
Nicotinamide Mononucleotide Supplementation Improves Mitochondrial Dysfunction and Rescues Cellular Senescence by NAD+/Sirt3 Pathway in Mesenchymal Stem Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
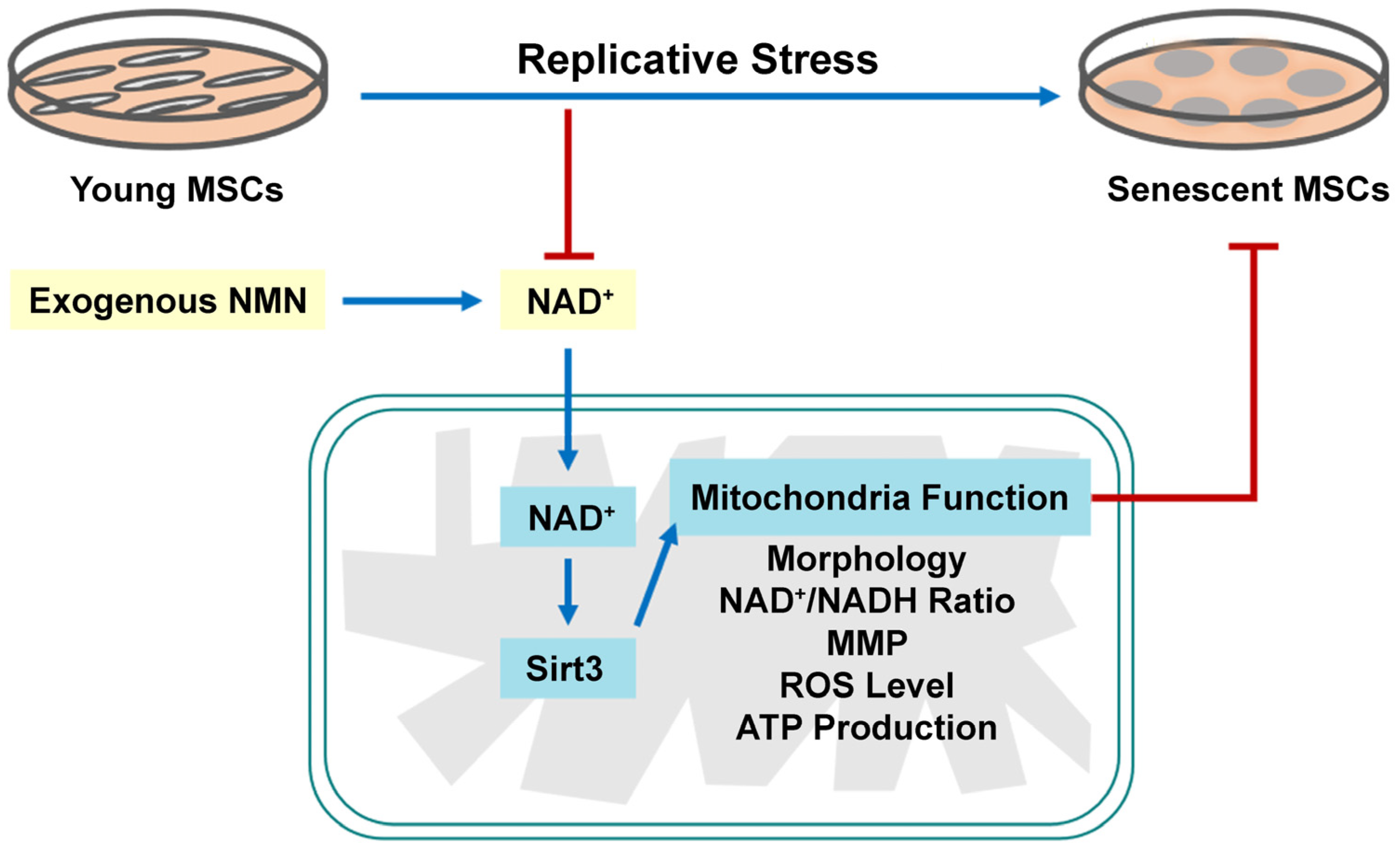
Abstract
:1. Introduction
2. Results
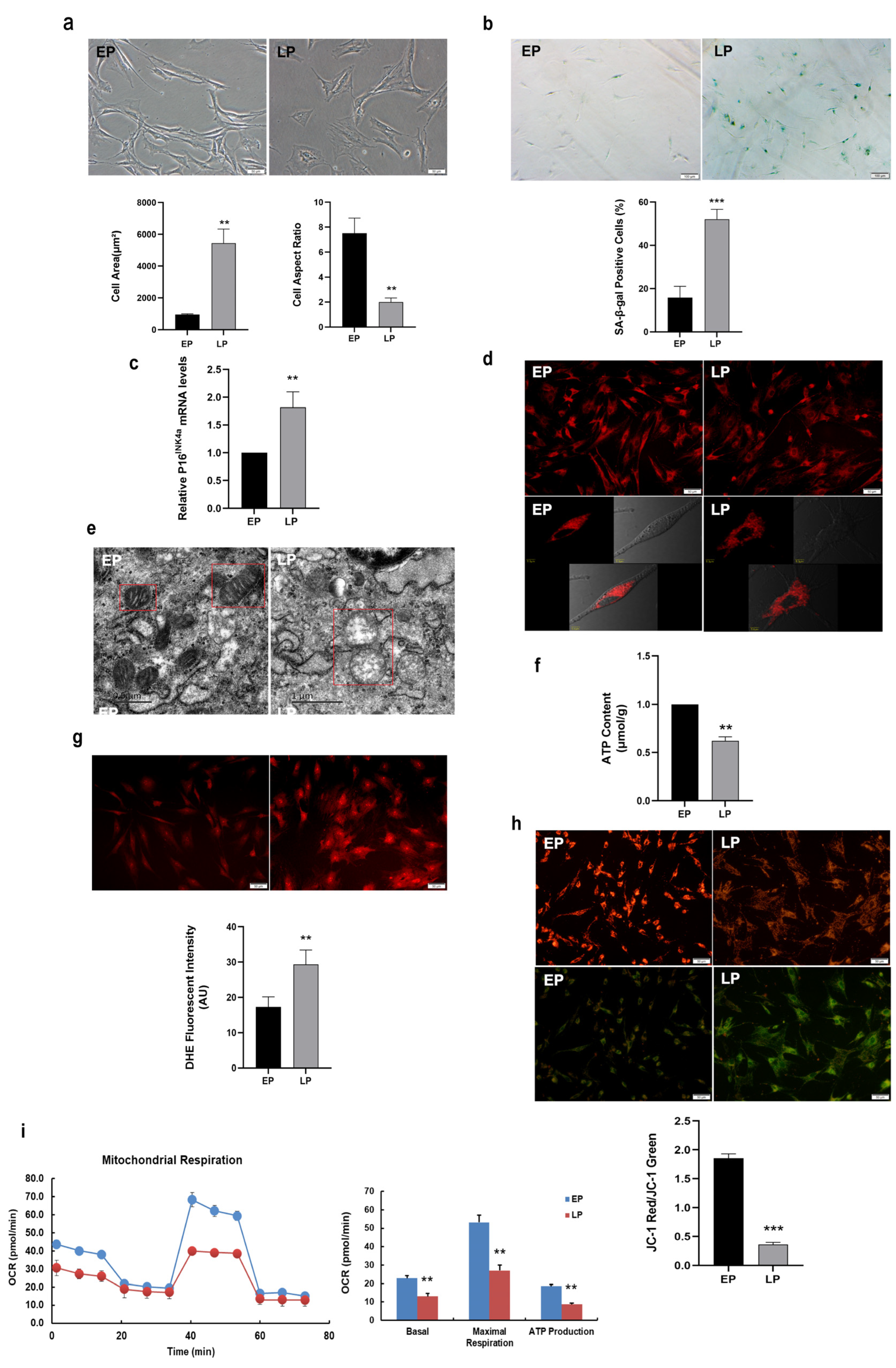
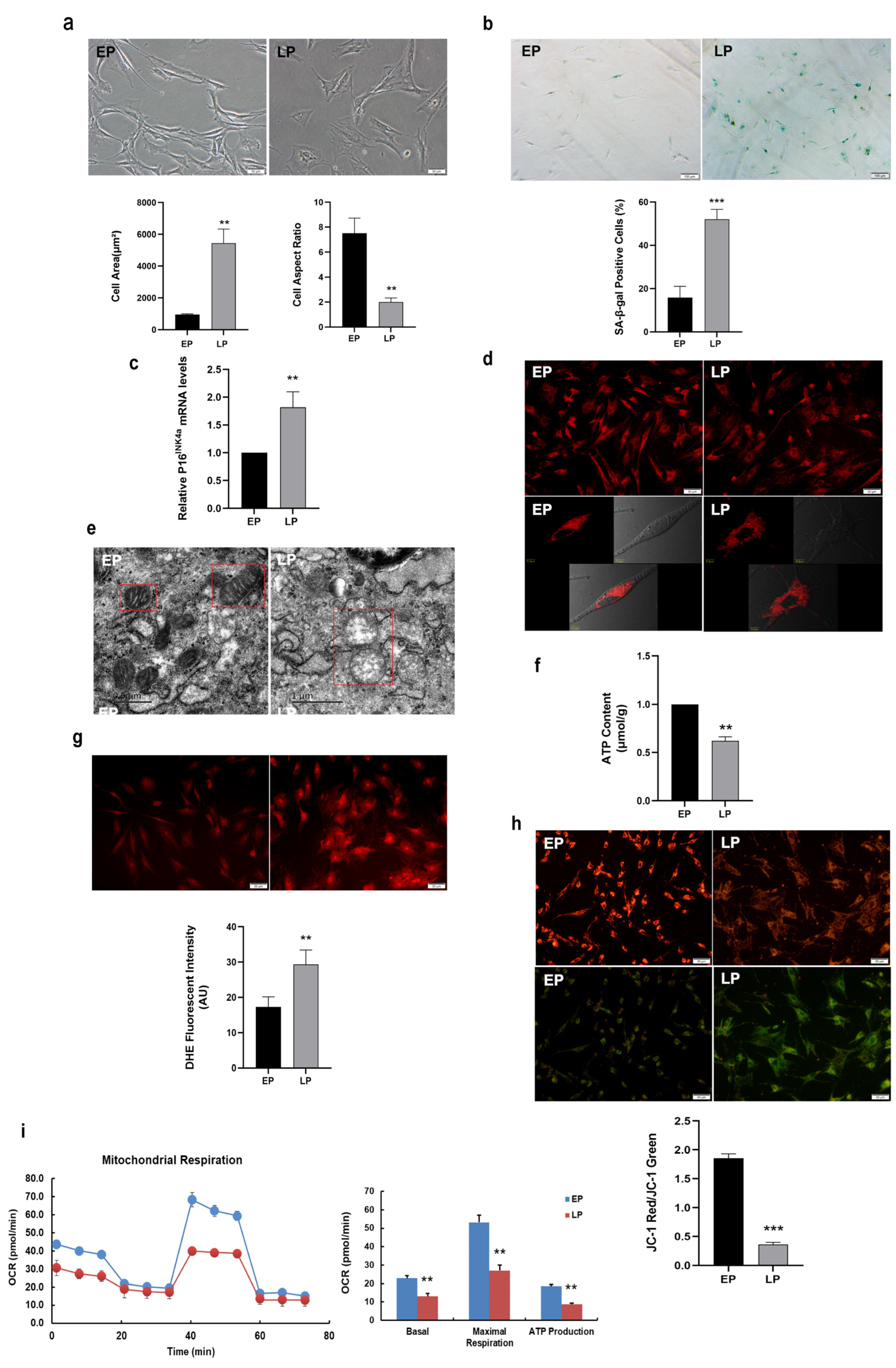
2.1. Mitochondrial Dysfunction in Replicative Senescent MSCs
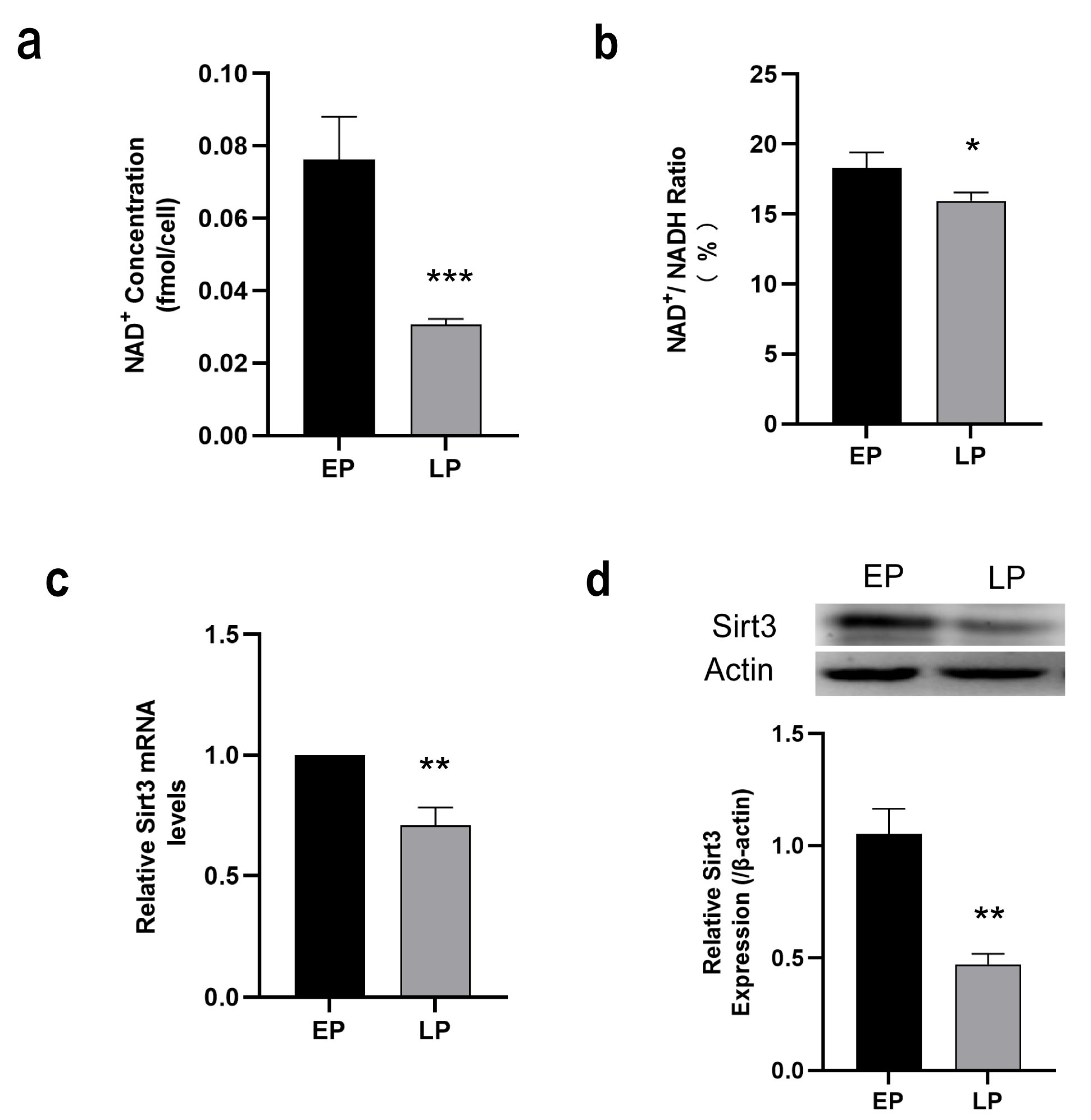
2.2. Down-Regulation of NAD+/Sirt3 Signaling Pathway in LP MSCs
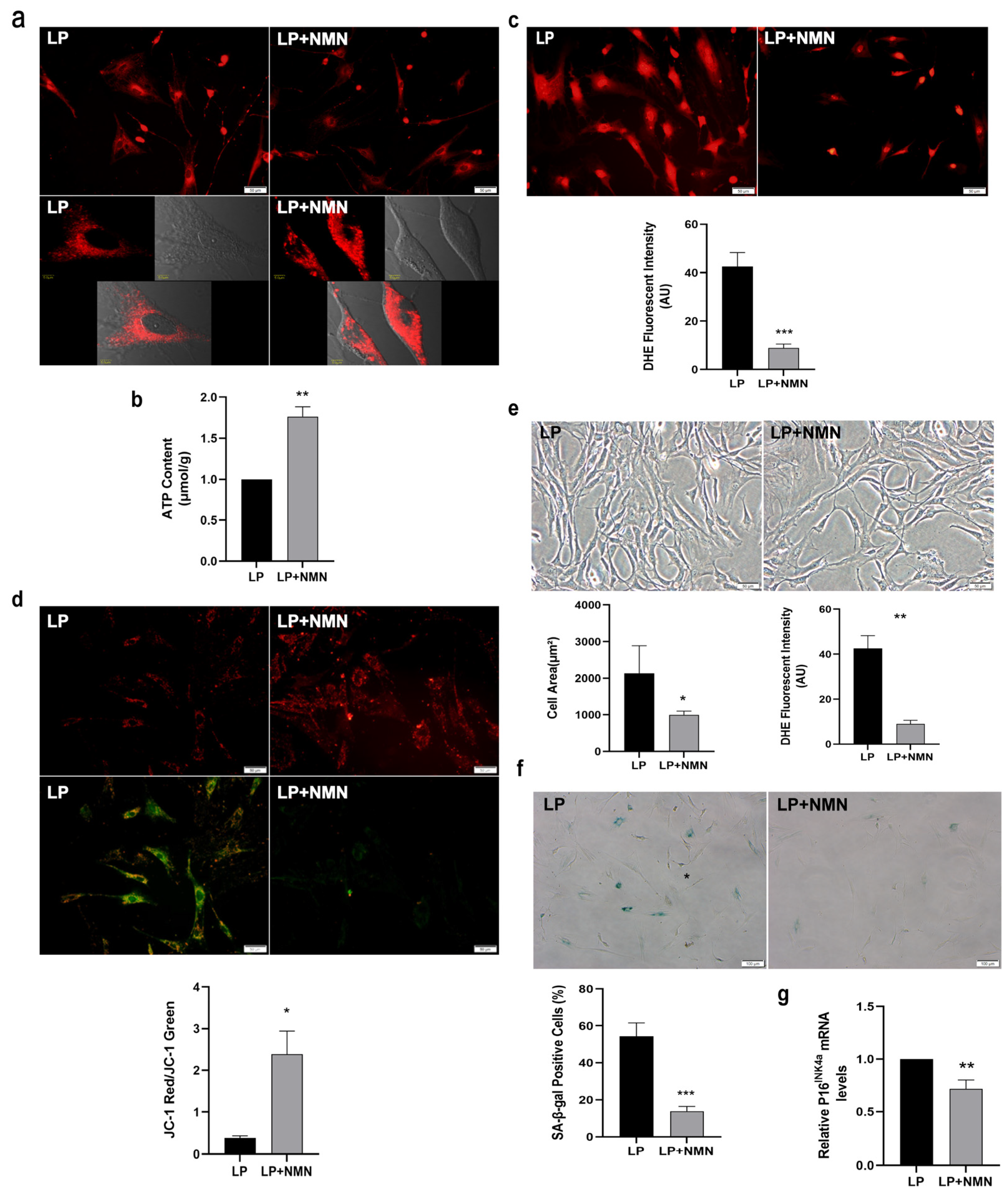
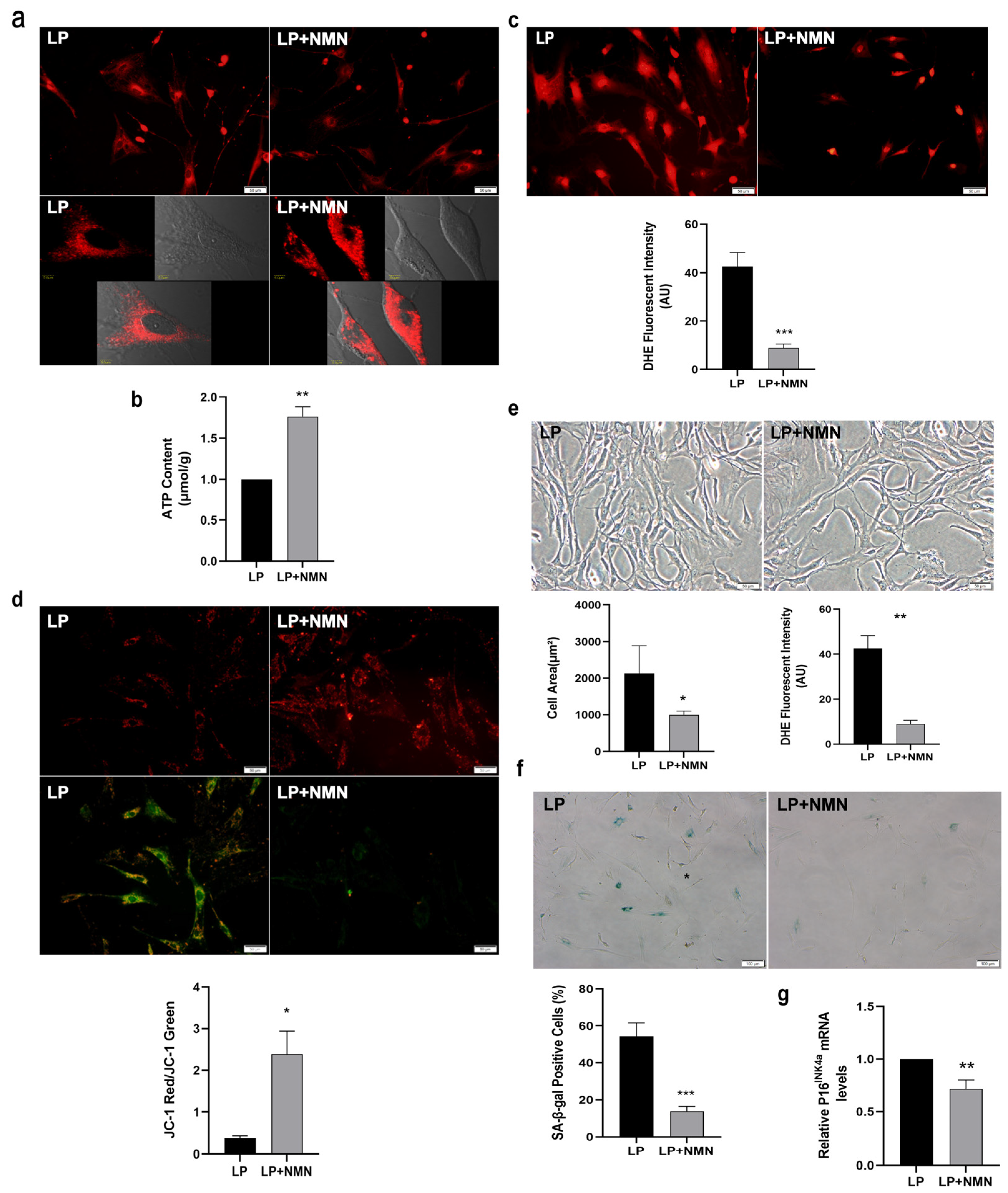
2.3. NMN Improves Mitochondrial Function and Inhibits Expansion-Mediated MSC Replicative Senescence
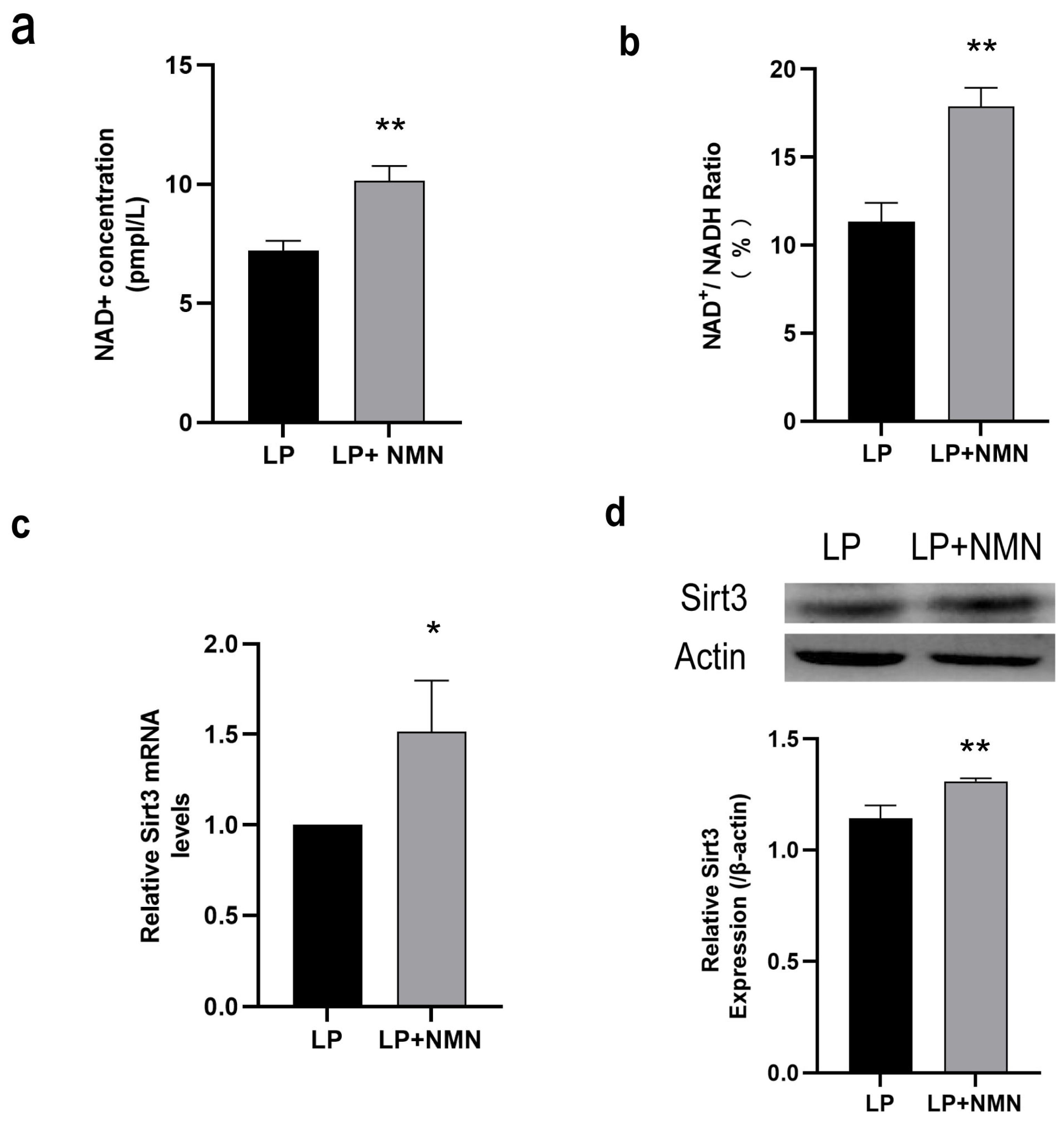
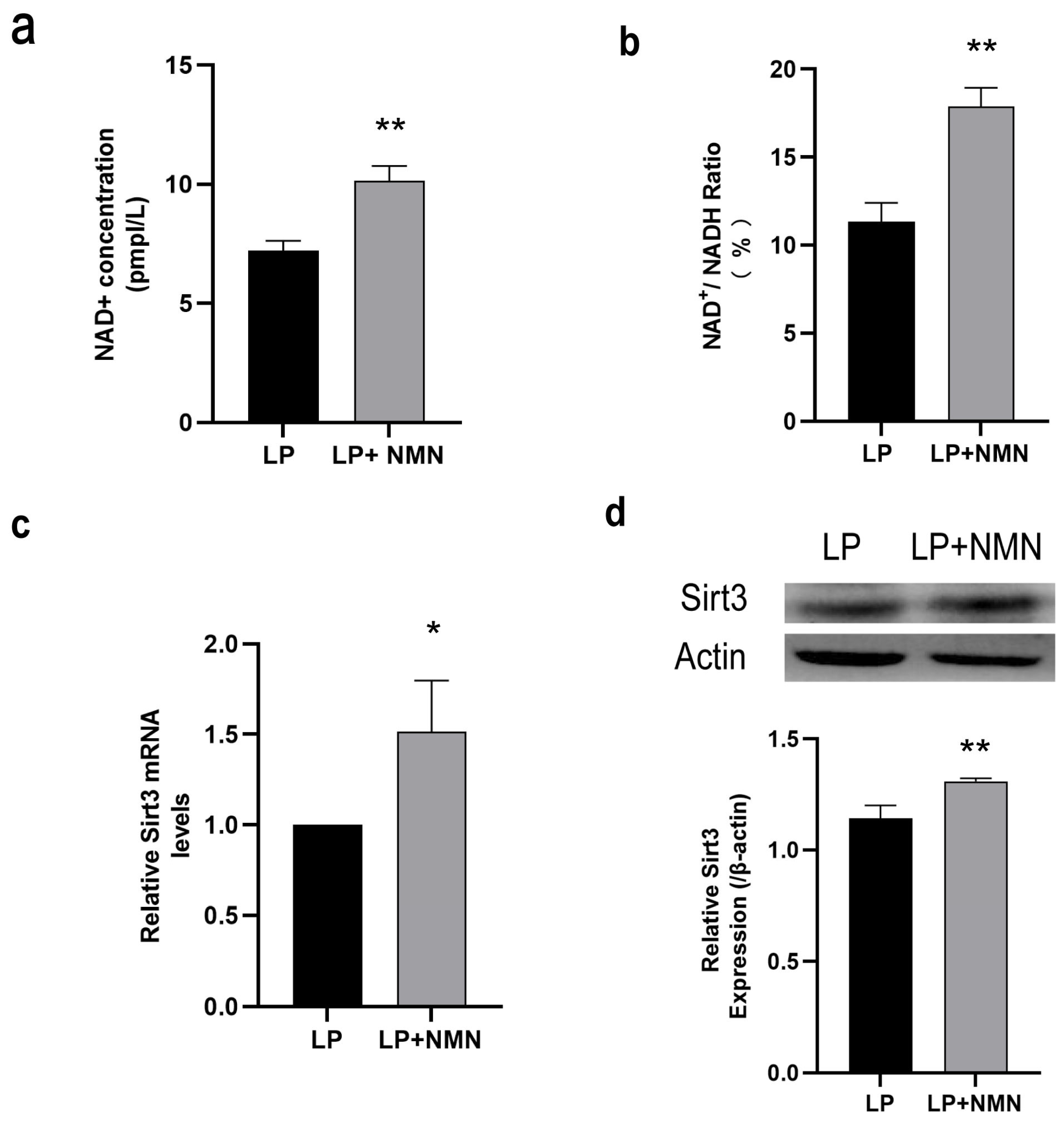
2.4. NMN Up-Regulates NAD+/Sirt3 Signaling Pathway in Replicative Senescent MSCs
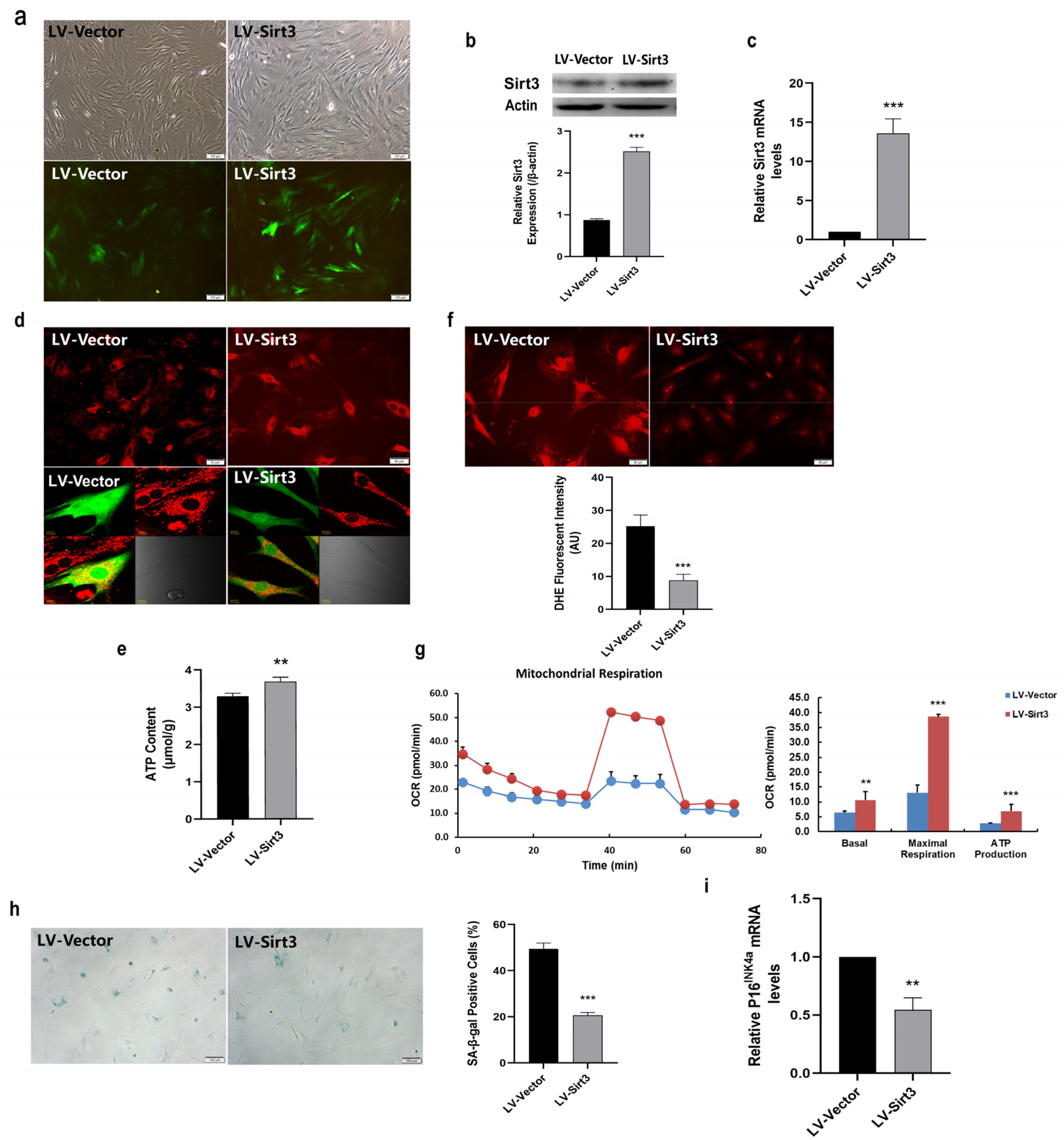
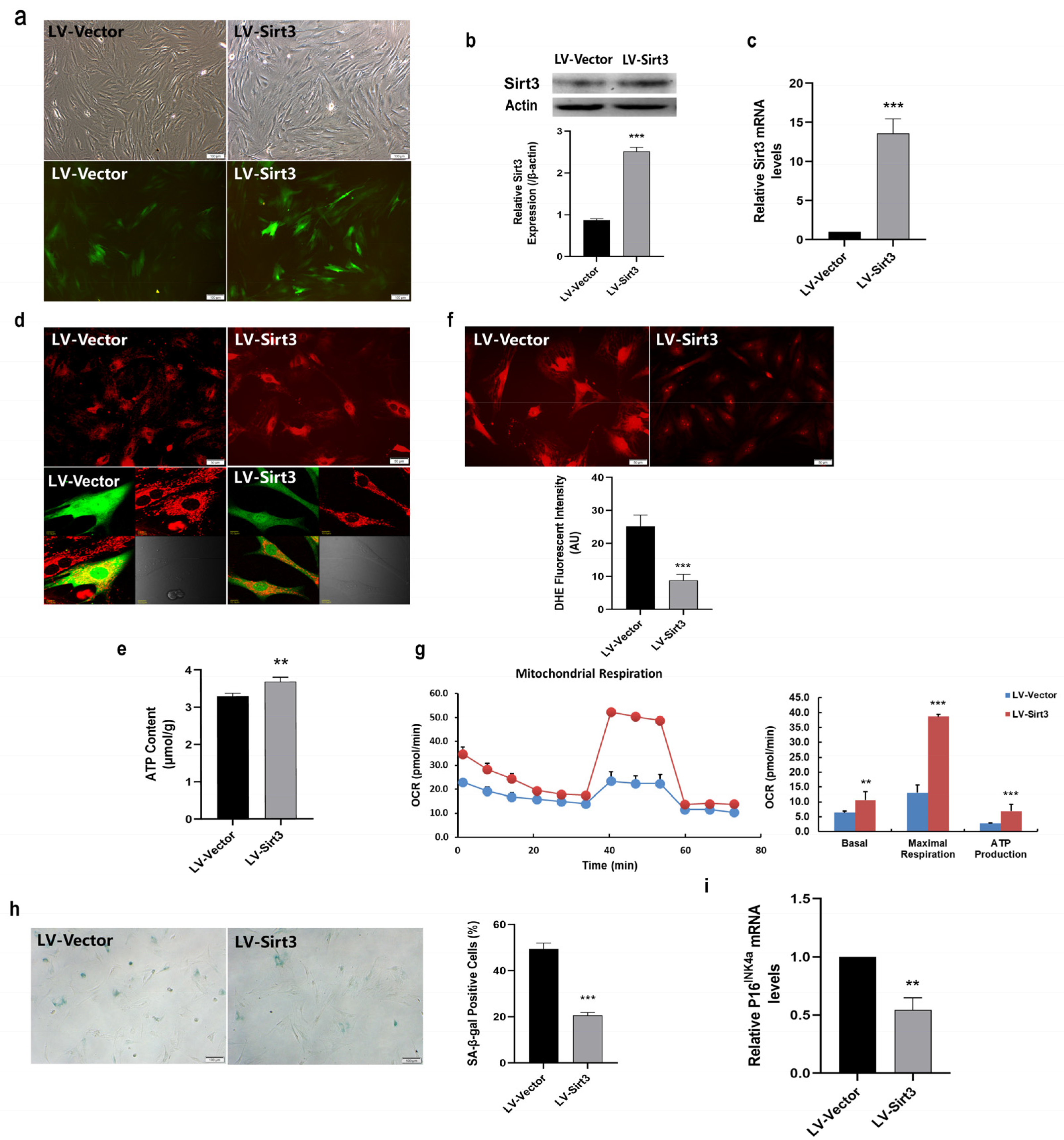
2.5. Sirt3 Over-Expression Ameliorates Mitochondrial Function and Rescues MSC Senescence
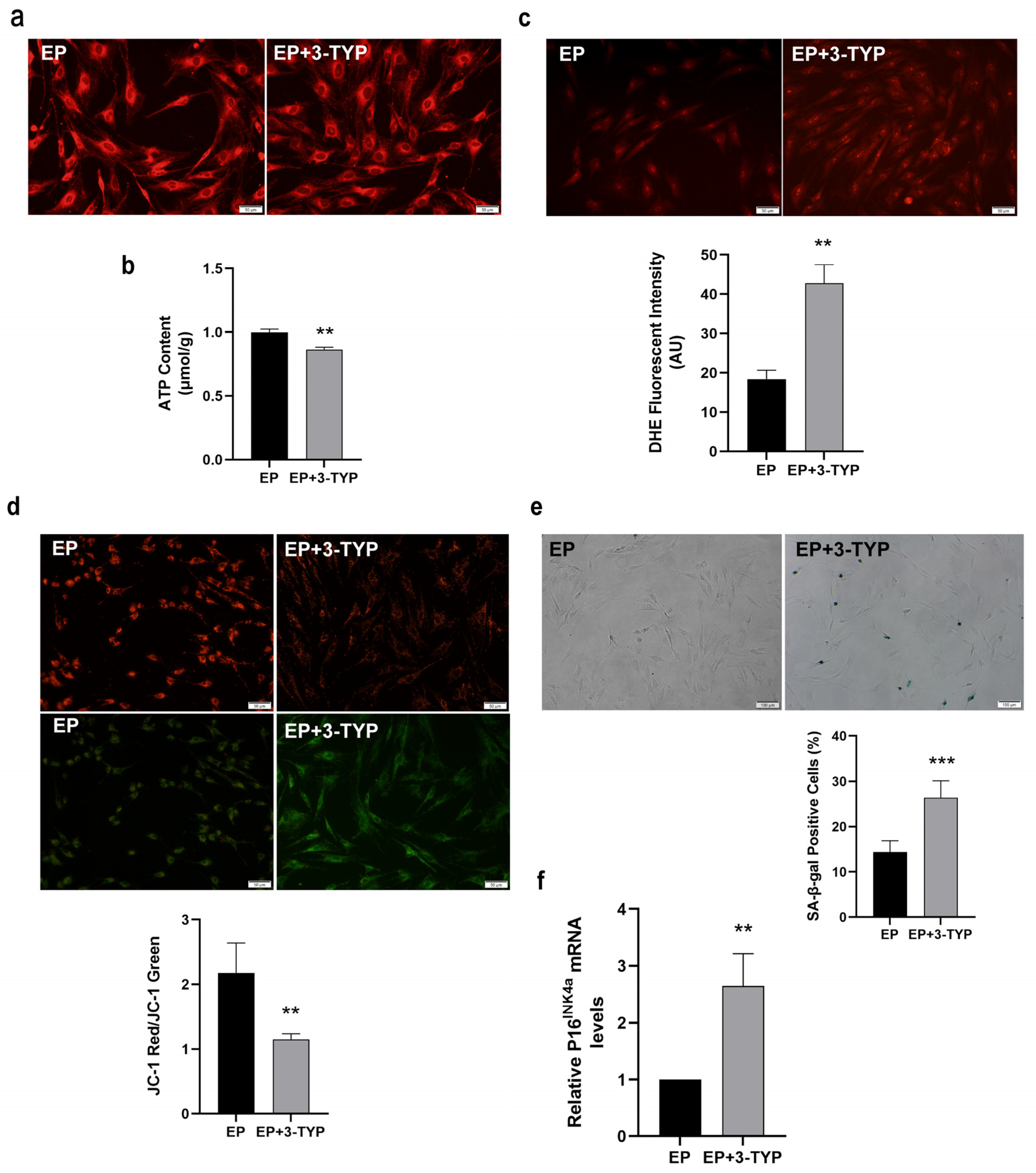
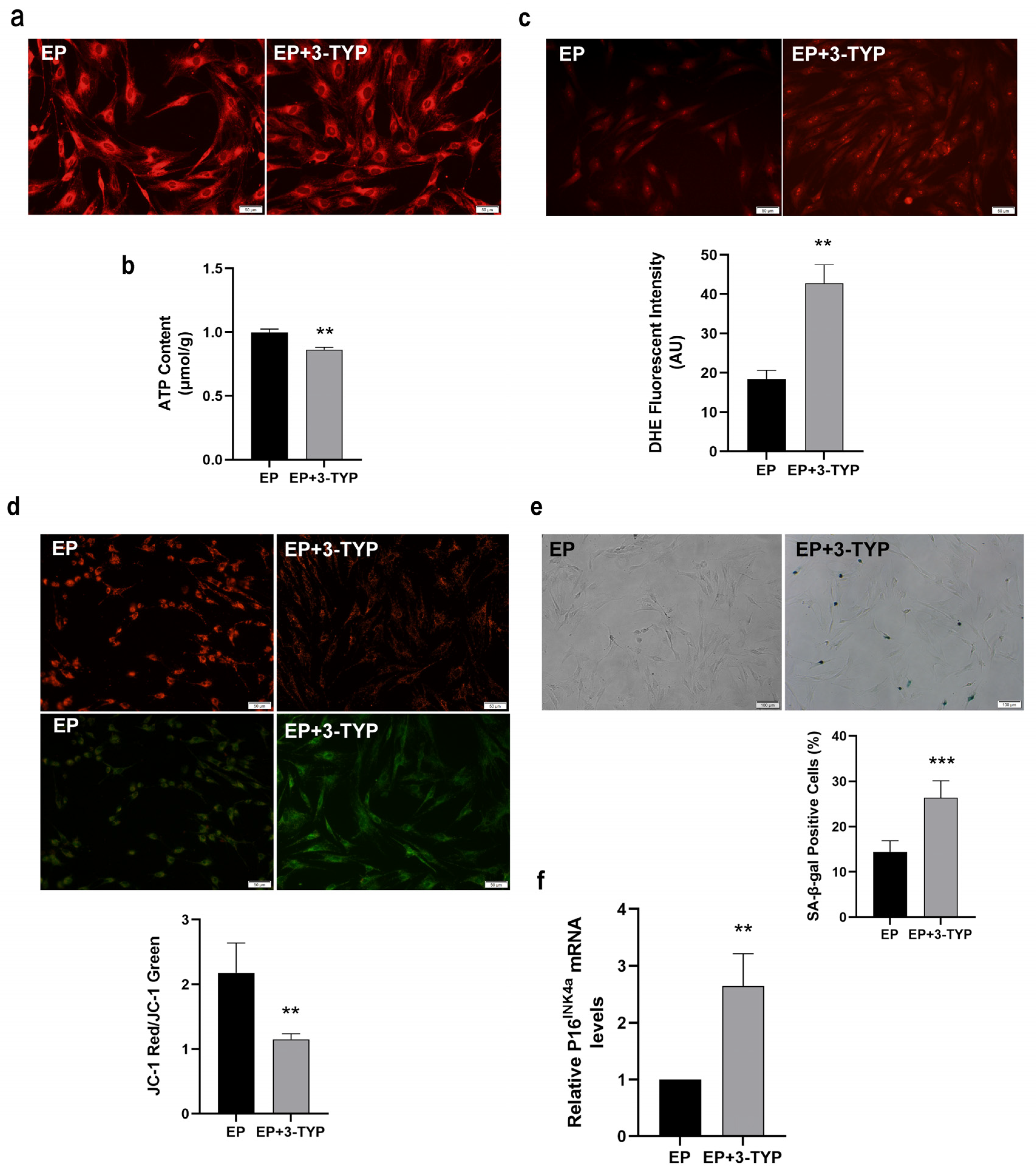
2.6. Sirt3 Selective Inhibitor 3-TYP Induces Mitochondrial Dysfunction and Accelerates MSC Replicative Senescence
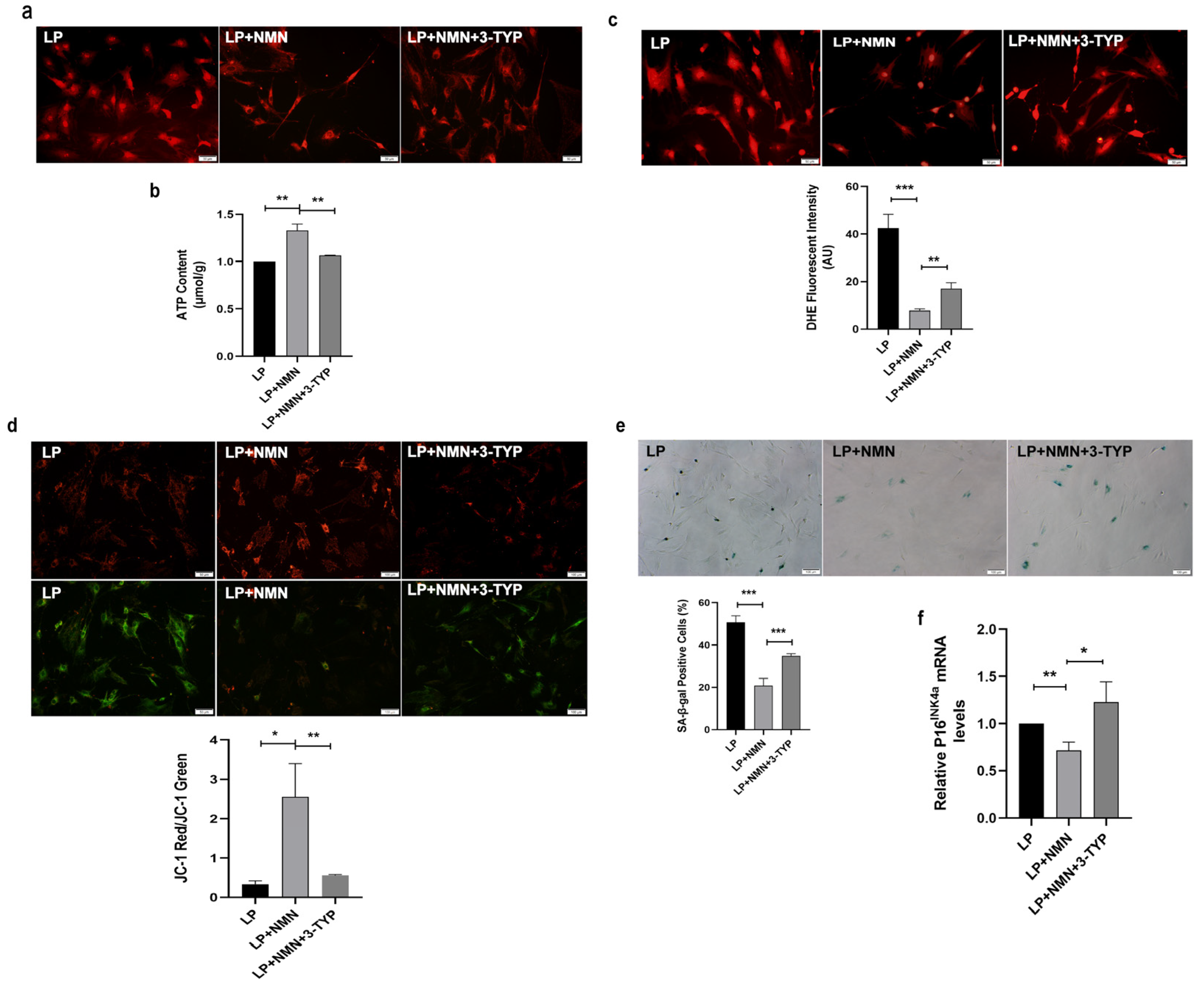
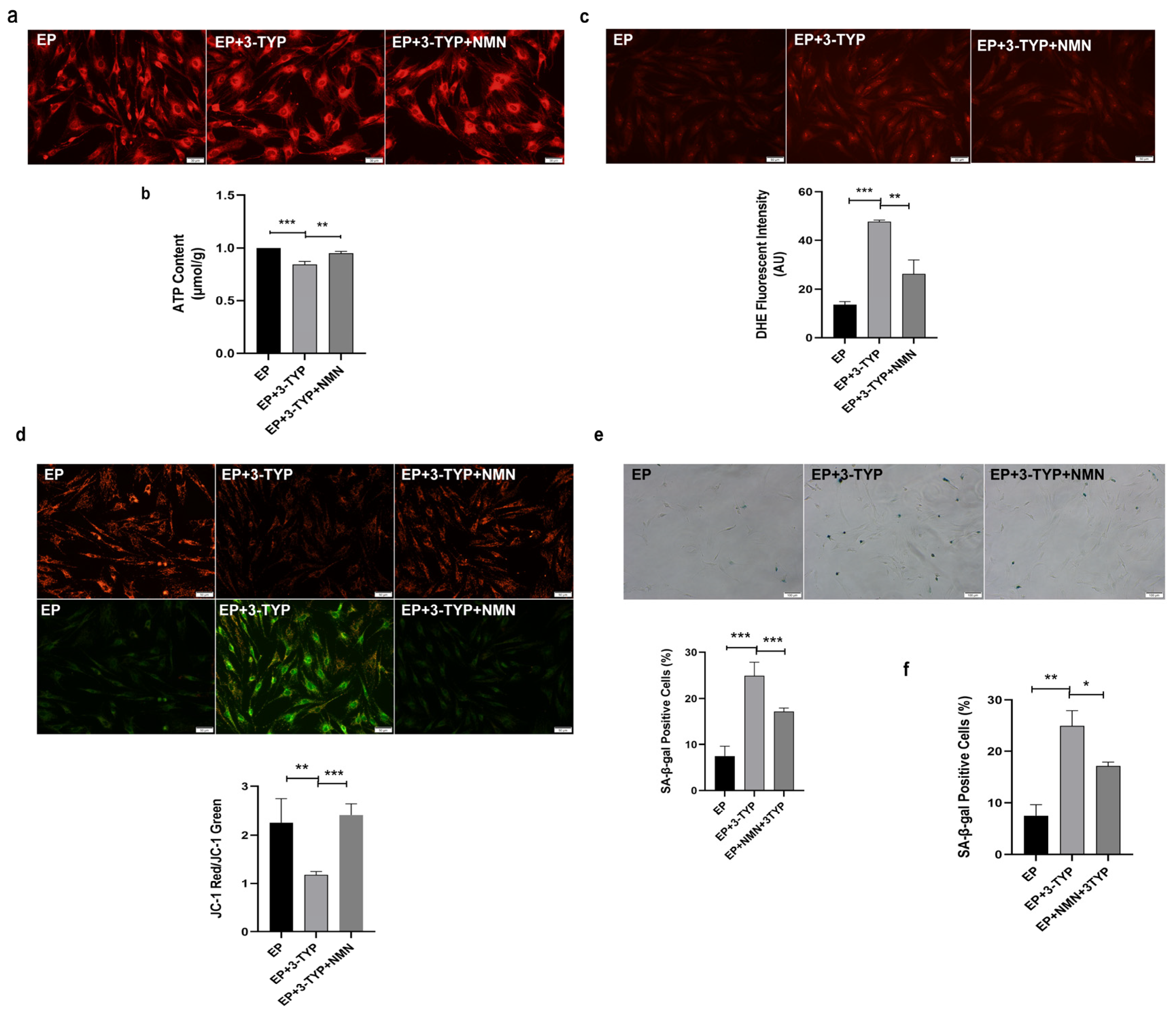
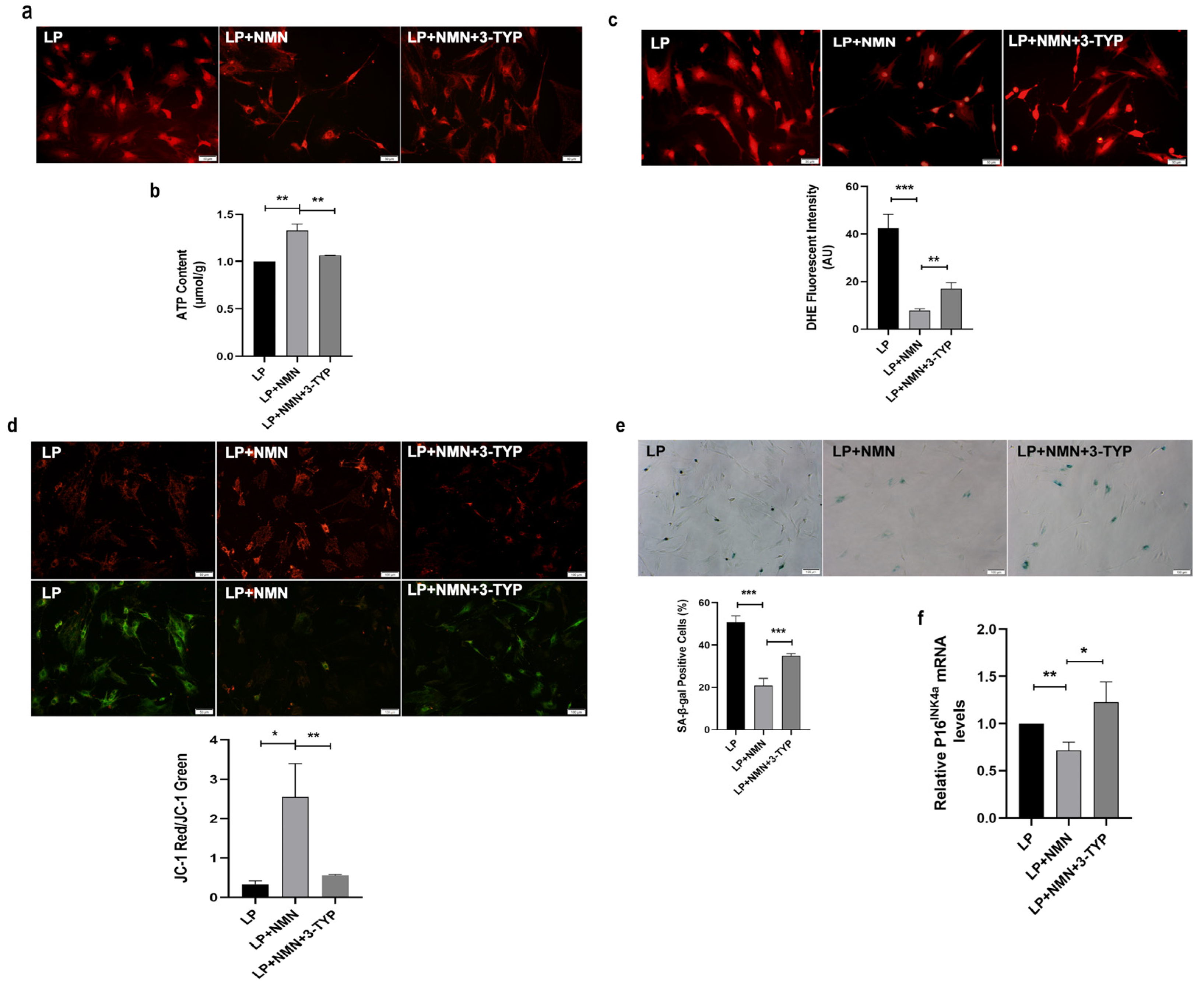
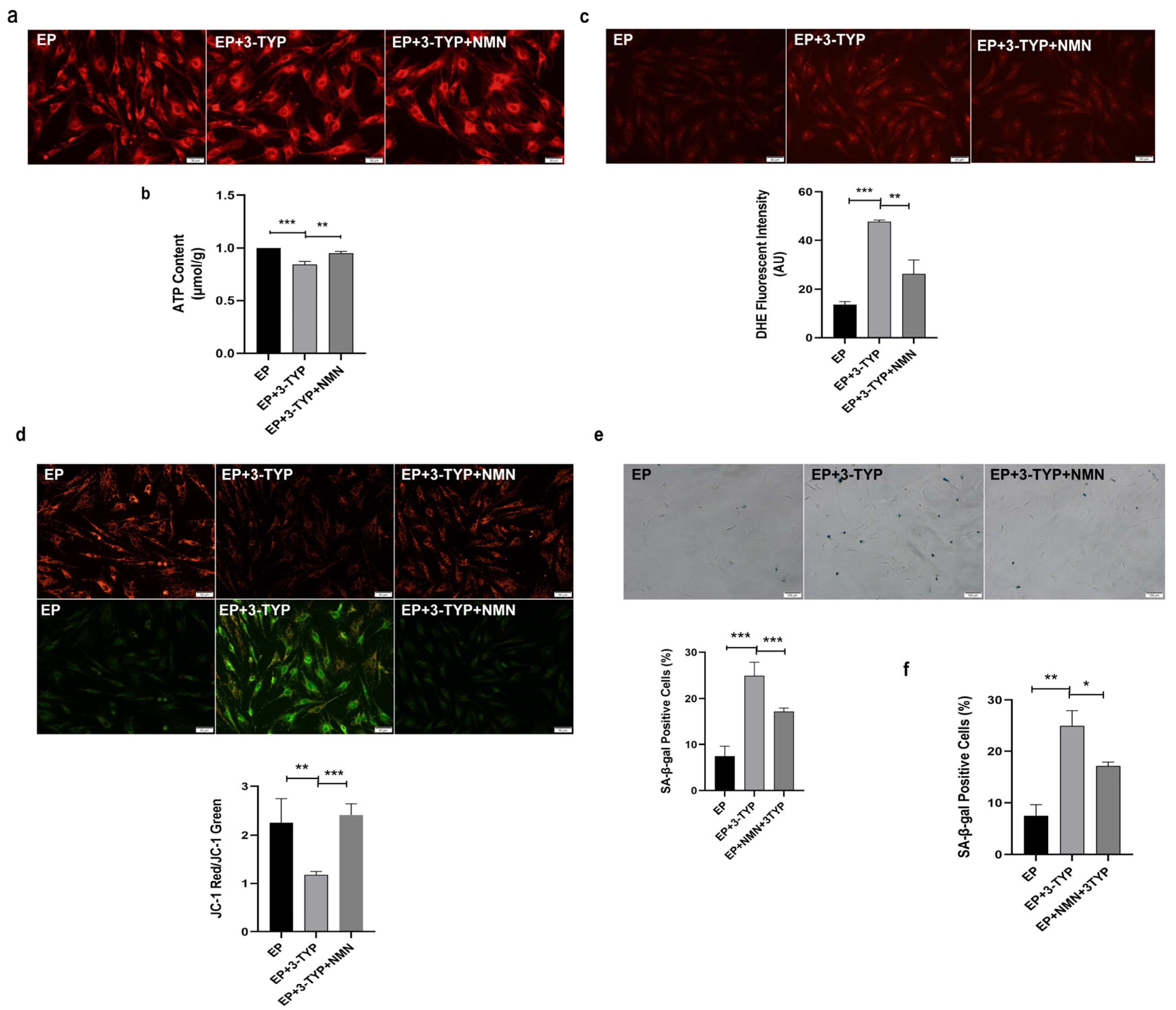
2.7. NMN Rejuvenates Mitochondrial Function and Delays MSC Senescence through Activating NAD+/Sirt3 Signaling Pathway
3. Discussion
4. Materials and Methods
4.1. MSC Isolation and Subculture
4.2. Quantitative Analysis of MSC Morphology
4.3. Senescence-Associated β-Galactosidase (SA-β-gal) Staining
4.4. Gene Expression Analysis
4.5. Mitochondrial MitoTracker Red Staining
4.6. Measurement of ATP Levels
4.7. Measurement of ROS Levels
4.8. Measurement of MMP
4.9. Oxygen Consumption Rate (OCR)
4.10. Determination of Intracellular NAD+ and NAD+/NADH Ratio
4.11. Lentiviral Transduction of MSCs
4.12. NMN and 3-TYP Intermediates Treatment
4.13. Western Blot Analysis
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Birch, J.; Barnes, P.J.; Passos, J.F. Mitochondria, telomeres and cell senescence: Implications for lung ageing and disease. Pharmacol. Ther. 2018, 183, 34–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correia-Melo, C.; Passos, J.F. Mitochondria: Are they causal players in cellular senescence? Biochim. Biophys. Acta 2015, 1847, 1373–1379. [Google Scholar] [CrossRef] [Green Version]
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence. Genes. Dev. 2010, 24, 2463–2479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Martin, N.; Beach, D.; Gil, J. Ageing as developmental decay: Insights from p16(INK4a.). Trends. Mol. Med. 2014, 20, 667–674. [Google Scholar] [CrossRef]
- Sun, Y.; Ma, C.; Sun, H.; Wang, H.; Peng, W.; Zhou, Z.; Wang, H.; Pi, C.; Shi, Y.; He, X. Metabolism: A Novel Shared Link between Diabetes Mellitus and Alzheimer’s Disease. J. Diabetes Res. 2020, 2020, 4981814. [Google Scholar] [CrossRef]
- Yu, E.P.; Reinhold, J.; Yu, H.; Starks, L.; Uryga, A.K.; Foote, K.; Finigan, A.; Figg, N.; Pung, Y.F.; Logan, A.; et al. Mitochondrial Respiration Is Reduced in Atherosclerosis, Promoting Necrotic Core Formation and Reducing Relative Fibrous Cap Thickness. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2322–2332. [Google Scholar] [CrossRef] [Green Version]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.U.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef] [Green Version]
- Mu, W.-C.; Ohkubo, R.; Widjaja, A.; Chen, D. The mitochondrial metabolic checkpoint in stem cell aging and rejuvenation. Mech. Ageing Dev. 2020, 188, 111254. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends. Biochem. Sci. 2010, 35, 505–513. [Google Scholar] [CrossRef]
- Dalton, C.M.; Carroll, J. Biased inheritance of mitochondria during asymmetric cell division in the mouse oocyte. J. Cell Sci. 2013, 126, 2955–2964. [Google Scholar] [PubMed] [Green Version]
- Singh, A.K.; Althoff, M.J.; Cancelas, J.A. Signaling Pathways Regulating Hematopoietic Stem Cell and Progenitor Aging. Curr. Stem. Cell Rep. 2018, 4, 166–181. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, X.; Zhang, C.; Wang, J.; Wang, S.; Hu, L. Dysfunction of metabolic activity of bone marrow mesenchymal stem cells in aged mice. Cell Prolif. 2022, 55, e13191. [Google Scholar] [CrossRef]
- Lee, J.H.; Yoon, Y.M.; Song, K.H.; Noh, H.; Lee, S.H. Melatonin suppresses senescence-derived mitochondrial dysfunction in mesenchymal stem cells via the HSPA1L-mitophagy pathway. Aging. Cell 2020, 19, e13111. [Google Scholar] [CrossRef] [Green Version]
- Belenky, P.; Bogan, K.L.; Brenner, C. NAD+ metabolism in health and disease. Trends. Biochem. Sci. 2007, 32, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Pi, C.; Yang, Y.; Sun, Y.; Wang, H.; Sun, H.; Ma, M.; Lin, L.; Shi, Y.; Li, Y.; Li, Y.; et al. Nicotinamide phosphoribosyltransferase postpones rat bone marrow mesenchymal stem cell senescence by mediating NAD+-Sirt1 signaling. Aging 2019, 11, 3505–3522. [Google Scholar] [CrossRef]
- Pi, C.; Ma, C.; Wang, H.; Sun, H.; Yu, X.; Gao, X.; Yang, Y.; Sun, Y.; Zhang, H.; Shi, Y.; et al. MiR-34a suppression targets Nampt to ameliorate bone marrow mesenchymal stem cell senescence by regulating NAD+-Sirt1 pathway. Stem Cell Res. Ther. 2021, 12, 271. [Google Scholar] [CrossRef]
- Gong, B.; Pan, Y.; Vempati, P.; Zhao, W.; Knable, L.; Ho, L.; Pasinetti, G.M. Nicotinamide riboside restores cognition through an upregulation of proliferator-activated receptor-γ coactivator 1α regulated β-secretase 1 degradation and mitochondrial gene expression in Alzheimer’s mouse models. Neurobiol. Aging 2013, 34, 1581–1588. [Google Scholar] [CrossRef] [Green Version]
- Kaeberlein, M.; McVey, M.; Guarente, L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes. Dev. 1999, 13, 2570–2580. [Google Scholar] [CrossRef] [Green Version]
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 2012, 13, 225–238. [Google Scholar] [CrossRef]
- Tissenbaum, H.A.; Guarente, L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature 2001, 410, 227–230. [Google Scholar] [CrossRef]
- Taylor, J.R.; Wood, J.G.; Mizerak, E.; Hinthorn, S.; Liu, J.; Finn, M.; Helfand, S.L. Sirt6 regulates lifespan in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 2022, 119, e2111176119. [Google Scholar] [CrossRef] [PubMed]
- Benigni, A.; Cassis, P.; Conti, S.; Perico, L.; Corna, D.; Cerullo, D.; Zentilin, L.; Zoja, C.; Perna, A.; Lionetti, V.; et al. Sirt3 Deficiency Shortens Life Span and Impairs Cardiac Mitochondrial Function Rescued by Opa1 Gene Transfer. Antioxid. Redox. Signal 2019, 31, 1255–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Z.; Song, J.; Wang, G.; Cui, X.; Zheng, J.; Tang, Y.; Yu, W. Deacetylation of serine hydroxymethyl-transferase 2 by SIRT3 promotes colorectal carcinogenesis. Nat. Commun. 2018, 9, 4468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Lombard, D.B. Mitochondrial sirtuins and their relationships with metabolic disease and cancer. Antioxid. Redox. Signal 2015, 22, 1060–1077. [Google Scholar] [CrossRef]
- Yang, W.; Nagasawa, K.; Münch, C.; Xu, Y.; Satterstrom, K.; Jeong, S.; Hayes, S.D.; Jedrychowski, M.P.; Vyas, F.S.; Zaganjor, E.; et al. Mitochondrial Sirtuin Network Reveals Dynamic SIRT3-Dependent Deacetylation in Response to Membrane Depolarization. Cell 2016, 167, 985–1000.e21. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.; Sun, Y.; Pi, C.; Wang, H.; Sun, H.; Yu, X.; Shi, Y.; He, X. Sirt3 Attenuates Oxidative Stress Damage and Rescues Cellular Senescence in Rat Bone Marrow Mesenchymal Stem Cells by Targeting Superoxide Dismutase 2. Front. Cell Dev. Biol. 2020, 8, 599376. [Google Scholar] [CrossRef]
- McDonnell, E.; Peterson, B.S.; Bomze, H.M.; Hirschey, M.D. SIRT3 regulates progression and development of diseases of aging. Trends. Endocrinol. Metab. 2015, 26, 486–492. [Google Scholar] [CrossRef] [Green Version]
- Almeida, M.; Porter, R.M. Sirtuins and FoxOs in osteoporosis and osteoarthritis. Bone 2019, 121, 284–292. [Google Scholar] [CrossRef]
- Zhang, H.; Ryu, D.; Wu, Y.; Gariani, K.; Wang, X.; Luan, P.; D’Amico, D.; Ropelle, E.R.; Lutolf, M.P.; Aebersold, R.; et al. NAD⁺ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 2016, 352, 1436–1443. [Google Scholar] [CrossRef]
- Son, M.J.; Kwon, Y.; Son, T.; Cho, Y.S. Restoration of Mitochondrial NAD+ Levels Delays Stem Cell Senescence and Facilitates Reprogramming of Aged Somatic Cells. Stem Cells 2016, 34, 2840–2851. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Yu, X.; Gao, X.; Zhang, C.; Sun, H.; Xu, K.; Wei, D.; Wang, Q.; Zhang, H.; Shi, Y.; et al. RNA sequencing profiles reveal dynamic signaling and glucose metabolic features during bone marrow mesenchymal stem cell senescence. Cell Biosci. 2022, 12, 62. [Google Scholar] [CrossRef] [PubMed]
- Camacho-Pereira, J.; Tarragó, M.G.; Chini, C.C.; Nin, V.; Escande, C.; Warner, G.M.; Puranik, A.S.; Schoon, R.A.; Reid, J.M.; Galina, A.; et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 2016, 23, 1127–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.; Sun, Y.; Yu, X.; Gao, X.; Wang, H.; Zhang, L.; Shi, Y.; He, X. Analysis of Age-Related Circular RNA Expression Profiles in Mesenchymal Stem Cells of Rat Bone Marrow. Front. Genet. 2021, 12, 600632. [Google Scholar] [CrossRef]
- Russell, O.M.; Gorman, G.S.; Lightowlers, R.N.; Turnbull, D.M. Mitochondrial Diseases: Hope for the Future. Cell 2020, 181, 168–188. [Google Scholar] [CrossRef]
- Miao, Y.; Cui, Z.; Gao, Q.; Rui, R.; Xiong, B. Nicotinamide Mononucleotide Supplementation Reverses the Declining Quality of Maternally Aged Oocytes. Cell Rep. 2020, 32, 107987. [Google Scholar] [CrossRef]
- Croteau, D.L.; Fang, E.F.; Nilsen, H.; Bohr, V.A. NAD+ in DNA repair and mitochondrial maintenance. Cell Cycle 2017, 16, 491–492. [Google Scholar] [CrossRef] [Green Version]
- Sverkeli, L.J.; Hayat, F.; Migaud, M.E.; Ziegler, M. Enzymatic and Chemical Syntheses of Vacor Analogs of Nicotinamide Riboside, NMN and NAD. Biomolecules 2021, 11, 1044. [Google Scholar] [CrossRef]
- Uddin, G.M.; Youngson, N.A.; Sinclair, D.A.; Morris, M.J. Head to Head Comparison of Short-Term Treatment with the NAD+ Precursor Nicotinamide Mononucleotide (NMN) and 6 Weeks of Exercise in Obese Female Mice. Front. Pharmacol. 2016, 7, 258. [Google Scholar] [CrossRef] [Green Version]
- Belenky, P.; Racette, F.G.; Bogan, K.L.; McClure, J.M.; Smith, J.S.; Brenner, C. Nicotinamide riboside promotes Sir2 silencing and extends lifespan via Nrk and Urh1/Pnp1/Meu1 pathways to NAD+. Cell 2007, 129, 473–484. [Google Scholar] [CrossRef]
- Song, J.; Li, J.; Yang, F.; Ning, G.; Zhen, L.; Wu, L.; Zheng, Y.; Zhang, Q.; Lin, D.; Xie, C.; et al. Nicotinamide mononucleotide promotes osteogenesis and reduces adipogenesis by regulating mesenchymal stromal cells via the SIRT1 pathway in aged bone marrow. Cell Death Dis. 2019, 10, 336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabibzadeh, S. From genoprotection to rejuvenation. Front. Biosci. 2021, 26, 97–162. [Google Scholar] [CrossRef] [PubMed]
- Denu, R.A. SIRT3 Enhances Mesenchymal Stem Cell Longevity and Differentiation. Oxid. Med. Cell Longev. 2017, 2017, 5841716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Z.; Liu, G.H.; Qu, J. Mitochondrial sirtuins, metabolism, and aging. J. Genet. Genom. 2022, 49, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Jia, X.; Cui, Y.; Song, Y.; Wang, S.; Geng, Y.; Li, R.; Gao, W.; Fu, D. Sirt3-mediated mitophagy regulates AGEs-induced BMSCs senescence and senile osteoporosis. Redox. Biol. 2021, 41, 101915. [Google Scholar] [CrossRef]
- Liu, S.-M.; Zhang, Y.-R.; Chen, Y.; Ji, D.-R.; Zhao, J.; Fu, S.; Jia, M.-Z.; Yu, Y.-R.; Tang, C.-S.; Huang, W.; et al. Intermedin Alleviates Vascular Calcification in CKD through Sirtuin 3-Mediated Inhibition of Mitochondrial Oxidative Stress. Pharmaceuticals 2022, 15, 1224. [Google Scholar] [CrossRef]
- Sawant Dessai, A.; Dominguez, M.P.; Chen, U.I.; Hasper, J.; Prechtl, C.; Yu, C.; Katsuta, E.; Dai, T.; Zhu, B.; Jung, S.Y.; et al. Transcriptional Repression of SIRT3 Potentiates Mitochondrial Aconitase Activation to Drive Aggressive Prostate Cancer to the Bone. Cancer Res. 2021, 81, 50–63. [Google Scholar] [CrossRef]
- Gao, J.; Feng, Z.; Wang, X.; Zeng, M.; Liu, J.; Han, S.; Xu, J.; Chen, L.; Cao, K.; Long, J.; et al. SIRT3/SOD2 maintains osteoblast differentiation and bone formation by regulating mitochondrial stress. Cell Death Differ. 2018, 25, 229–240. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Quan, Y.; Xia, W. SIRT3 inhibits prostate cancer metastasis through regulation of FOXO3A by suppressing Wnt/β-catenin pathway. Exp. Cell Res. 2018, 364, 143–151. [Google Scholar] [CrossRef]
- Zhang, J.; Xiang, H.; Liu, J.; Chen, Y.; He, R.R.; Liu, B. Mitochondrial Sirtuin 3, New emerging biological function and therapeutic target. Theranostics 2020, 10, 8315–8342. [Google Scholar] [CrossRef]
- Klimova, N.; Fearnow, A.; Long, A.; Kristian, T. NAD+ precursor modulates post-ischemic mitochondrial fragmentation and reactive oxygen species generation via SIRT3 dependent mechanisms. Exp. Neurol. 2020, 325, 113144. [Google Scholar] [CrossRef]
- Ralto, K.M.; Rhee, E.P.; Parikh, S.M. NAD+ homeostasis in renal health and disease. Nat. Rev. Nephrol. 2020, 16, 99–111. [Google Scholar] [CrossRef]
- Li, K.; Deng, Y.; Deng, G.; Chen, P.; Wang, Y.; Wu, H.; Ji, Z.; Yao, Z.; Zhang, X.; Yu, B.; et al. High cholesterol induces apoptosis and autophagy through the ROS-activated AKT/FOXO1 pathway in tendon-derived stem cells. Stem Cell Res. Ther. 2020, 11, 131. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Sun, Y.; Pi, C.; Yu, X.; Gao, X.; Zhang, C.; Sun, H.; Zhang, H.; Shi, Y.; He, X. Nicotinamide Mononucleotide Supplementation Improves Mitochondrial Dysfunction and Rescues Cellular Senescence by NAD+/Sirt3 Pathway in Mesenchymal Stem Cells. Int. J. Mol. Sci. 2022, 23, 14739. https://doi.org/10.3390/ijms232314739
Wang H, Sun Y, Pi C, Yu X, Gao X, Zhang C, Sun H, Zhang H, Shi Y, He X. Nicotinamide Mononucleotide Supplementation Improves Mitochondrial Dysfunction and Rescues Cellular Senescence by NAD+/Sirt3 Pathway in Mesenchymal Stem Cells. International Journal of Molecular Sciences. 2022; 23(23):14739. https://doi.org/10.3390/ijms232314739
Chicago/Turabian StyleWang, Huan, Yanan Sun, Chenchen Pi, Xiao Yu, Xingyu Gao, Chang Zhang, Hui Sun, Haiying Zhang, Yingai Shi, and Xu He. 2022. "Nicotinamide Mononucleotide Supplementation Improves Mitochondrial Dysfunction and Rescues Cellular Senescence by NAD+/Sirt3 Pathway in Mesenchymal Stem Cells" International Journal of Molecular Sciences 23, no. 23: 14739. https://doi.org/10.3390/ijms232314739