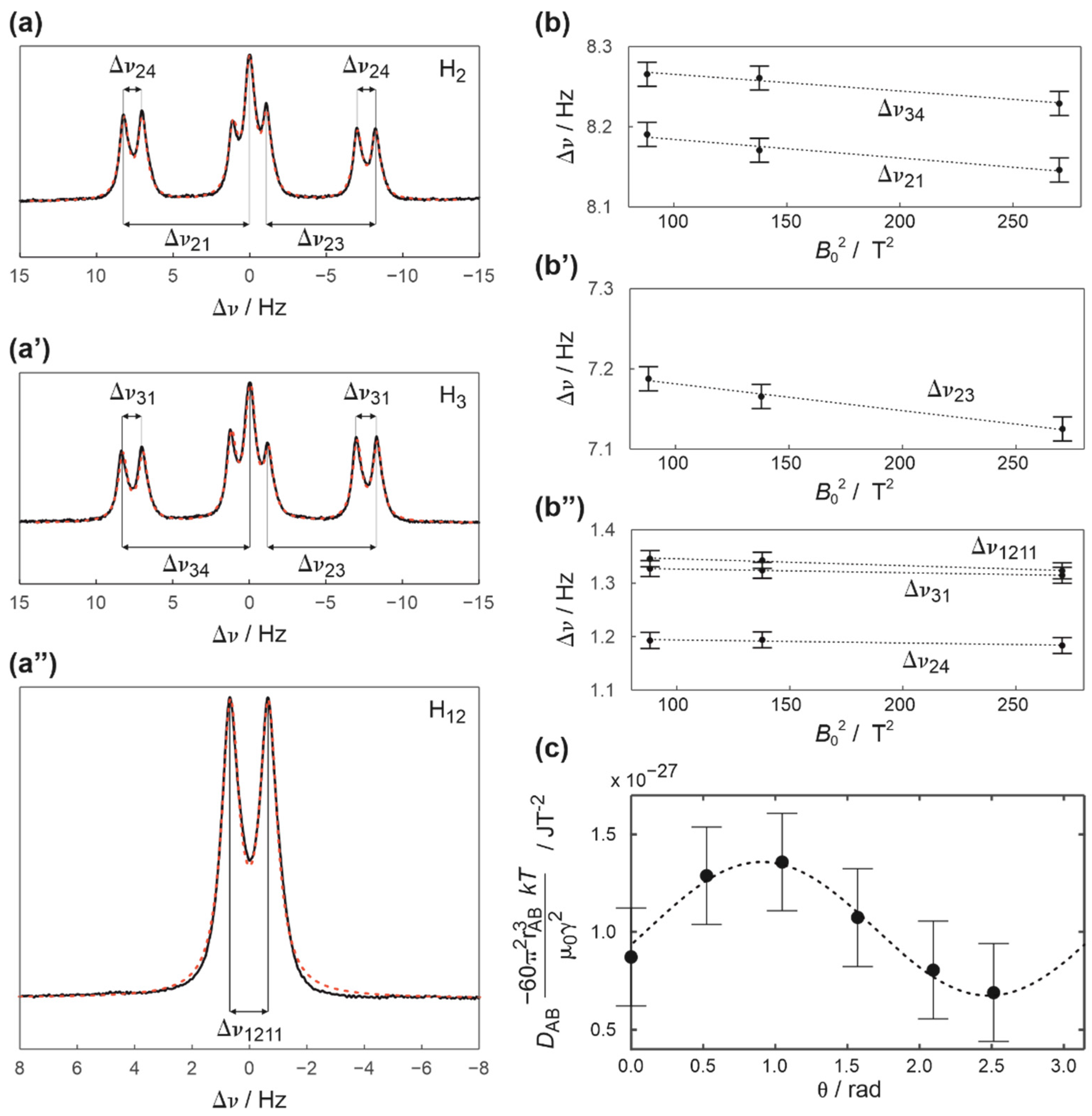
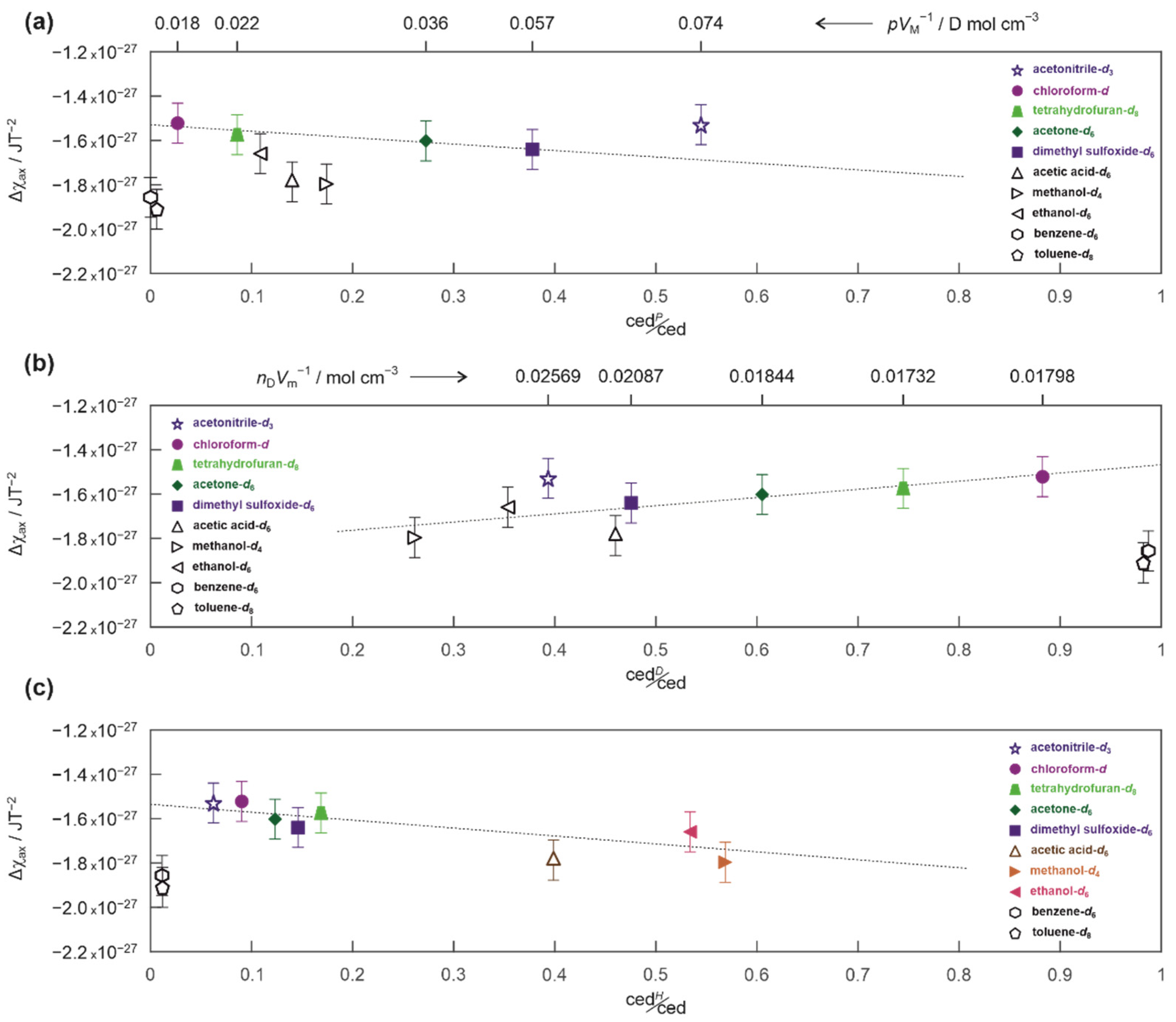
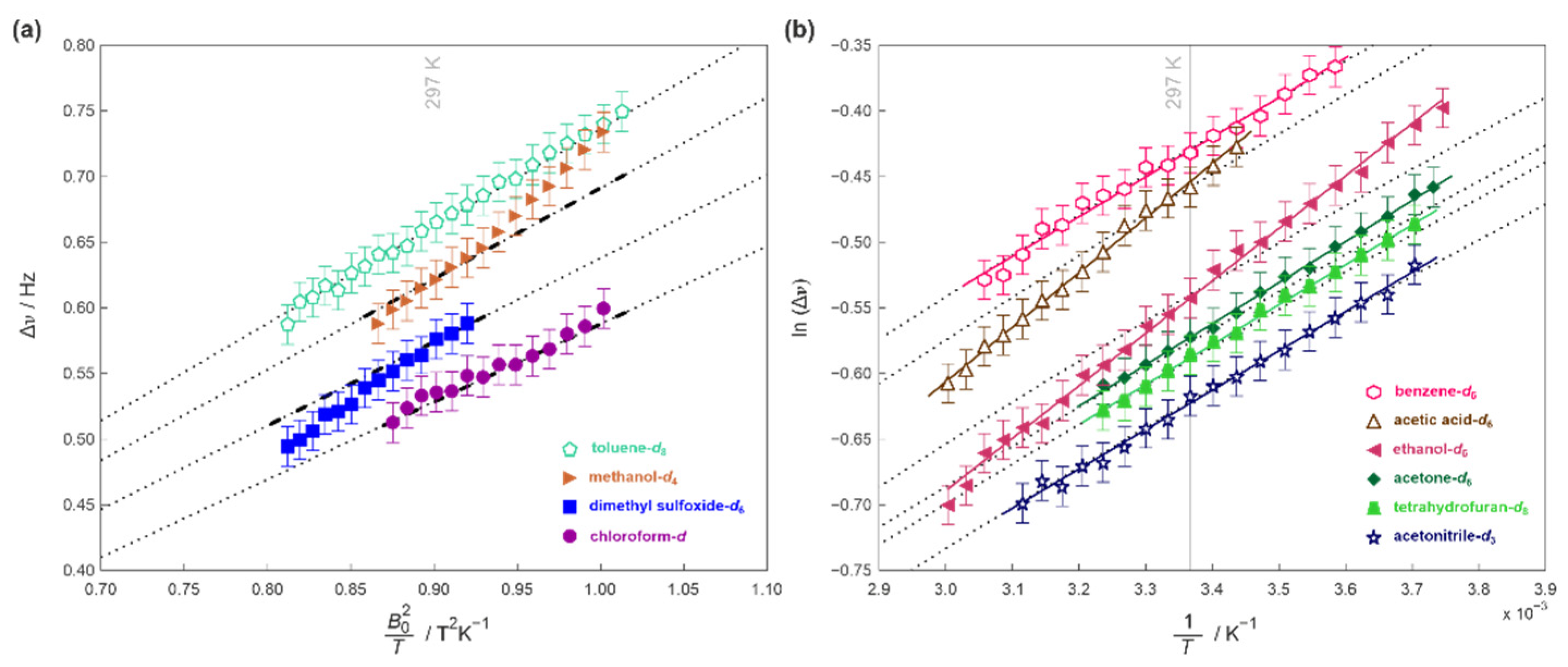
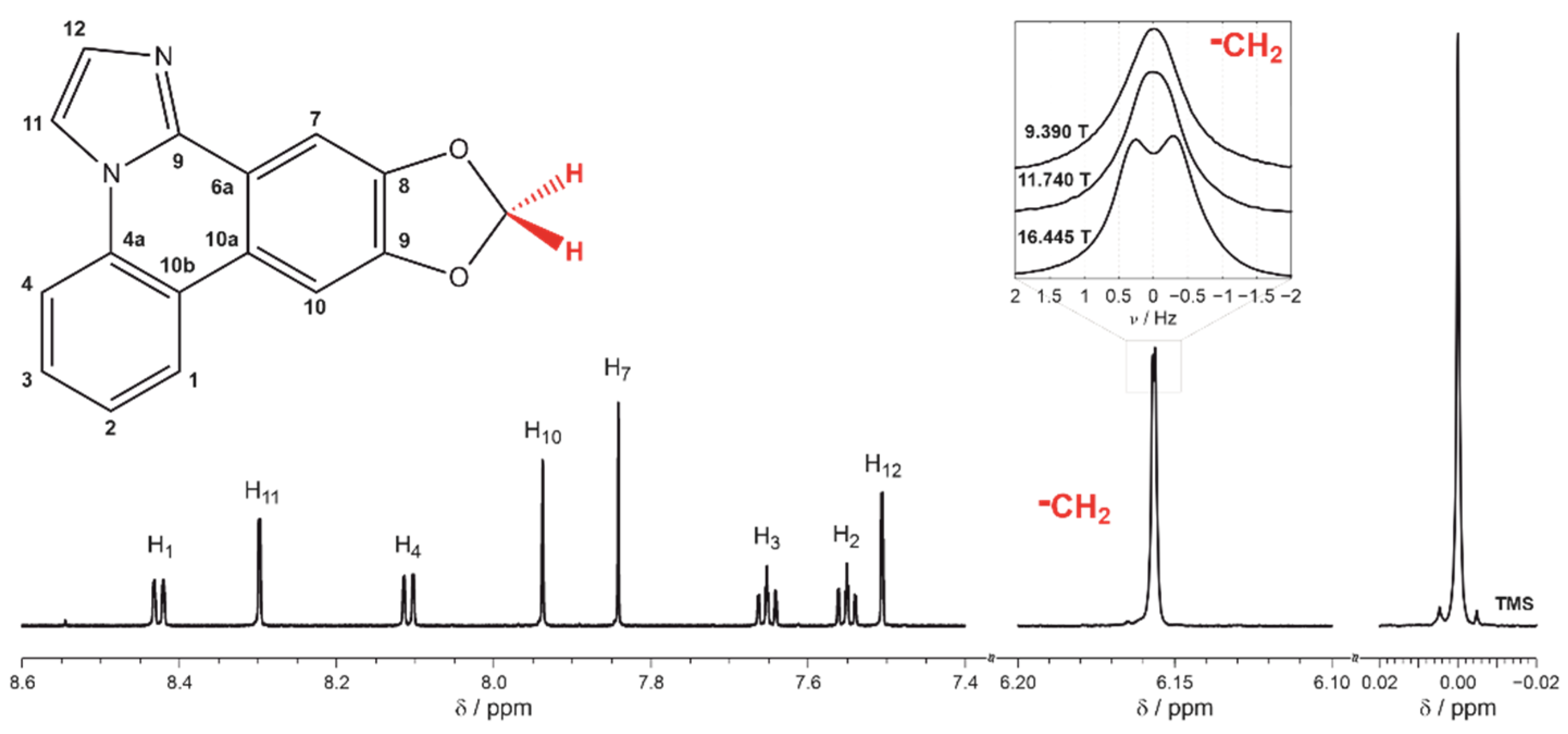
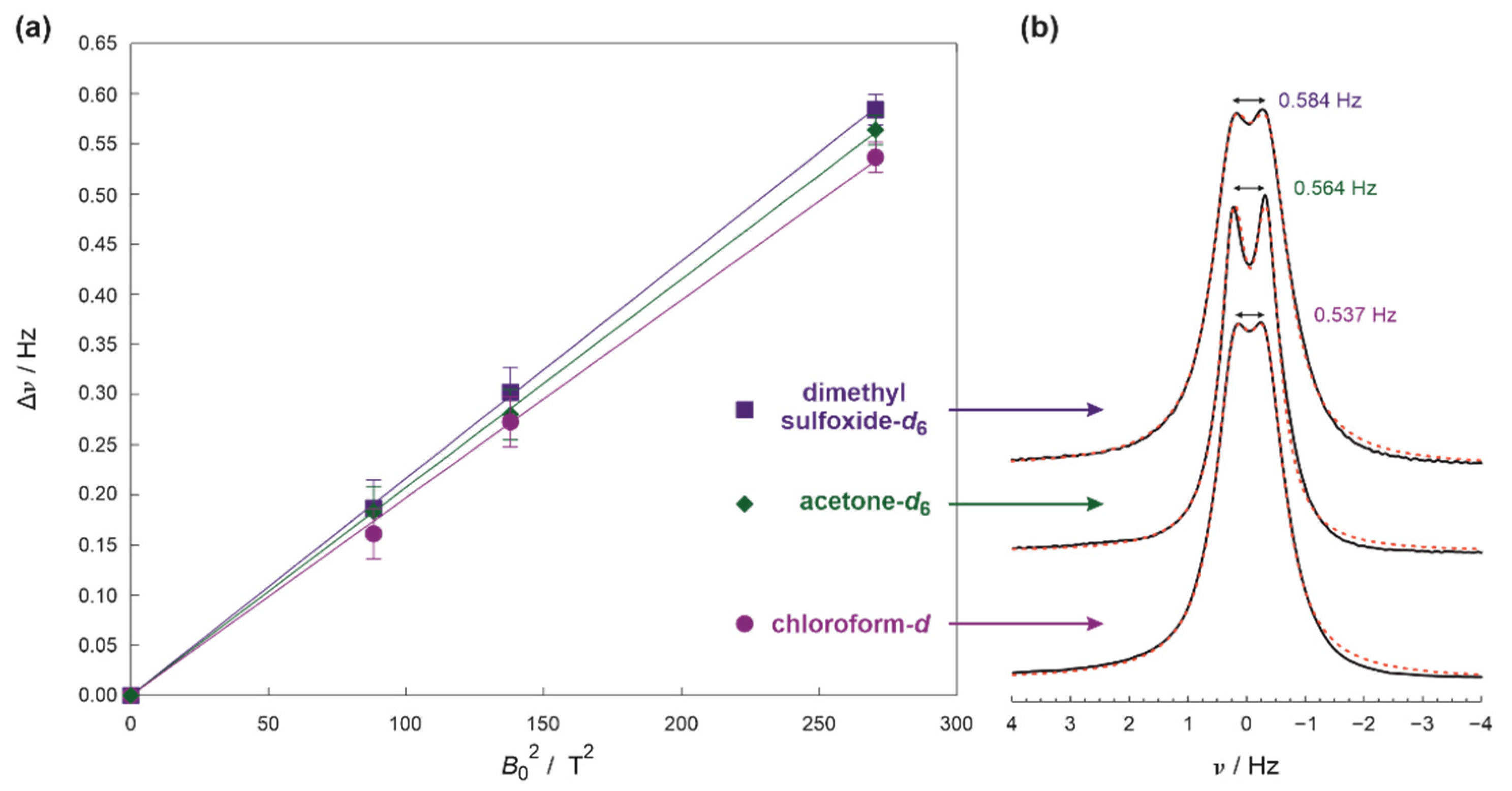
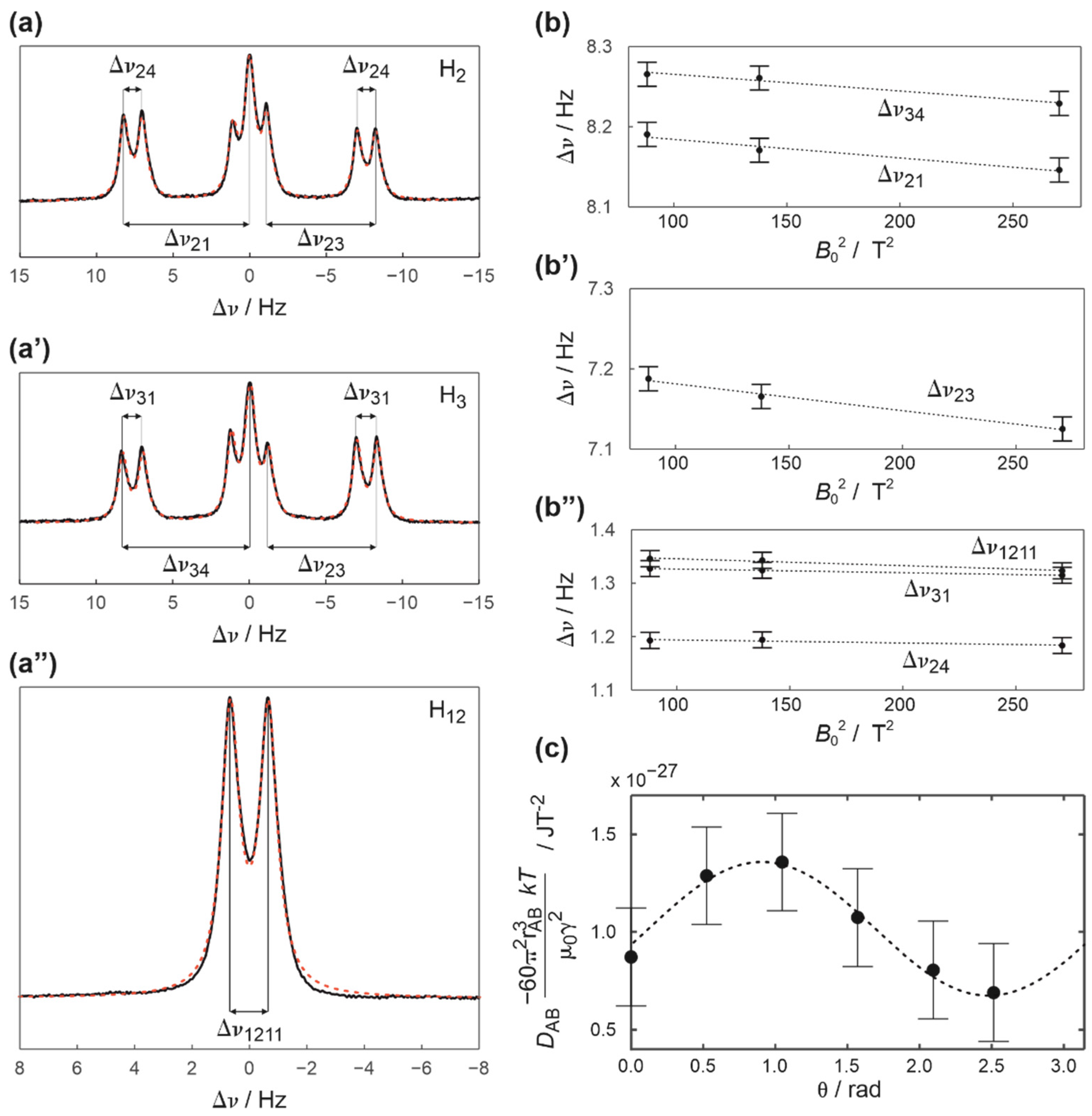
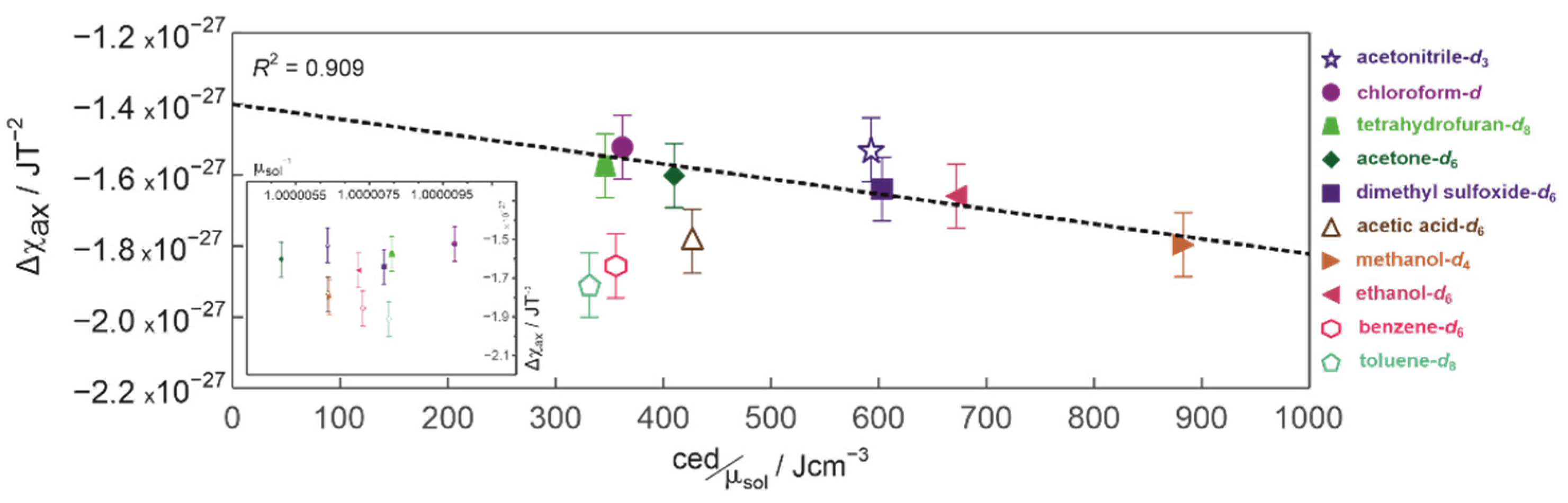
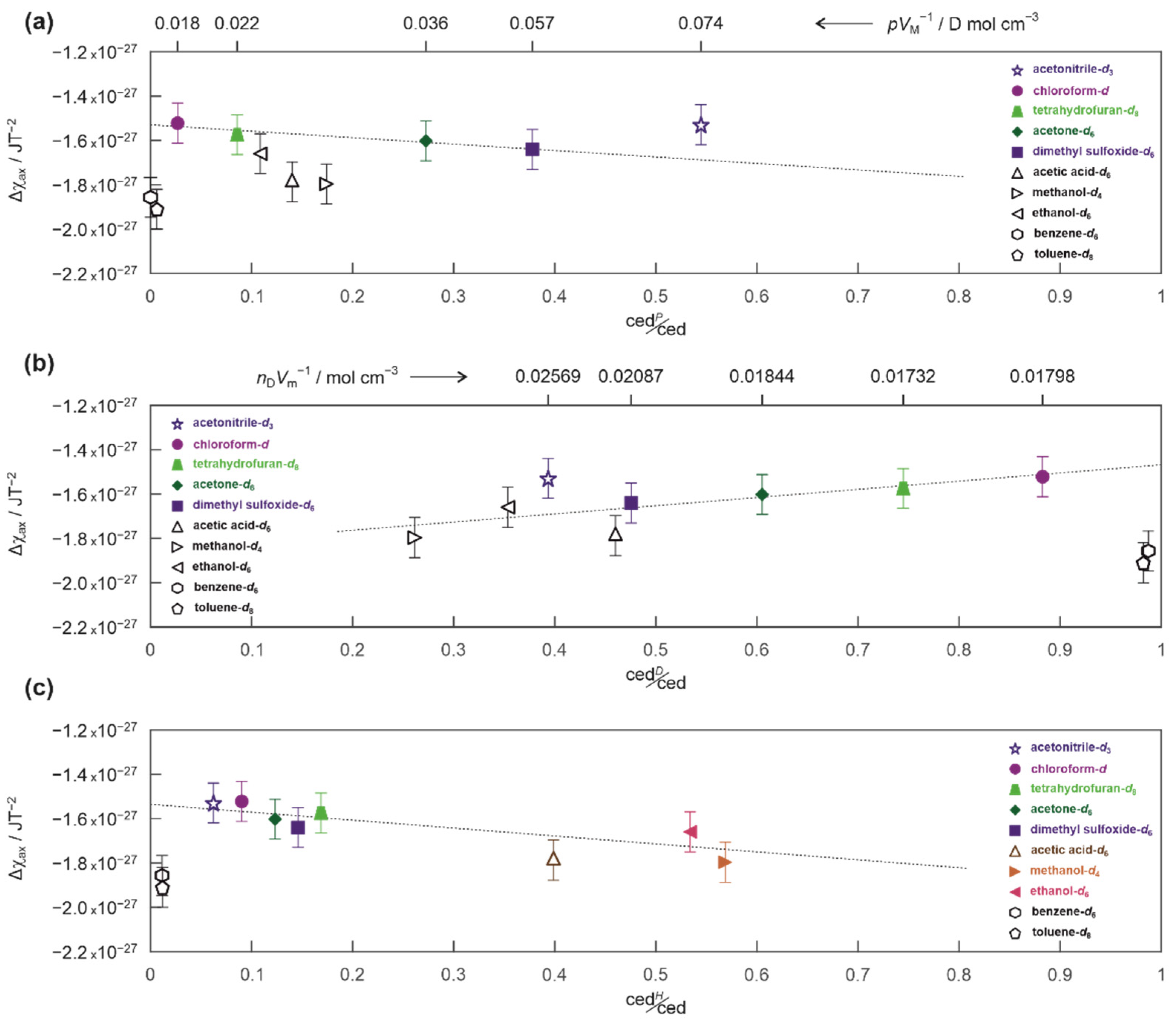
2. Results and Discussion
3. Materials and Methods
3.1. Mathematical Description of Magnetic Field Induced Self-Orientation
3.2. Sample Solutions and NMR Instrumentation
3.3. Elucidation of RDC from NMR Spectra and Experimental Errors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protons Interacting | θ/deg | |
|---|---|---|
| methylene | 1.81 | - |
| H2-H1 | 2.45 | 0 |
| H2-H3 | 2.51 | 60 |
| H2-H4 | 4.18 | 90 |
| H3-H4 | 2.47 | 30 |
| H3-H1 | 4.17 | 120 |
| H12-H11 | 2.76 | 144 |
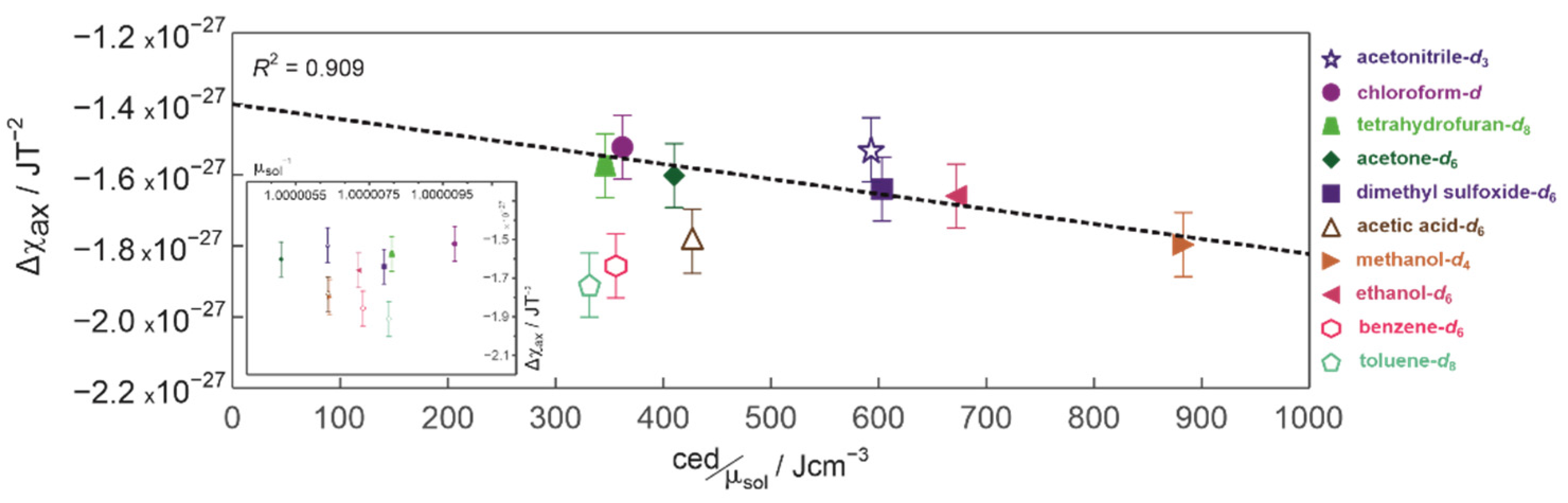
3.4. Magnetic Susceptibilities and Cohesion Energy Densities of the Solvents
3.5. Details of the Energy Barrier Evaluation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, G.W.; Liu, H.; Qiu, F.; Wang, X.J.; Lei, X.X. Residual dipolar couplings in structure determination of natural products. Nat. Prod. Bioprospecting 2018, 8, 279–295. [Google Scholar] [CrossRef] [Green Version]
- Boettcher, B.; Thiele, C.M. Determining the stereochemistry of molecules form residual dipolar couplings (RDCs). eMagRes 2012, 1, 169–180. [Google Scholar]
- Chen, K.; Tjandra, N. The use of residual dipolar coupling in studying proteins by NMR. Top. Curr. Chem. 2012, 326, 47–68. [Google Scholar] [PubMed] [Green Version]
- Prestegard, J.H.; Bougault, C.M.; Kishore, A.I. Residual dipolar couplings in structure determinations of biomolecules. Chem. Rev. 2004, 104, 3519–3540. [Google Scholar] [CrossRef] [PubMed]
- Kramer, F.; Deshmukh, M.V.; Kessler, H.; Glaser, S.J. Residual dipolar coupling constants: An elementary derivation of key equations. Concepts Magn. Reson. Part A 2004, 21, 10–21. [Google Scholar] [CrossRef]
- Bertini, I.; Luchinat, C.; Parigi, G.; Ravera, E. The effect of partial orientation: Residual dipolar couplings. In NMR of Para-Magnetic Molecules, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 61–67. [Google Scholar]
- Lei, X.; Sun, H.; Bai, L.; Wang, W.X.; Xiang, W.; Xiao, H. A self-assembled oligopeptide as a versatile NMR alignment medium for the measurements of residual dipolar couplings in methanol. Angew. Chem. Int. Ed. 2017, 56, 12857–12861. [Google Scholar] [CrossRef]
- Naumann, C.; Kuchel, P.W. Prochiral and chiral resolution in 2H NMR spectra: Solutes in stretched and compressed gelatin gels. J. Phys. Chem. A 2008, 112, 8659–8664. [Google Scholar] [CrossRef]
- Kummerlöwe, G.; Halbach, F.; Laufer, B.; Luy, B. Precise measurement of RDCs in water and DMSO based gels using a silicone rubber tube for tunable stretching. Open Spectrosc. J. 2008, 2, 29–33. [Google Scholar] [CrossRef]
- Emsley, J.W.; Rauzah, H.; Luckhurst, G.R.; Rumbles, G.N.; Viloria, F.R. The Saupe ordering matrices for solutes in uniaxial liquid crystals. Experiment and theory. Mol. Phys. 1983, 6, 1321–1335. [Google Scholar] [CrossRef]
- Buckingham, A.D. Angular correlation in liquids. Discuss. Faraday Soc. 1967, 43, 205–211. [Google Scholar] [CrossRef]
- Saupe, A. Kernresonanzen in kristallinen Flüssigkeiten und in kristallinflüssigen lösungen. Z. Naturforschg. 1964, 19, 161–171. [Google Scholar] [CrossRef]
- Anet, F.A.L. Magnetic alignment in the 500 MHz 1H NMR spectrum of o-dichlorobenzene in acetone-d6. J. Am. Chem. Soc. 1986, 108, 1354–1355. [Google Scholar] [CrossRef]
- Lisicki, M.A.; Mishra, P.K.; Bothner-By, A.A.; Lindsey, J.S. Solution conformation of a porphyrin-quinone cage molecule determined by dipolar magnetic field effects in ultra-high-field NMR. J. Phys. Chem. 1988, 92, 3400–3403. [Google Scholar] [CrossRef]
- Laatikainen, R.; Ratilainen, J.; Sebastian, R.; Santa, H. NMR study of aromatic-aromatic interactions for benzene and some other fundamental aromatic systems using alignment of aromatics in strong magnetic field. J. Am. Chem. Soc. 1995, 117, 11006–11010. [Google Scholar] [CrossRef]
- Heist, L.M.; Poon, C.-D.; Samulski, E.T.; Photinos, D.J.; Jokisaari, J.; Vaara, J.; Emsley, J.W.; Mamone, S.; Lelli, M. Benzene at 1 GHz. Magnetic field-induced fine structure. J. Magn. Reson. 2015, 258, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Karschin, N.; Wolkenstein, K.; Griesinger, C. Magnetically induced alignment of natural products for stereochemical structure determination via NMR. Angew. Chem. Int. Ed. 2020, 59, 15860–15864. [Google Scholar] [CrossRef]
- Van Zijl, P.C.M.; Ruessink, B.H.; Bulthuis, J.; MacLean, C. NMR of partially aligned liquids: Magnetic susceptibility anisotropies and dielectric properties. Acc. Chem. Res. 1984, 17, 172–180. [Google Scholar] [CrossRef]
- Shestakova, A.K.; Makarkina, A.V.; Smirnova, O.V.; Shtern, M.M.; Chertkov, V.A. Orientation of molecules by magnetic field as a new source of information on their structures. Russ. Chem. Bull. 2006, 55, 1359–1367. [Google Scholar] [CrossRef]
- Jokisaari, J.; Kantola, A.M.; Vaara, J. Magnetic field-induced effects on NMR properties. J. Magn. Reson. 2017, 281, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Dracinsky, M.; Boul, P.J. Computational analysis of solvent effects in NMR spectroscopy. Chem. Theory Comput. 2010, 6, 288–299. [Google Scholar] [CrossRef]
- Zhan, G.; Qu, X.; Liu, J.; Tong, Q.; Zhou, Q.; Sun, B.; Yao, G. Zephycandidine A, the first naturally occurring imidazo[1,2-f]phenanthridine alkaloid from Zephyranthes candida, exhibits significant anti-tumor and anti-acetylcholinesterase activities. Sci. Rep. 2016, 6, 33990. [Google Scholar] [CrossRef]
- Murphy, P.J.; Tibble-Howlings, J.; Kowalczyk, R.M.; Stevens, K. Synthesis of zephycandidine A from haemanthamine. Tetrahedron Lett. 2020, 61, 151785. [Google Scholar] [CrossRef]
- Sheng, B.; Zeng, C.; Chen, J.; Ye, W.-C.; Tang, W.; Lan, P.; Banwell, M.G. Total syntheses of the imidazo[1,2-f]phenanthridine containing alkaloid Zephycandidine A. Eur. J. Org. Chem. 2022, 2022, e202101511. [Google Scholar] [CrossRef]
- Whitesides, G.M.; Holtz, D.; Roberts, J.D. Nuclear Magnetic Resonance Spectroscopy. The effect of structure on magnetic nonequivalence due to molecular asymmetry. J. Am. Chem. Soc. 1964, 86, 2628–2634. [Google Scholar] [CrossRef]
- Ault, A. Classification of spin systems in NMR spectroscopy. J. Chem. Educ. 1970, 47, 812–818. [Google Scholar] [CrossRef]
- Petrakis, L. Spectra line shapes. Gaussian and Lorentzian functions in magnetic resonance. J. Chem. Educ. 1967, 44, 432–436. [Google Scholar] [CrossRef]
- Zerbe, O.; Jurt, S. Applied NMR Spectroscopy for Chemists and Life Scientists; Willey: Weinheim, Germany, 2014; pp. 17–20. [Google Scholar]
- Levitt, M.H. Spin Dynamics, 2nd ed.; Wiley: Chichester, UK, 2015; pp. 51, 211–223. [Google Scholar]
- Johannesen, R.B.; Feretti, J.A.; Harris, R.K. UEAITR: A new computer program for analysis of NMR spectra analysis of the proton spectrum of triisopropylphosphine. J. Magn. Reson. 1970, 3, 84–93. [Google Scholar] [CrossRef]
- Vaara, J.; Jokisaari, J.; Wasylishen, R.E.; Bryce, D.L. Spin-spin coupling tensors as determined by experiment and computational chemistry. Nucl. Magn. Reson. Spectrosc. 2002, 41, 233–304. [Google Scholar] [CrossRef]
- Pauling, L. The diamagnetic anisotropy of aromatic molecules. J. Chem. Phys. 1936, 4, 673–677. [Google Scholar] [CrossRef] [Green Version]
- Dack, M.R.J. The importance of solvent internal pressure and cohesion to solution phenomena. Chem. Soc. Rev. 1975, 4, 211–229. [Google Scholar] [CrossRef]
- Marcus, Y. The properties of organic liquids that are relevant to their use as solvating solvents. Chem. Soc. Rev. 1993, 6, 409–416. [Google Scholar] [CrossRef]
- Hoffman, R.E. Variations on the chemical shift of TMS. J. Magn. Reson. 2003, 163, 325–331. [Google Scholar] [CrossRef]
- Hoffman, R.E.; Becker, E.D. Temperature dependence of the 1H chemical shift of tetramethylsilane in chloroform, methanol and dimethylsulfoxide. J. Magn. Reson. 2005, 176, 87–98. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, R.E. Standardization of chemical shifts of TMS and solvent signals in NMR solvents. Magn. Reson. Chem. 2006, 44, 606–616. [Google Scholar] [CrossRef]
- Hoffman, R.E. Magnetic susceptibility measurement by NMR: 2. The magnetic susceptibility of NMR solvents and their chemical shifts. J. Magn. Reson. 2022, 335, 107105. [Google Scholar] [CrossRef] [PubMed]
- Barton, A.F.M. Solubility parameters. Chem. Rev. 1975, 75, 731–753. [Google Scholar] [CrossRef]
- Pirika.com. Available online: https://pirika.com/index.html (accessed on 9 June 2022).
- Faubel, M.; Kisters, T. Non-equilibrium molecular evaporation of carboxylic acid dimers. Nature 1989, 339, 527–529. [Google Scholar] [CrossRef]
- Slavchov, R.I.; Novev, J.K.; Mosbach, S.; Kraft, M. Vapor Pressure and Heat of Vaporization of Molecules That Associate in the Gas Phase. Ind. Eng. Chem. Res. 2018, 57, 5722–5731. [Google Scholar] [CrossRef]
- Belmares, M.; Blanco, M.; Goddard, W.A., III; Ross, R.B.; Caldwell, G.; Chou, S.-H.; Pham, J.; Olofson, P.M.; Thomas, C. Hildebrand and Hansen Solubility Parameters from Molecular dynamics with application to electronic nose polymer sensors. J. Comput. Chem. 2004, 25, 1814–1826. [Google Scholar] [CrossRef] [Green Version]
- D’Amelia, R.P.; Tomic, J.C.; Nirode, W.F. The determination of the solubility parameter (δ) and the Mark-Houwink constants (K & α) of food grade polyvinyl acetate (PVAc). J. Polym. Biopolym. Phys. Chem. 2014, 2, 67–72. [Google Scholar]
- Hübel, H.; Faux, D.A.; Jones, R.B.; Dunstan, D.J. Solvation pressure in chloroform. J. Chem. Phys. 2006, 124, 204506-1–204506-7. [Google Scholar] [CrossRef] [Green Version]
- Konicek, J.; Wadsö, I. Enthalpies of vaporization of organic compounds. Acta Chem. Scand. 1970, 24, 2612–2616. [Google Scholar] [CrossRef]
- Chickos, J.S.; Acree, W.E., Jr. Enthalpies of vaporization of organic and organometallic compounds 1880–2002. J. Phys. Chem. Ref. Data 2003, 32, 519–878. [Google Scholar] [CrossRef] [Green Version]
- Hansen, C.M. Hansen Solubility Parameters. A User’s Handbook, 2nd ed.; CRC Press: London, UK; New York, NJ, USA, 2007; pp. 4–6, 13–17, 45–59. [Google Scholar]
- Atkins, P.W. Physical Chemistry, 3rd ed.; Oxford University Press: Oxford, UK, 1986; pp. 584–586. [Google Scholar]
- Takamuku, T.; Tabaya, M.; Yamaguchi, A.; Nishimoto, J.; Kumamoto, M.; Wakita, H.; Yamaguchi, T. Liquid structure of acetonitrile-water mixtures by x-ray diffraction and infrared spectroscopy. J. Phys. Chem. B 1998, 102, 8880–8888. [Google Scholar] [CrossRef]
- Timerghazin, Q.K.; Peslherbe, G.H. Electronic structure of the acetonitrile and acetonitrile dimer anions: A topological investigation. J. Phys. Chem. B 2008, 112, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Ford, T.A.; Glasser, L. Ab inito calculations of the structural, energetic, and vibrational properties of some hydrogen bonded and van der Waals Dimers. Part 4. The acetonitrile dimer. Int. J. Quantum Chem. 2001, 84, 226–240. [Google Scholar] [CrossRef]
- Siebers, J.G.; Buck, U.; Beu, T.A. Calculation of structures and vibrational spectra of acetonitrile clusters. Chem. Phys. 1998, 239, 549–560. [Google Scholar] [CrossRef]
- Reimers, J.R.; Hall, L.E. The solvation of acetonitrile. J. Am. Chem. Soc. 1999, 121, 3730–3744. [Google Scholar] [CrossRef]
- Vukovic, V.; Pitesa, T.; Jelsch, C.; Wenger, E.; Molcanov, K. An unusual intermolecular interaction between a lone pair and an electron-rich π electron system of a quinoid dianion. Cryst. Growth Des. 2021, 21, 5651–5658. [Google Scholar] [CrossRef]
- Schottel, B.L.; Chifotides, H.T.; Dunbar, K.R. Anion-π interactions. Chem. Soc. Rev. 2008, 37, 68–83. [Google Scholar] [CrossRef]
- Hogben, H.J.; Krzystyniak, M.; Charnock, G.T.P.; Hore, P.J.; Kuprov, I. Spinach—A software library for simulation of spin dynamics in large spin systems. J. Magn. Reson. 2011, 208, 179–194. [Google Scholar] [CrossRef] [PubMed]
- MATLAB, version R2021a; The Mathworks, Inc.: Natick, MA, USA, 2021.
- Deshpande, V.T.; Pathki, K.G. Magnetic susceptibility of acetic acid + pyridine mixtures. Trans. Faraday Soc. 1962, 58, 2134–2138. [Google Scholar] [CrossRef]
- Sriraman, S.; Shanmugasundaram, V.; Sabesan, R. The diamagnetic study of the nature of the association of acetic acid. II. The binary systems of acetic acid with Triethylamine, pyridine and Aniline. Bull. Chem. Soc. Jpn. 1965, 38, 1391–1393. [Google Scholar] [CrossRef]
- Available online: https://nmr.chem.umn.edu/ResReports/NMR002.html (accessed on 12 June 2022).
- Christian Reichardt, C.; Welton, T. Solvents and Solvent Effects in Organic Chemistry, 4th ed.; Appendix A; Willey: Weinheim, Germany, 2011; pp. 549–553. [Google Scholar]
- Bruker Almanac 2012; Bruker Corporation: Billerica, MA, USA, 2012; NMR tables p15.

| Solvent | |||||
|---|---|---|---|---|---|
| aromatic | benzene-d6 | −1.86 × 10−27 | −1.41 × 10−27 | −0.35 × 10−27 | 0.00 |
| toluene-d8 | −1.91 × 10−27 | −1.51 × 10−27 | −0.57 × 10−27 | 0.13 | |
| protic | acetic acid-d6 | −1.79 × 10−27 | −1.33 × 10−27 | −0.41 × 10−27 | 0.87 |
| methanol-d4 | −1.80 × 10−27 | −1.32 × 10−27 | −0.28 × 10−27 | 1.22 | |
| ethanol-d6 | −1.66 × 10−27 | −1.21 × 10−27 | −0.29 × 10−27 | 0.85 | |
| aprotic | dimethyl sulfoxide-d6 | −1.64 × 10−27 | −1.21 × 10−27 | −0.63 × 10−27 | 1.08 |
| acetone-d6 | −1.60 × 10−27 | −1.16 × 10−27 | −0.38 × 10−27 | 0.33 | |
| tetrahydrofuran-d8 | −1.57 × 10−27 | −1.01 × 10−27 | −0.21 × 10−27 | 0.13 | |
| acetonitrile-d3 | −1.53 × 10−27 | −1.01 × 10−27 | −0.23 × 10−27 | 0.05 | |
| chloroform-d | −1.52 × 10−27 | −1.00 × 10−27 | −0.27 × 10−27 | 0.02 | |
| standard errors (confidence level 95.4%) | 0.06 × 10−27 | 0.14 × 10−27 | 0.12 × 10−27 | 0.28 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kowalczyk, R.M.; Murphy, P.J.; Tibble-Howlings, J. Sensing Magnetic Field and Intermolecular Interactions in Diamagnetic Solution Using Residual Dipolar Couplings of Zephycandidine. Int. J. Mol. Sci. 2022, 23, 15118. https://doi.org/10.3390/ijms232315118
Kowalczyk RM, Murphy PJ, Tibble-Howlings J. Sensing Magnetic Field and Intermolecular Interactions in Diamagnetic Solution Using Residual Dipolar Couplings of Zephycandidine. International Journal of Molecular Sciences. 2022; 23(23):15118. https://doi.org/10.3390/ijms232315118
Chicago/Turabian StyleKowalczyk, Radoslaw M., Patrick J. Murphy, and Jamie Tibble-Howlings. 2022. "Sensing Magnetic Field and Intermolecular Interactions in Diamagnetic Solution Using Residual Dipolar Couplings of Zephycandidine" International Journal of Molecular Sciences 23, no. 23: 15118. https://doi.org/10.3390/ijms232315118
APA StyleKowalczyk, R. M., Murphy, P. J., & Tibble-Howlings, J. (2022). Sensing Magnetic Field and Intermolecular Interactions in Diamagnetic Solution Using Residual Dipolar Couplings of Zephycandidine. International Journal of Molecular Sciences, 23(23), 15118. https://doi.org/10.3390/ijms232315118