


Ligand-Based Discovery of a Small Molecule as Inhibitor of α-Synuclein Amyloid Formation

 , , , ,
, , , , 
Abstract
:
1. Introduction
2. Results and Discussion
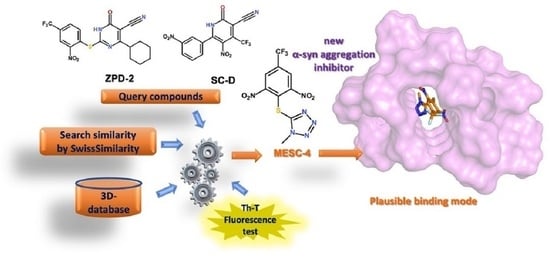
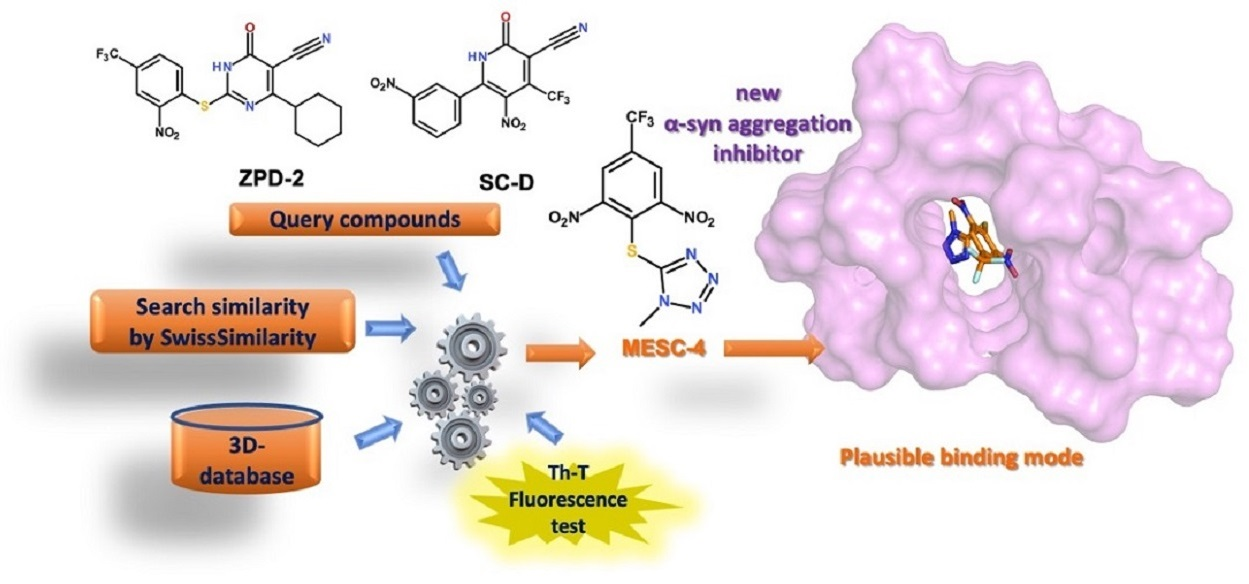
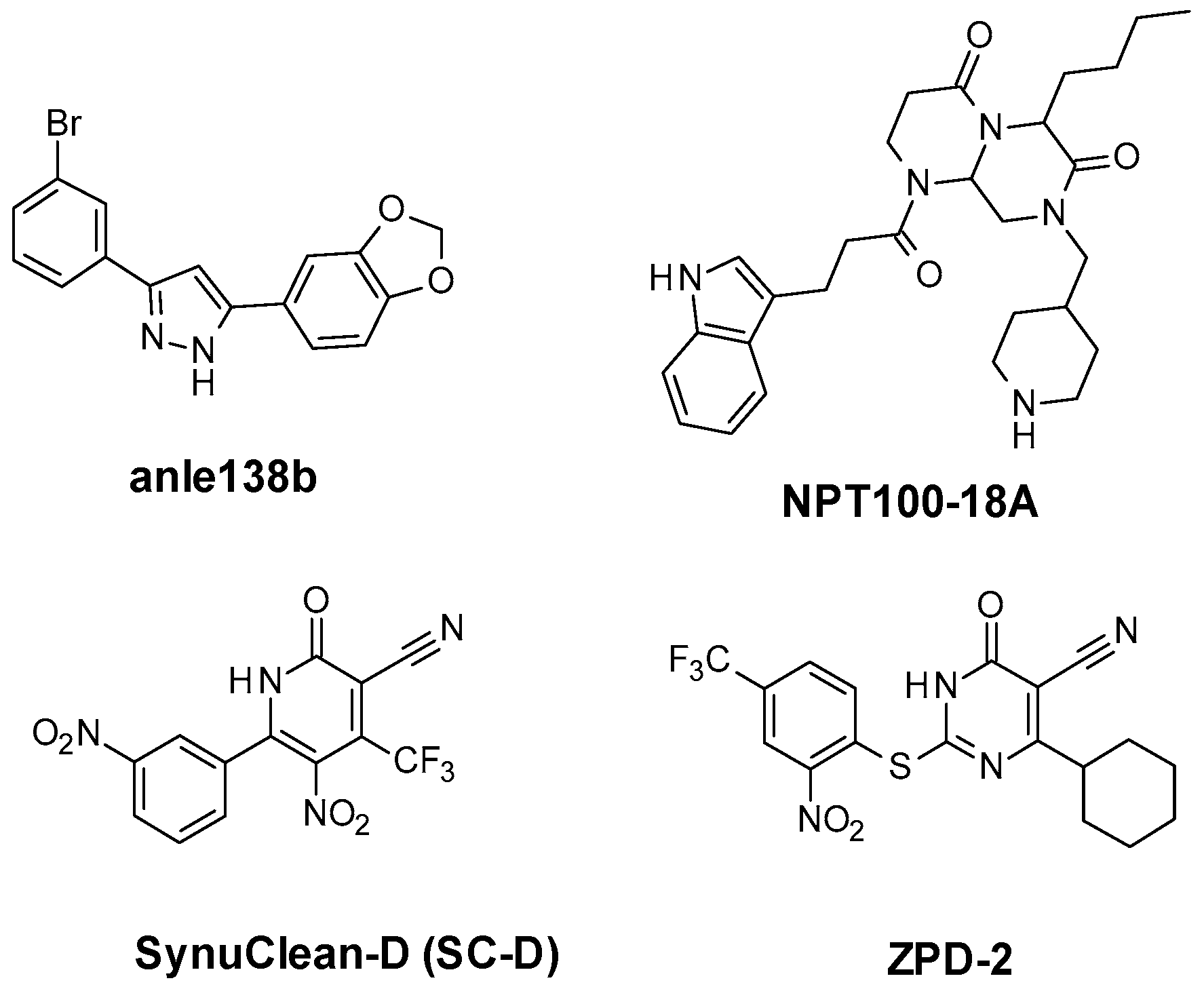
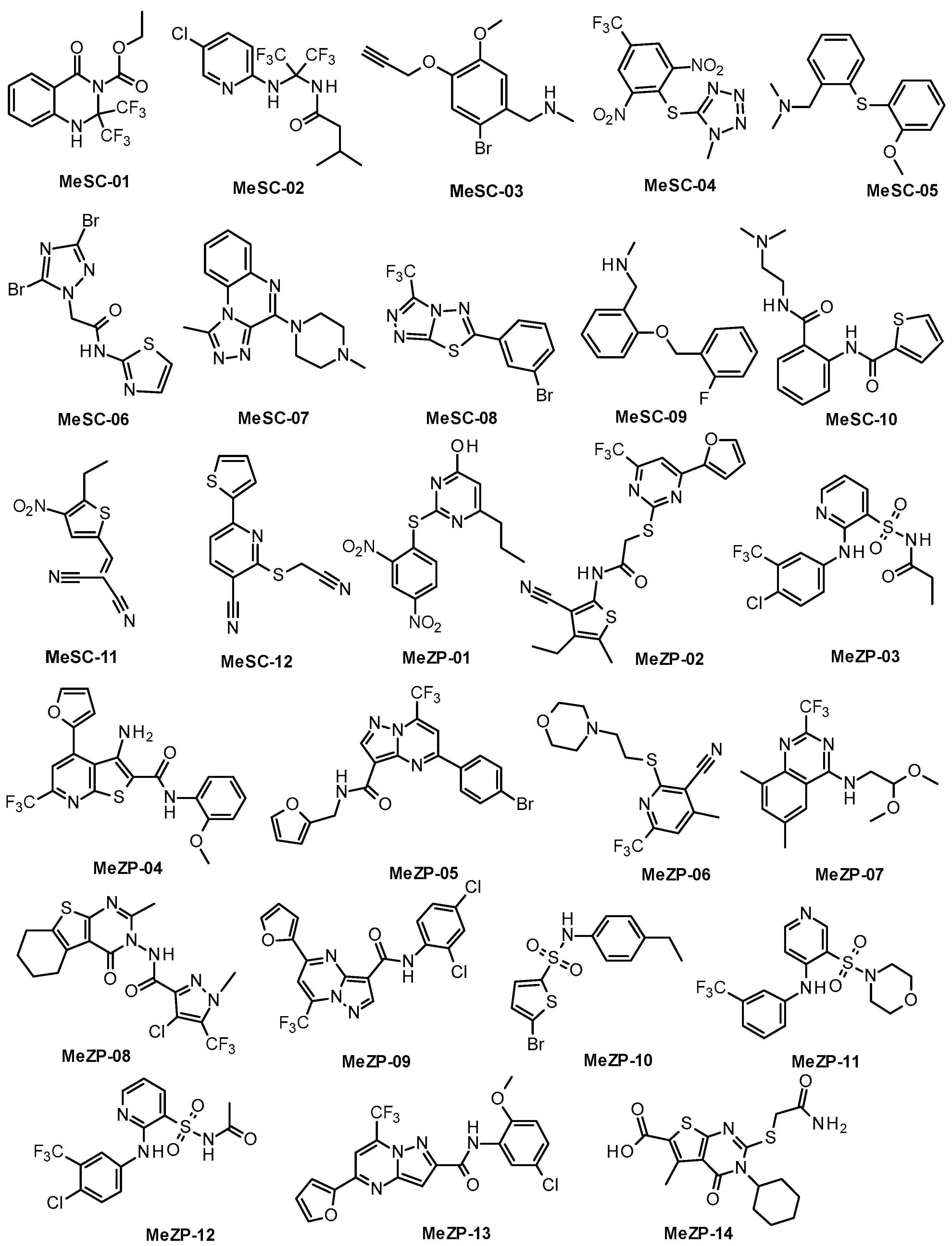
2.1. Similarity-Based Virtual Screening (VS)
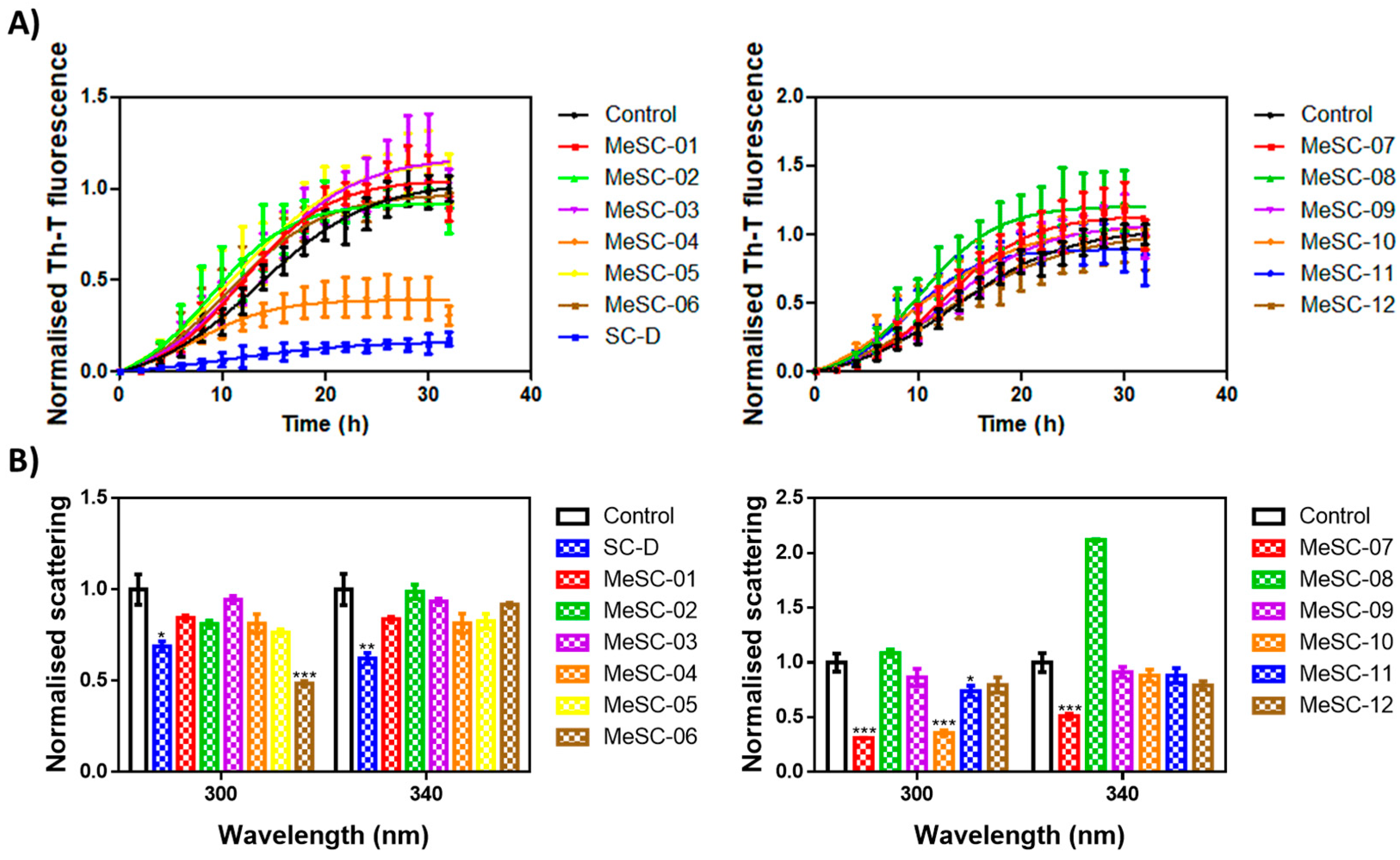
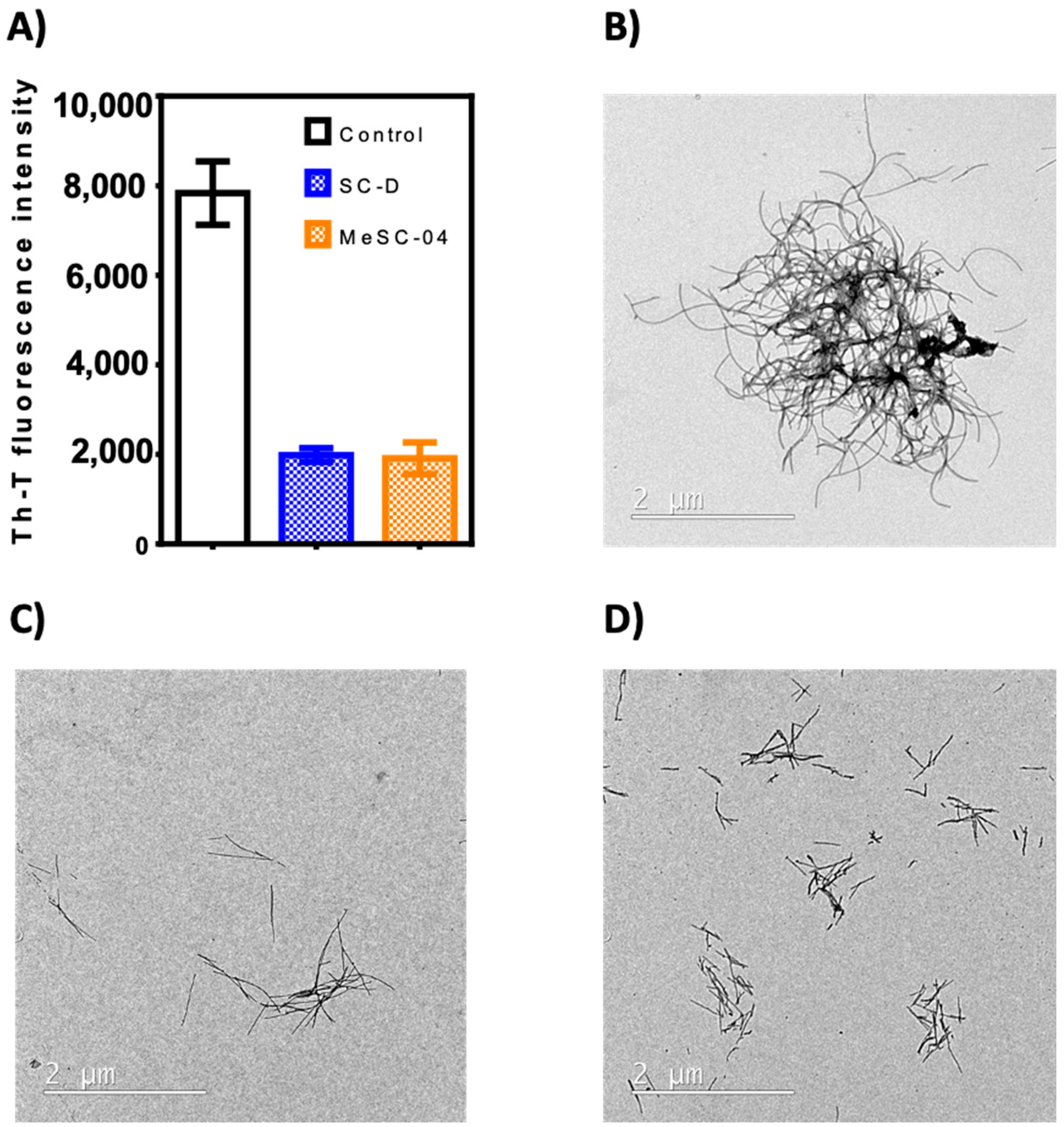
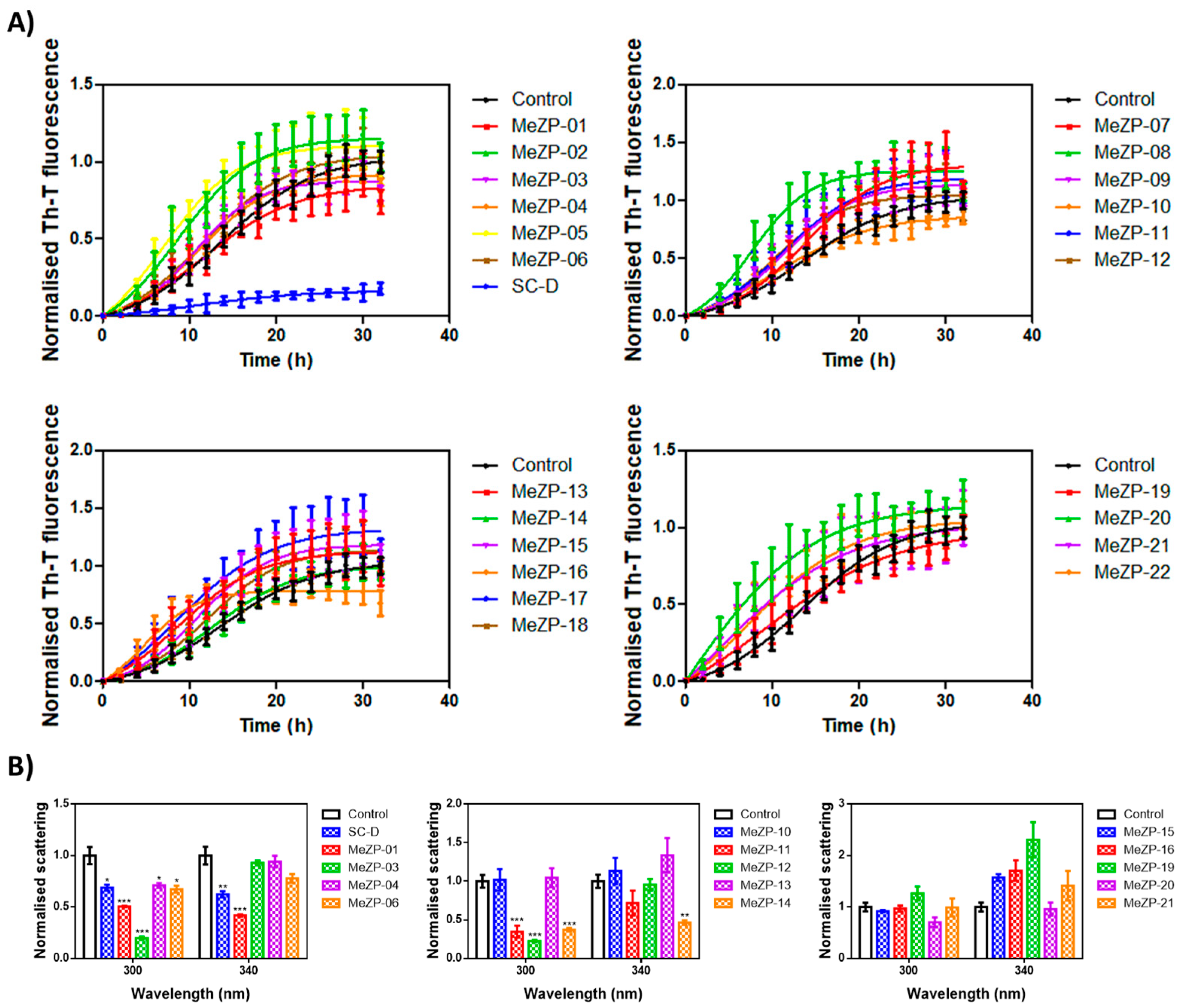
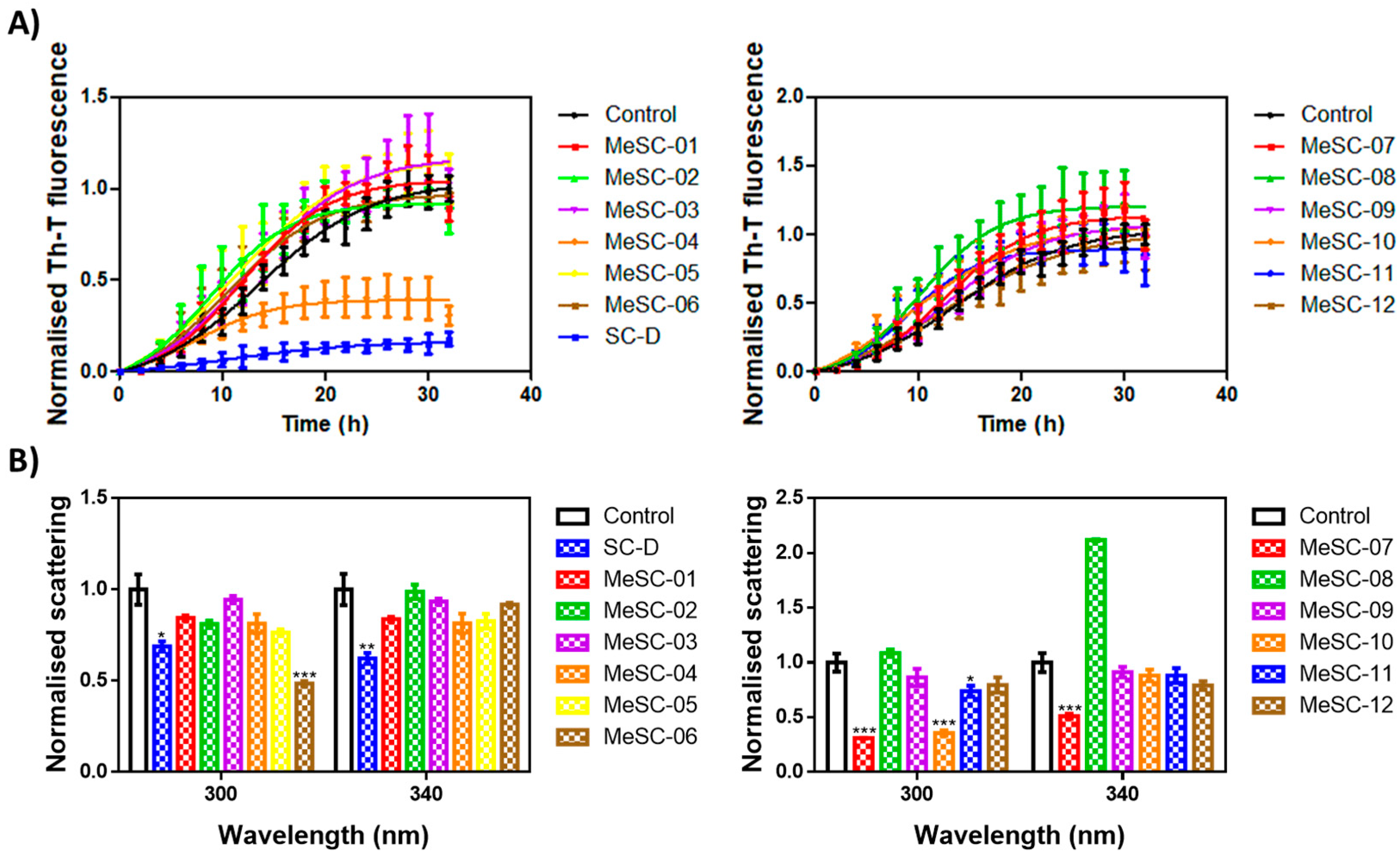
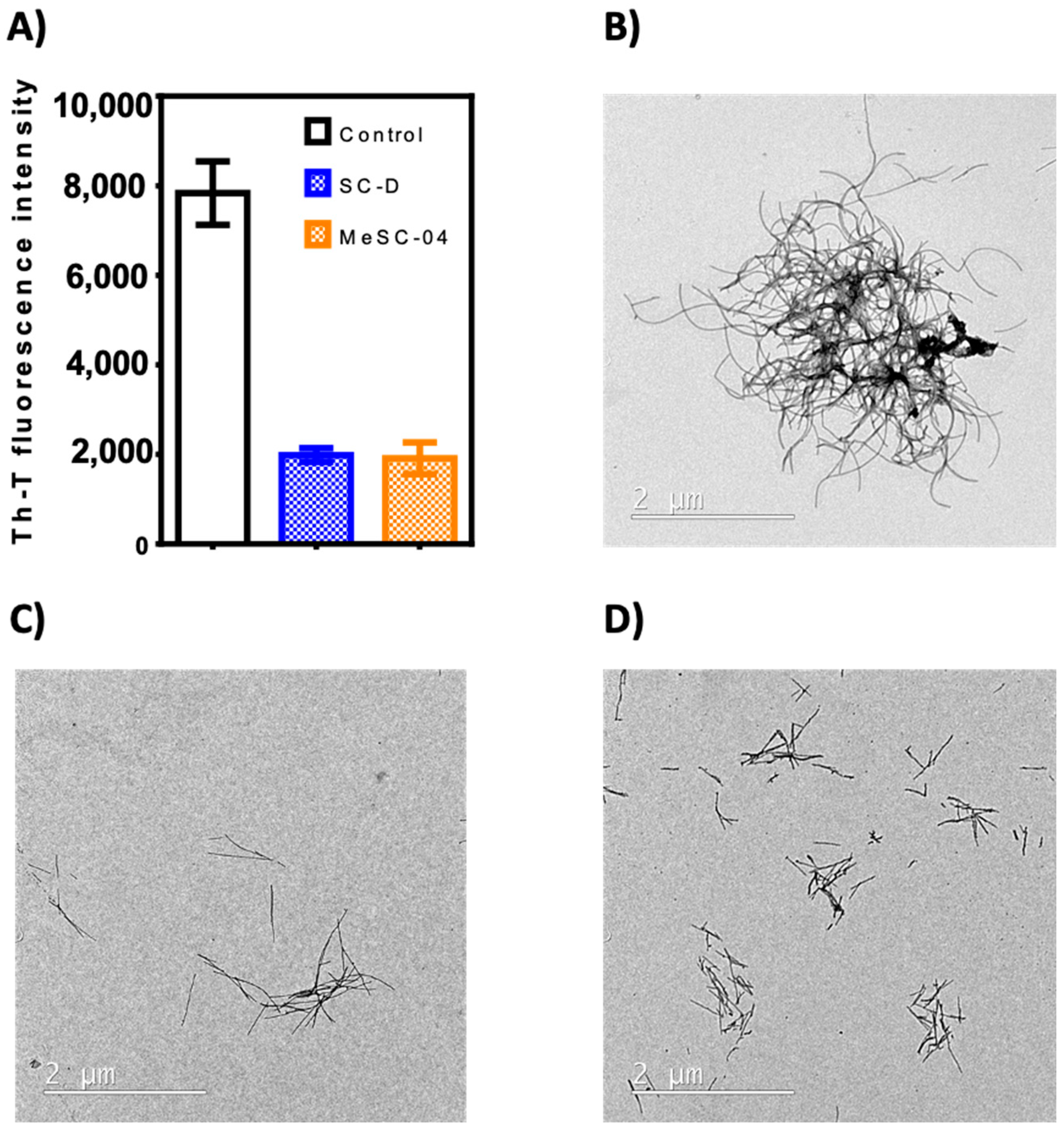
2.2. Thioflavin-T Fluorescence Test
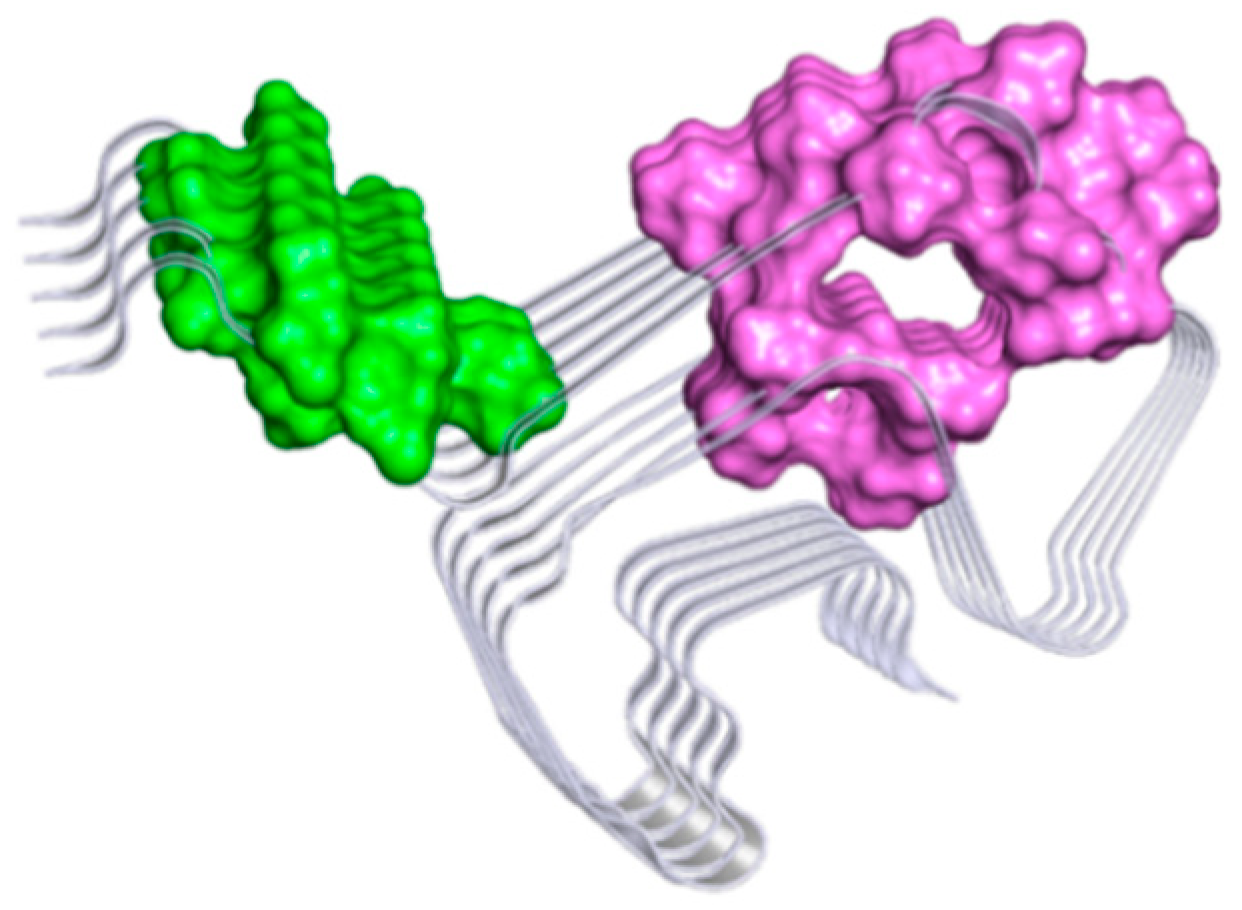
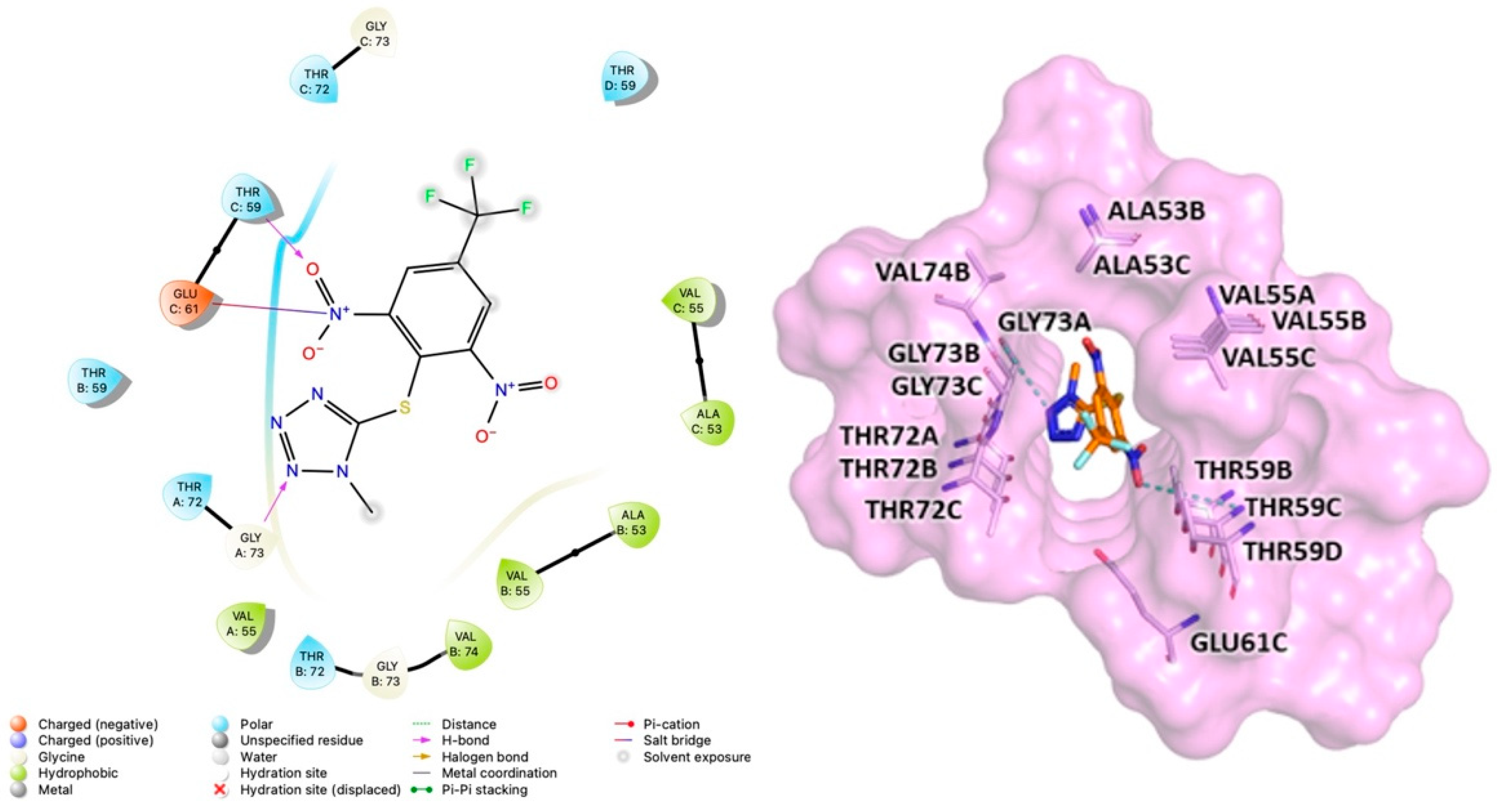
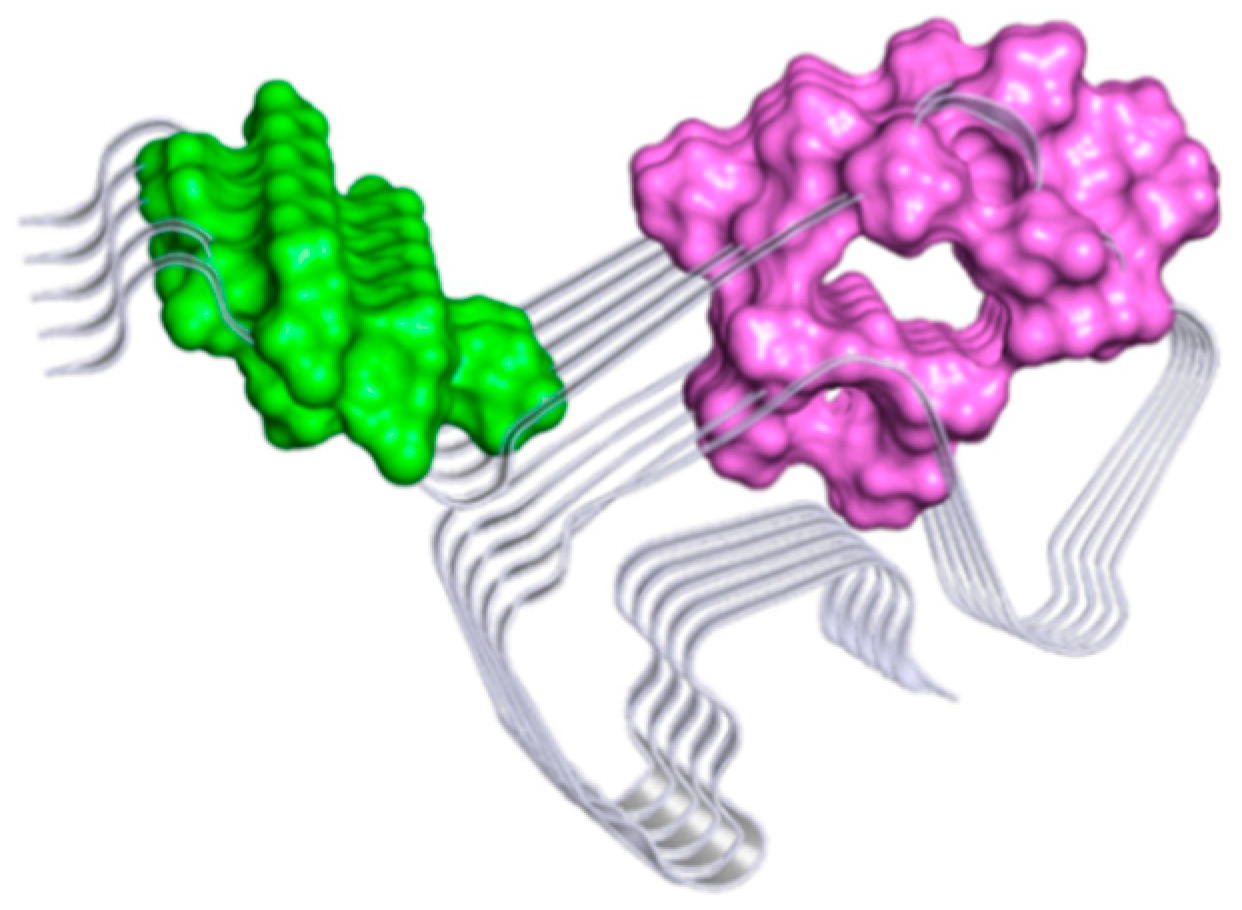
2.3. Computational Studies to Predict the Binding Site
3. Materials and Methods
3.1. Ligand-Based Virtual Screening
3.2. In Vitro Testing (Th-T Fluorescence, Light Scattering, and Electron Microscopy Assays)
3.3. Analysis of Potential Binding Sites
3.4. Molecular Docking Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Martinelli, A.H.S.; Lopes, F.C.; John, E.B.O.; Carlini, C.R.; Ligabue-Braun, R. Modulation of Disordered Proteins with a Focus on Neurodegenerative Diseases and Other Pathologies. Int. J. Mol. Sci. 2019, 20, 1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grosso Jasutkar, H.; Oh, S.E.; Mouradian, M.M. Therapeutics in the Pipeline Targeting alpha-Synuclein for Parkinson’s Disease. Pharmacol. Rev. 2022, 74, 207–237. [Google Scholar] [CrossRef]
- Fields, C.R.; Bengoa-Vergniory, N.; Wade-Martins, R. Targeting Alpha-Synuclein as a Therapy for Parkinson’s Disease. Front. Mol. Neurosci. 2019, 12, 299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.K.; Dutta, A.; Modi, G. alpha-Synuclein aggregation modulation: An emerging approach for the treatment of Parkinson’s disease. Future Med. Chem. 2017, 9, 1039–1053. [Google Scholar] [CrossRef] [PubMed]
- AlNajjar, Y.T.; Gabr, M.; ElHady, A.K.; Salah, M.; Wilms, G.; Abadi, A.H.; Becker, W.; Abdel-Halim, M.; Engel, M. Discovery of novel 6-hydroxybenzothiazole urea derivatives as dual Dyrk1A/alpha-synuclein aggregation inhibitors with neuroprotective effects. Eur. J. Med. Chem. 2022, 227, 113911. [Google Scholar] [CrossRef] [PubMed]
- Gitto, R.; Vittorio, S.; Bucolo, F.; Pena-Diaz, S.; Siracusa, R.; Cuzzocrea, S.; Ventura, S.; Di Paola, R.; De Luca, L. Discovery of Neuroprotective Agents Based on a 5-(4-Pyridinyl)-1,2,4-triazole Scaffold. ACS Chem. Neurosci. 2022, 13, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Dhouafli, Z.; Cuanalo-Contreras, K.; Hayouni, E.A.; Mays, C.E.; Soto, C.; Moreno-Gonzalez, I. Inhibition of protein misfolding and aggregation by natural phenolic compounds. Cell. Mol. Life Sci. 2018, 75, 3521–3538. [Google Scholar] [CrossRef] [PubMed]
- Mahia, A.; Pena-Diaz, S.; Navarro, S.; Jose Galano-Frutos, J.; Pallares, I.; Pujols, J.; Diaz-de-Villegas, M.D.; Galvez, J.A.; Ventura, S.; Sancho, J. Design, synthesis and structure-activity evaluation of novel 2-pyridone-based inhibitors of alpha-synuclein aggregation with potentially improved BBB permeability. Bioorg. Chem. 2021, 117, 105472. [Google Scholar] [CrossRef] [PubMed]
- Pena, D.S.; Ventura, S. One ring is sufficient to inhibit alpha-synuclein aggregation. Neural Regen. Res. 2022, 17, 508–511. [Google Scholar] [CrossRef]
- Pena-Diaz, S.; Pujols, J.; Pinheiro, F.; Santos, J.; Pallares, I.; Navarro, S.; Conde-Gimenez, M.; Garcia, J.; Salvatella, X.; Dalfo, E.; et al. Inhibition of alpha-Synuclein Aggregation and Mature Fibril Disassembling with a Minimalistic Compound, ZPDm. Front. Bioeng. Biotechnol. 2020, 8, 588947. [Google Scholar] [CrossRef]
- Price, D.L.; Koike, M.A.; Khan, A.; Wrasidlo, W.; Rockenstein, E.; Masliah, E.; Bonhaus, D. The small molecule alpha-synuclein misfolding inhibitor, NPT200-11, produces multiple benefits in an animal model of Parkinson’s disease. Sci. Rep. 2018, 8, 16165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pujols, J.; Pena-Diaz, S.; Conde-Gimenez, M.; Pinheiro, F.; Navarro, S.; Sancho, J.; Ventura, S. High-Throughput Screening Methodology to Identify Alpha-Synuclein Aggregation Inhibitors. Int. J. Mol. Sci. 2017, 18, 478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pujols, J.; Pena-Diaz, S.; Lazaro, D.F.; Peccati, F.; Pinheiro, F.; Gonzalez, D.; Carija, A.; Navarro, S.; Conde-Gimenez, M.; Garcia, J.; et al. Small molecule inhibits alpha-synuclein aggregation, disrupts amyloid fibrils, and prevents degeneration of dopaminergic neurons. Proc. Natl. Acad. Sci. USA 2018, 115, 10481–10486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vittorio, S.; Adornato, I.; Gitto, R.; Pena-Diaz, S.; Ventura, S.; De Luca, L. Rational design of small molecules able to inhibit alpha-synuclein amyloid aggregation for the treatment of Parkinson’s disease. J. Enzyme Inhib. Med. Chem. 2020, 35, 1727–1735. [Google Scholar] [CrossRef]
- Heras-Garvin, A.; Weckbecker, D.; Ryazanov, S.; Leonov, A.; Griesinger, C.; Giese, A.; Wenning, G.K.; Stefanova, N. Anle138b modulates alpha-synuclein oligomerization and prevents motor decline and neurodegeneration in a mouse model of multiple system atrophy. Mov. Disord. 2019, 34, 255–263. [Google Scholar] [CrossRef] [Green Version]
- Wagner, J.; Ryazanov, S.; Leonov, A.; Levin, J.; Shi, S.; Schmidt, F.; Prix, C.; Pan-Montojo, F.; Bertsch, U.; Mitteregger-Kretzschmar, G.; et al. Anle138b: A novel oligomer modulator for disease-modifying therapy of neurodegenerative diseases such as prion and Parkinson’s disease. Acta Neuropathol. 2013, 125, 795–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pena-Diaz, S.; Pujols, J.; Conde-Gimenez, M.; Carija, A.; Dalfo, E.; Garcia, J.; Navarro, S.; Pinheiro, F.; Santos, J.; Salvatella, X.; et al. ZPD-2, a Small Compound That Inhibits alpha-Synuclein Amyloid Aggregation and Its Seeded Polymerization. Front. Mol. Neurosci. 2019, 12, 306. [Google Scholar] [CrossRef]
- Bragina, M.E.; Daina, A.; Perez, M.A.S.; Michielin, O.; Zoete, V. The SwissSimilarity 2021 Web Tool: Novel Chemical Libraries and Additional Methods for an Enhanced Ligand-Based Virtual Screening Experience. Int. J. Mol. Sci. 2022, 23, 811. [Google Scholar] [CrossRef]
- Zoete, V.; Daina, A.; Bovigny, C.; Michielin, O. SwissSimilarity: A Web Tool for Low to Ultra High Throughput Ligand-Based Virtual Screening. J. Chem. Inf. Model 2016, 56, 1399–1404. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.S.; Morris, G.M.; Finn, P.W.; Sharma, R.; Moretti, L.; Cooper, R.I.; Richards, W.G. ElectroShape: Fast molecular similarity calculations incorporating shape, chirality and electrostatics. J. Comput. Aided Mol. Des. 2010, 24, 789–801. [Google Scholar] [CrossRef]
- Gladysz, R.; Dos Santos, F.M.; Langenaeker, W.; Thijs, G.; Augustyns, K.; De Winter, H. Spectrophores as one-dimensional descriptors calculated from three-dimensional atomic properties: Applications ranging from scaffold hopping to multi-target virtual screening. J. Cheminform. 2018, 10, 9. [Google Scholar] [CrossRef] [Green Version]
- Tuttle, M.D.; Comellas, G.; Nieuwkoop, A.J.; Covell, D.J.; Berthold, D.A.; Kloepper, K.D.; Courtney, J.M.; Kim, J.K.; Barclay, A.M.; Kendall, A.; et al. Solid-state NMR structure of a pathogenic fibril of full-length human α-synuclein. Nat. Struct. Mol. Biol. 2016, 23, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Ferreira, R.; Taylor, N.M.; Mona, D.; Ringler, P.; Lauer, M.E.; Riek, R.; Britschgi, M.; Stahlberg, H. Cryo-EM structure of alpha-synuclein fibrils. eLife 2018, 7, e36402. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System, Version 1.8; Schrödinger, L.L.C.: New York, NY, USA, 2015.
- Wang, W.; Wang, X.; Gao, W.; Cui, Z.; Zhang, H.; Lu, F.; Liu, F. Ulvan inhibits alpha-synuclein fibrillation and disrupts the mature fibrils: In vitro and in vivo studies. Int. J. Biol. Macromol. 2022, 211, 580–591. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.J.; Ferrie, J.J.; Xu, K.; Lee, I.; Graham, T.J.A.; Tu, Z.; Yu, J.; Dhavale, D.; Kotzbauer, P.; Petersson, E.J.; et al. Alpha Synuclein Fibrils Contain Multiple Binding Sites for Small Molecules. ACS Chem. Neurosci. 2018, 9, 2521–2527. [Google Scholar] [CrossRef]
- Bian, J.; Liu, Y.Q.; He, J.; Lin, X.; Qiu, C.Y.; Yu, W.B.; Shen, Y.; Zhu, Z.Y.; Ye, D.Y.; Wang, J.; et al. Discovery of styrylaniline derivatives as novel alpha-synuclein aggregates ligands. Eur. J. Med. Chem. 2021, 226, 113887. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Macarthur, M.W.; Moss, D.S.; Thornton, J.M. Procheck—A Program to Check the Stereochemical Quality of Protein Structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Grove, L.E.; Hall, D.R.; Bohnuud, T.; Mottarella, S.E.; Luo, L.; Xia, B.; Beglov, D.; Vajda, S. The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat. Protoc. 2015, 10, 733–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halgren, T. New method for fast and accurate binding-site identification and analysis. Chem. Biol. Drug Des. 2007, 69, 146–148. [Google Scholar] [CrossRef]
- Schmidtke, P.; Le Guilloux, V.; Maupetit, J.; Tuffery, P. fpocket: Online tools for protein ensemble pocket detection and tracking. Nucleic Acids Res. 2010, 38, W582–W589. [Google Scholar] [CrossRef] [Green Version]
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinform. 2009, 10, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrödinger Release 2021-4: Maestro; Schrödinger, L.L.C.: New York, NY, USA, 2021.
- Pedretti, A.; Mazzolari, A.; Gervasoni, S.; Fumagalli, L.; Vistoli, G. The VEGA suite of programs: An versatile platform for cheminformatics and drug design projects. Bioinformatics 2021, 37, 1174–1175. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Query Cmpds | FP2 | Electroshape a | Spectrophores b |
|---|---|---|---|
| SC-D | 3 (3) | 33 (33) | 400 (108) |
| ZPD-2 | 2 (2) | 400 (178) | 400 (39) |
| Putative Binding Sites | Residues |
|---|---|
| SITE A | VAL52A; ALA53A; THR54A; VAL55A; ALA56A; GLU57A; LYS58A; THR59A; LYS60A; GLU61A; VAL70A; VAL71A; THR72A; GLY73A; VAL74A; THR75A; THR92A; GLY51B; VAL52B; ALA53B; THR54B; VAL55B; ALA56B; GLU57B; LYS58B; THR59B; LYS60B; GLU61B; GLN62B; THR64B; VAL66B; VAL70B; VAL71B; THR72B; GLY73B; VAL74B; THR75B; ALA76B; THR92B; HIS50C; GLY51C; VAL52C; ALA53C; THR54C; VAL55C; ALA56C; GLU57C; LYS58C; THR59C; LYS60C; GLU61C; GLN62C; THR54C; VAL66C; ALA69C; VAL70C; VAL71C; THR72C; GLY73C; VAL74C; THR75C; ALA76C; THR92C; GLY93C; GLY51D; VAL52D; ALA53D; THR54D; VAL55D; ALA56D; GLU57D; LYS58D; THR59D; LYS60D; GLU61D; GLN62D; THR64D; VAL70D; VAL71D; THR72D; GLY73D; VAL74D; THR75D; ALA76D; THR92D; GLY51E; VAL52E; ALA53E; THR54E; VAL55E; ALA56E; GLU57E; LYS58E; THR59E; LYS60E; GLU61E; VAL70E; VAL71E; THR72E; GLY73E; VAL74E; THR75E |
| SITE B | VAL40A; GLY41A; SER42A; LYS43A; THR44A; LYS45A; GLU46A; VAL48A; TYR39B; VAL40B; GLY41B; SER42B; LYS43B; THR44B; LYS45B; GLU46B; GLY47B; VAL48B; VAL49B; HIS50B; TYR39C; VAL40C; GLY41C; SER42C; LYS43C; THR44C; LYS45C; GLU46C; GLY47C; VAL48C; VAL49C; HIS50C; TYR39D; VAL40D; GLY41D; SER42D; LYS43D; THR44D; LYS45D; GLU46D; GLY47D; VAL48D; VAL49D; HIS50D; GLY41E; SER42E; LYS43E; THR44E; LYS45E; GLU46E; VAL48E; VAL49E; HIS50E. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Luca, L.; Vittorio, S.; Peña-Díaz, S.; Pitasi, G.; Fornt-Suñé, M.; Bucolo, F.; Ventura, S.; Gitto, R. Ligand-Based Discovery of a Small Molecule as Inhibitor of α-Synuclein Amyloid Formation. Int. J. Mol. Sci. 2022, 23, 14844. https://doi.org/10.3390/ijms232314844
De Luca L, Vittorio S, Peña-Díaz S, Pitasi G, Fornt-Suñé M, Bucolo F, Ventura S, Gitto R. Ligand-Based Discovery of a Small Molecule as Inhibitor of α-Synuclein Amyloid Formation. International Journal of Molecular Sciences. 2022; 23(23):14844. https://doi.org/10.3390/ijms232314844
Chicago/Turabian StyleDe Luca, Laura, Serena Vittorio, Samuel Peña-Díaz, Giovanna Pitasi, Marc Fornt-Suñé, Federica Bucolo, Salvador Ventura, and Rosaria Gitto. 2022. "Ligand-Based Discovery of a Small Molecule as Inhibitor of α-Synuclein Amyloid Formation" International Journal of Molecular Sciences 23, no. 23: 14844. https://doi.org/10.3390/ijms232314844
APA StyleDe Luca, L., Vittorio, S., Peña-Díaz, S., Pitasi, G., Fornt-Suñé, M., Bucolo, F., Ventura, S., & Gitto, R. (2022). Ligand-Based Discovery of a Small Molecule as Inhibitor of α-Synuclein Amyloid Formation. International Journal of Molecular Sciences, 23(23), 14844. https://doi.org/10.3390/ijms232314844