

Preventing Surgery-Induced NK Cell Dysfunction Using Anti-TGF-β Immunotherapeutics

 , , , and
, , , and
Abstract
1. Introduction
2. Results
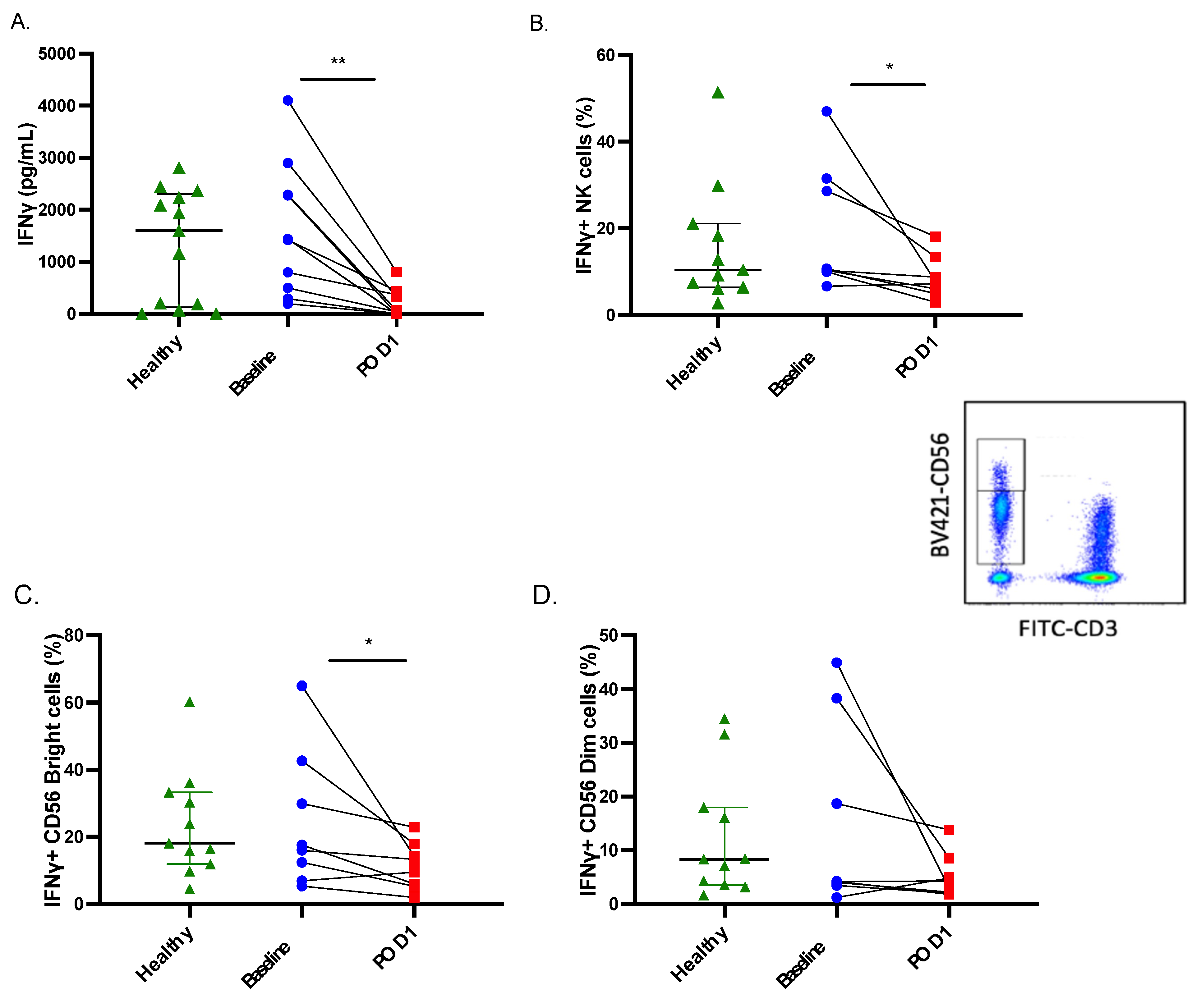
2.1. Both IFNγ Production and Secretion Are Impaired in Postoperative NK Cells
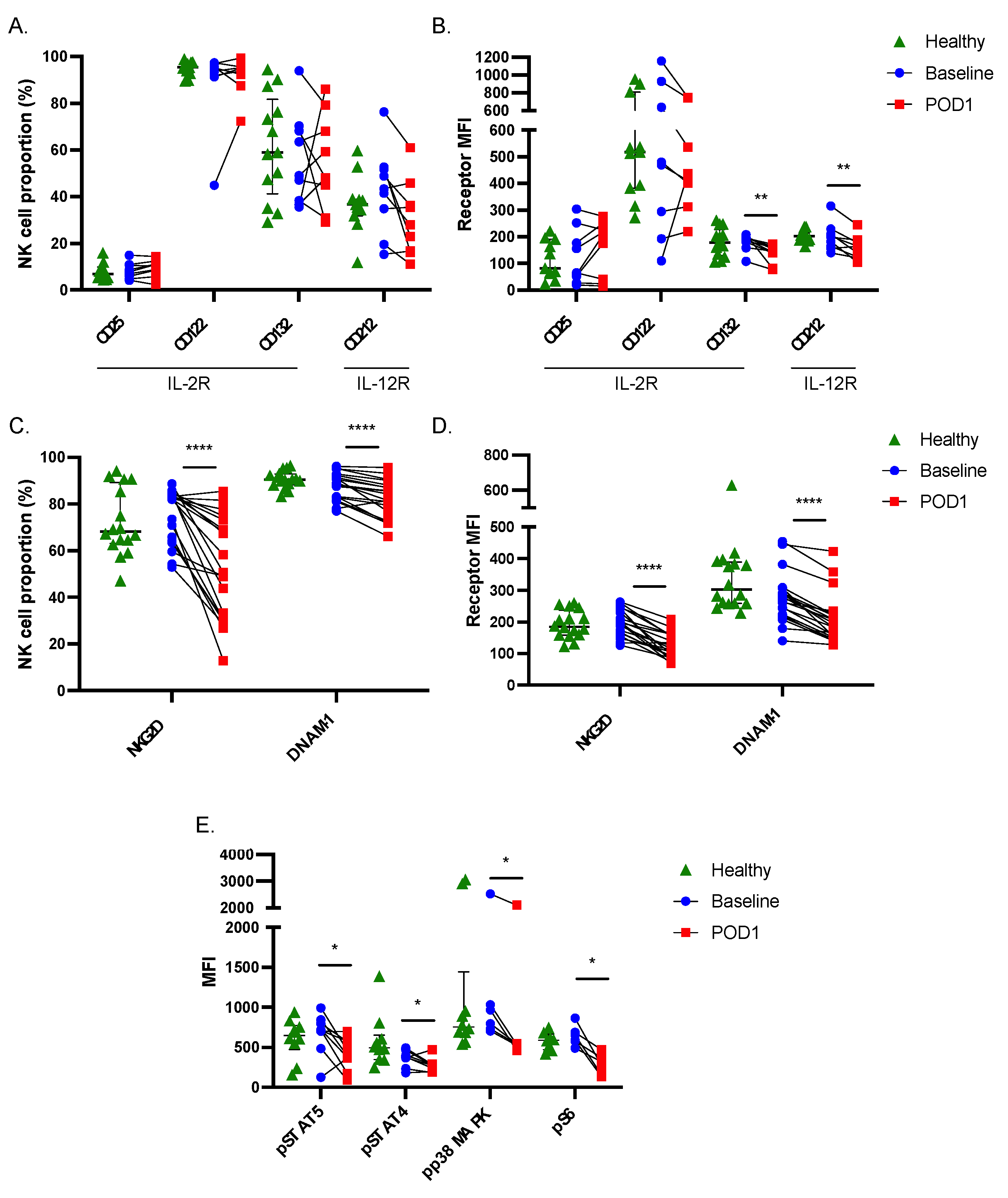
2.2. Postoperative NK Cells Have Reduced Receptor Expression and Downstream Signaling Activity
2.3. Signal Transduction in Response to Cytokine Stimulation Is Impaired in Postoperative NK Cells
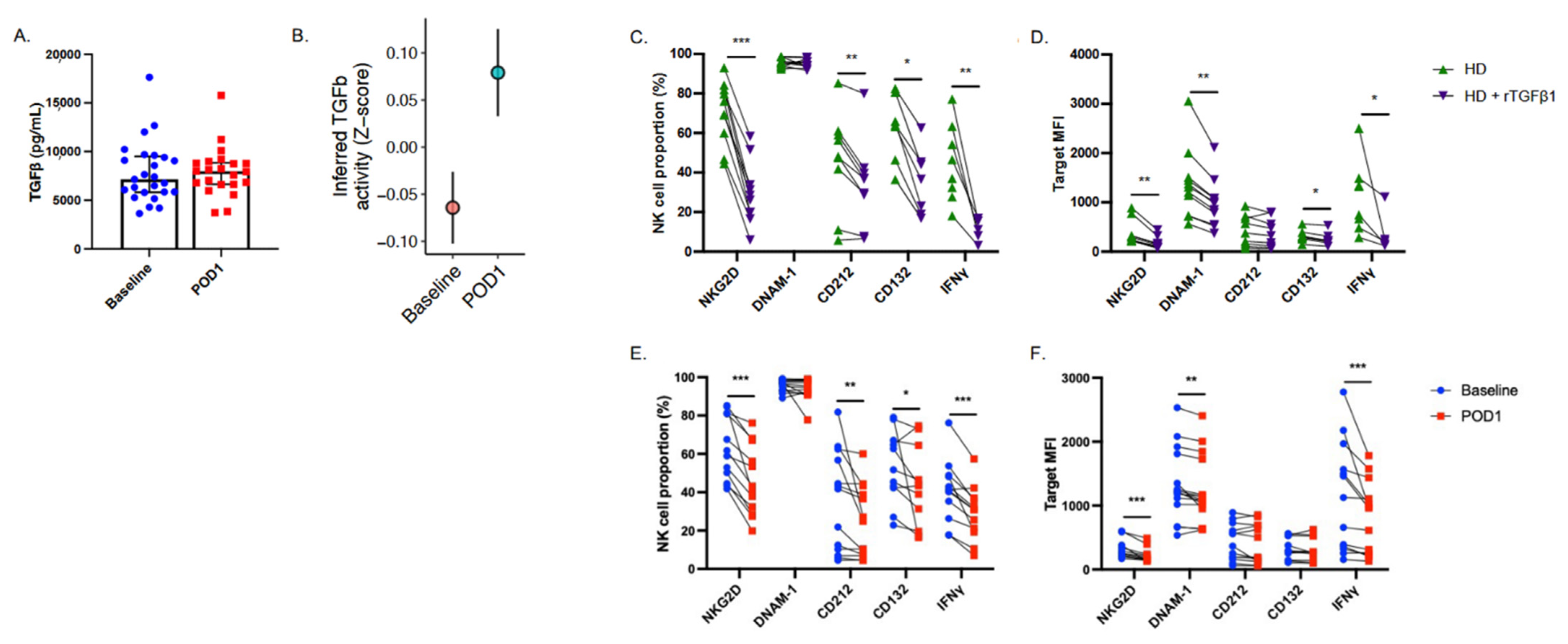
2.4. NK Cell Suppressive Factors Are Present in Postoperative Patient Plasma
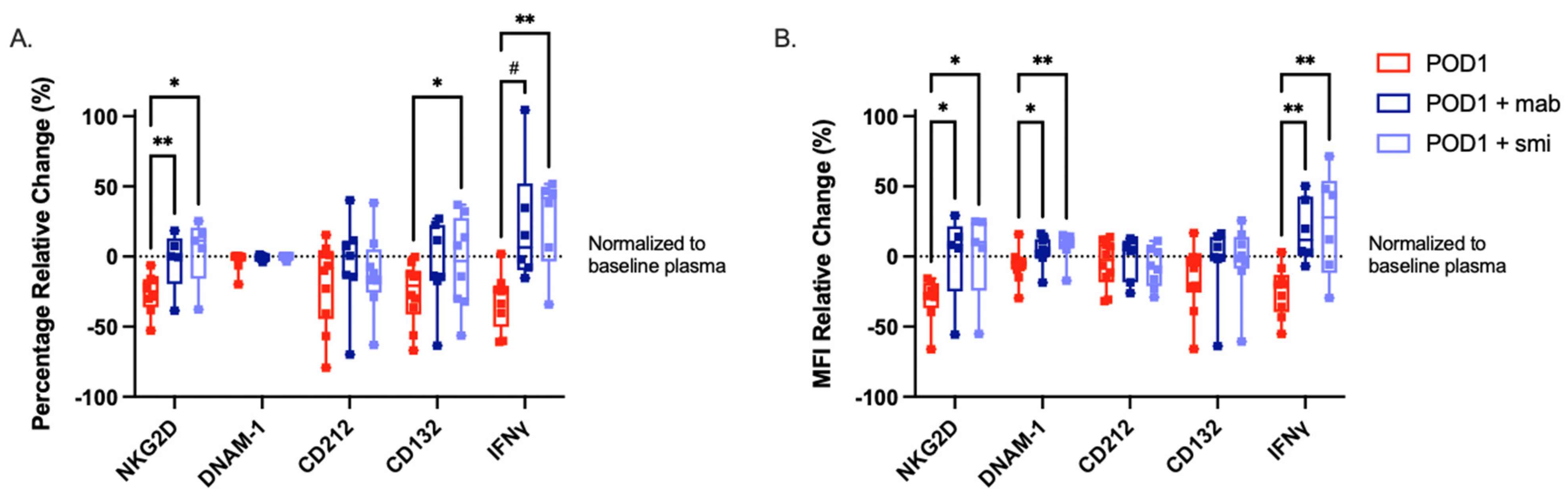
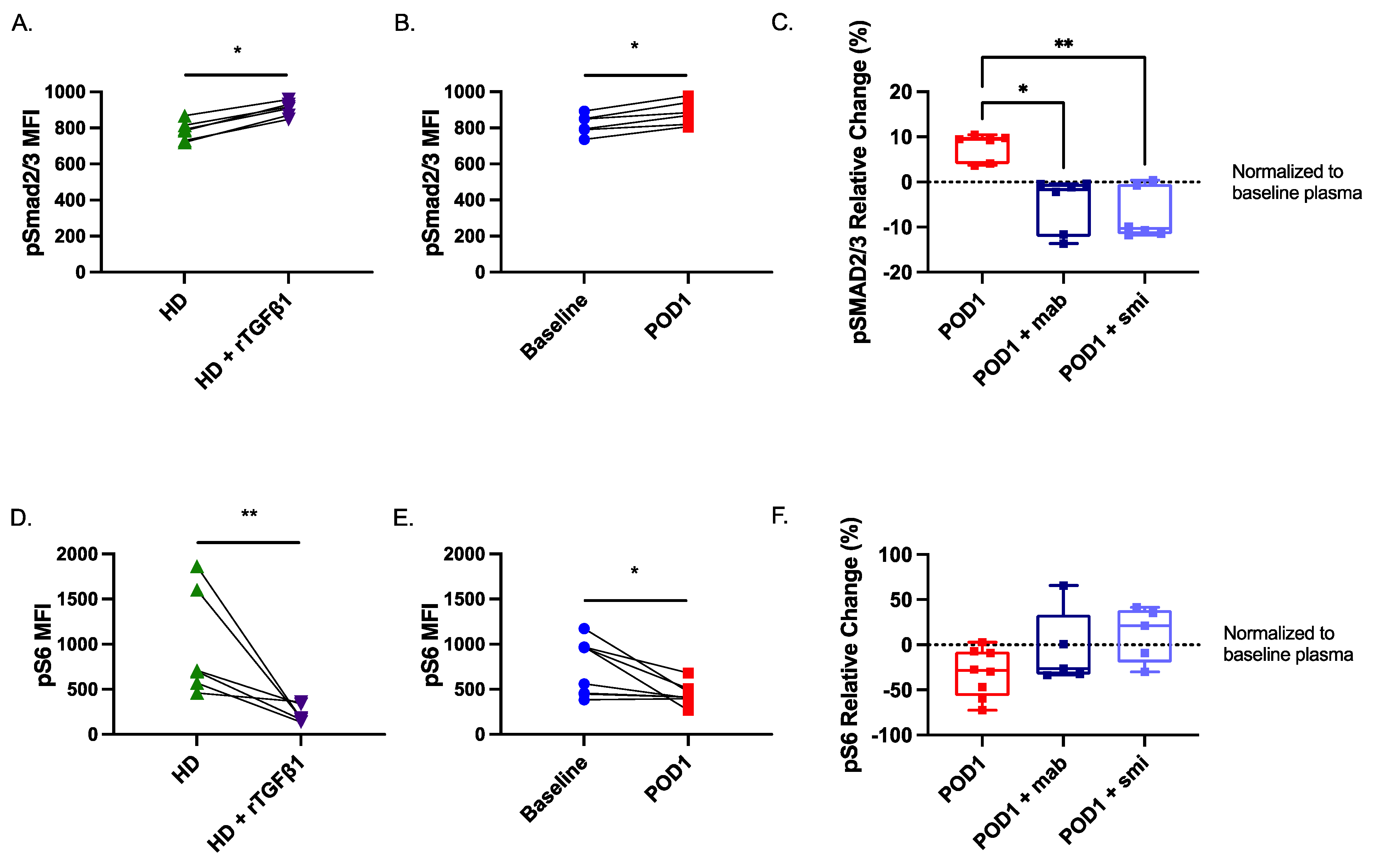
2.5. Targeted Inhibition of TGF-β Prevents NK Cell Dysfunction When Cultured with Postoperative Plasma
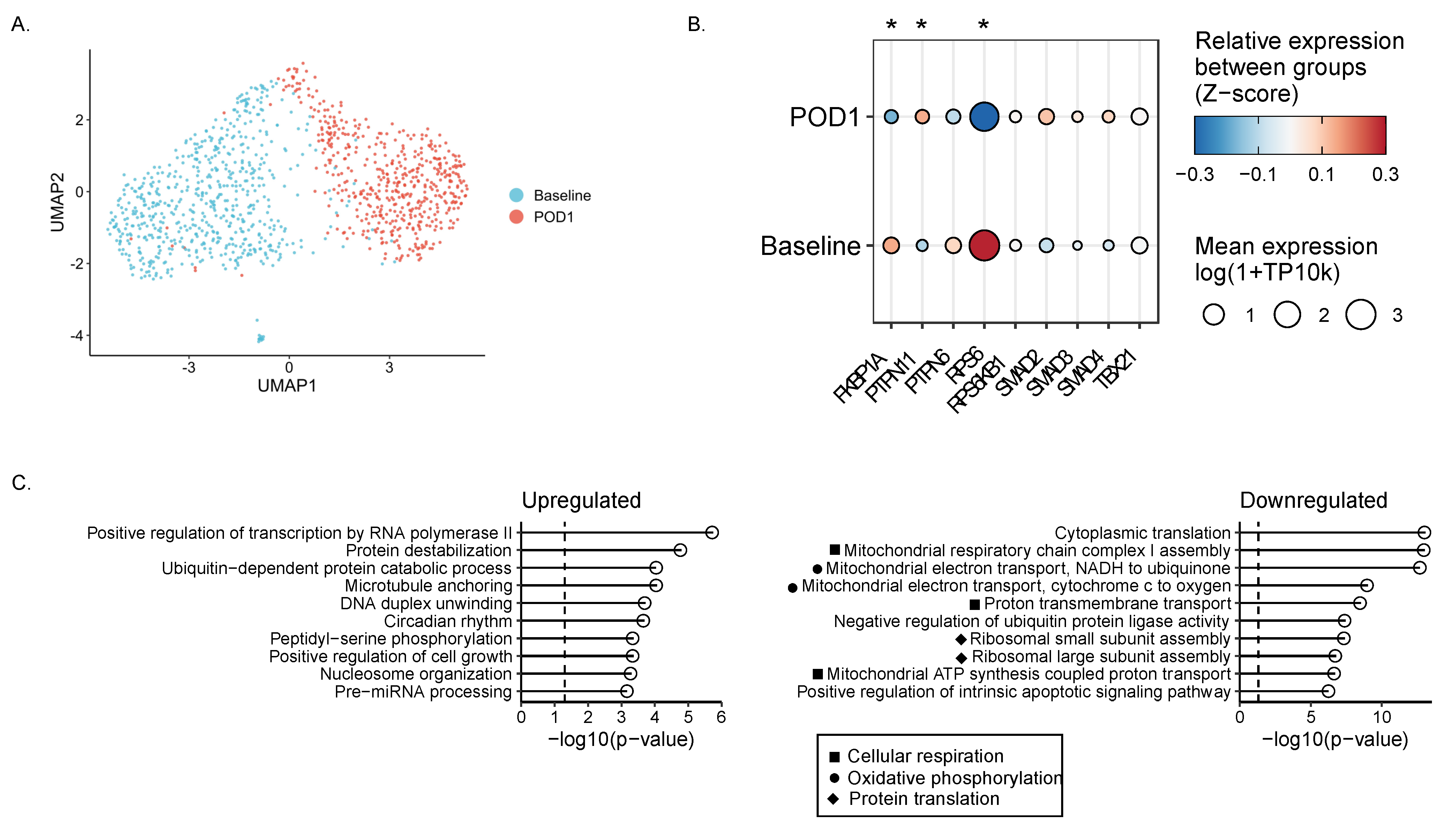
2.6. Single-Cell RNA Sequencing Reveals Impaired Metabolism in Surgically Stressed NK Cells
2.7. Smad2/3 Phosphorylation Is Increased While S6 Phosphorylation Is Reduced in Postoperative NK Cells
3. Materials and Methods
3.1. Clinical Protocols
3.1.1. Perioperative Human Blood and Tissue Specimen Collection Program (PHBSP) Protocol No. 2011884-01H
3.1.2. Single-Cell RNA Sequencing Patient Sample Protocol (NCT02987296)
3.2. Patient Demographics
3.3. Antibodies and FACS Analysis
3.4. Whole Blood Phenotyping and Functionality Assays
3.5. Platelet-Free Plasma Collection
3.6. ELISA
3.7. Cell Isolation
3.8. Combined Plasma Culture Assays: Measuring Effects of Patient Plasma on NK Cell Function and Phenotype Ex Vivo
3.9. In Vitro Staining Protocol
3.10. Single-Cell RNA Sequencing
3.11. Statistics
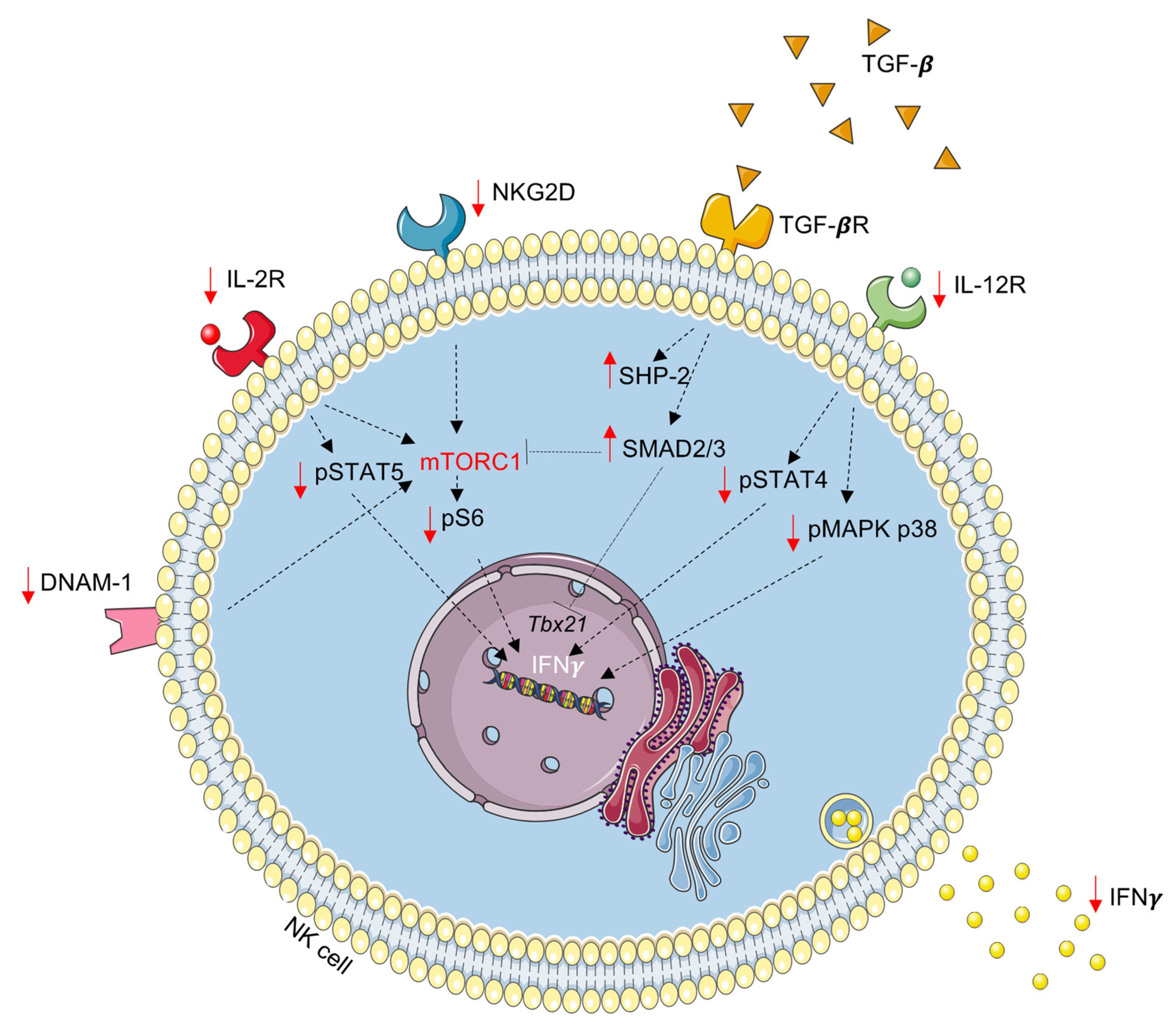
4. Discussion
4.1. Limitations
4.1.1. Inter-Patient Variability
4.1.2. Measuring Soluble TGF-β
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Angka, L.; Martel, A.B.; Kilgour, M.; Jeong, A.; Sadiq, M.; de Souza, C.T.; Baker, L.; Kennedy, M.A.; Kekre, N.; Auer, R.C. Natural Killer Cell IFNγ Secretion is Profoundly Suppressed Following Colorectal Cancer Surgery. Ann. Surg. Oncol. 2018, 25, 3747–3754. [Google Scholar] [CrossRef] [PubMed]
- Tai, L.H.; De Souza, C.T.; Bélanger, S.; Ly, L.; Alkayyal, A.A.; Zhang, J.; Rintoul, J.L.; Ananth, A.A.; Lam, T.; Breitbach, C.J.; et al. Preventing postoperative metastatic disease by inhibiting surgery-induced dysfunction in natural killer cells. Cancer Res. 2013, 73, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, H.; Old, L.J.; Schreiber, R.D. The roles of IFNy in protection against tumor development and cancer immunoediting. Cytokine Growth Factor Rev. 2002, 13, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, M.R.; Merlino, G. The two faces of interferon-γ in cancer. Clin. Cancer Res. 2011, 17, 6118–6124. [Google Scholar] [CrossRef]
- Cooper, M.A.; Fehniger, T.A.; Caligiuri, M.A. The biology of human natural killer-cell subsets. Trends Immunol. 2001, 17, 6118–6124. [Google Scholar] [CrossRef]
- Paust, S.; Senman, B.; Von Andrian, U.H. Adaptive immune responses mediated by natural killer cells. Immunol. Rev. 2010, 235, 286–296. [Google Scholar] [CrossRef]
- Chester, C.; Fritsch, K.; Kohrt, H.E. Natural killer cell immunomodulation: Targeting activating, inhibitory, and co-stimulatory receptor signaling for cancer immunotherapy. Front. Immunol. 2015, 6, 601. [Google Scholar] [CrossRef] [PubMed]
- Waldhauer, I.; Steinle, A. NK cells and cancer immunosurveillance. Oncogene 2008, 27, 5932–5943. [Google Scholar] [CrossRef]
- Lanier, L.L. NK cell recognition. Annu. Rev. Immunol. 2005, 23, 225–274. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef]
- Wu, Y.; Tian, Z.; Wei, H. Developmental and functional control of natural killer cells by cytokines. Front. Immunol. 2017, 8, 930. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, R.; Pohlmann, S.; Kleinertz, H.; Hepner-Schefczyk, M.; Paul, A.; Flohé, S.B. Invasive Surgery Impairs the Regulatory Function of Human CD56 bright Natural Killer Cells in Response to Staphylococcus aureus. Suppression of Interferon-γ Synthesis. PLoS ONE 2015, 10, e0130155. [Google Scholar] [CrossRef] [PubMed]
- Market, M.; Baxter, K.E.; Angka, L.; Kennedy, M.A.; Auer, R.C. The potential for cancer immunotherapy in targeting surgery-induced natural killer cell dysfunction. Cancers 2019, 11, 2. [Google Scholar] [CrossRef] [PubMed]
- Angka, L.; Khan, S.T.; Kilgour, M.; Xu, R.; Kennedy, M.A.; Auer, R.C. Dysfunctional Natural Killer cells in the Aftermath of Cancer Surgery. Int. J. Mol. Sci. 2017, 18, 1787. [Google Scholar] [CrossRef] [PubMed]
- Shariat, S.F.; Shalev, M.; Menesses-Diaz, A.; Kim, I.Y.; Kattan, M.W.; Wheeler, T.M.; Slawin, K.M. Preoperative plasma levels of transforming growth factor beta1 (TGF-β1) strongly predict progression in patients undergoing radical prostatectomy. J. Clin. Oncol. 2001, 19, 2856–2864. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Chen, Z.; Zhu, S.; Liu, T.; Wen, Y.; Su, Y.; Xi, X.; Hu, Y.; Lian, L.; Liu, F.; et al. Prognostic value of transforming growth factor-beta in patients with colorectal cancer who undergo surgery: A meta-analysis. BMC Cancer 2017, 17, 240. [Google Scholar] [CrossRef]
- Li, J.; Shen, C.; Wang, X.; Lai, Y.; Zhou, K.; Li, P.; Liu, L.; Che, G. Prognostic value of TGF-β in lung cancer: Systematic review and meta-analysis. BMC Cancer 2019, 19, 691. [Google Scholar] [CrossRef]
- Katz, L.H.; Li, Y.; Chen, J.S.; Muñoz, N.M.; Majumdar, A.; Chen, J.; Mishra, L. Targeting TGF-β signaling in cancer. Expert Opin. Ther. Targets 2013, 17, 743–760. [Google Scholar] [CrossRef]
- Lazarova, M.; Steinle, A. Impairment of NKG2D-Mediated Tumor Immunity by TGF-β. Front. Immunol. 2019, 10, 2689. [Google Scholar] [CrossRef]
- Castriconi, R.; Cantoni, C.; Della Chiesa, M.; Vitale, M.; Marcenaro, E.; Conte, R.; Biassoni, R.; Bottino, C.; Moretta, L.; Moretta, A. Transforming growth factor β1 inhibits expression of NKP30 and NKG2d receptors: Consequences for the NK-mediated killing of dendritic cells. Proc. Natl. Acad. Sci. USA 2003, 100, 4120–4125. [Google Scholar] [CrossRef]
- Viel, S.; Marçais, A.; Guimaraes, F.S.F.; Loftus, R.; Rabilloud, J.; Grau, M.; Degouve, S.; Djebali, S.; Sanlaville, A.; Charrier, E.; et al. TGF-β inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci. Signal. 2016, 9, ra19. [Google Scholar] [CrossRef] [PubMed]
- Zaiatz-Bittencourt, V.; Finlay, D.K.; Gardiner, C.M. Canonical TGF-β Signaling Pathway Represses Human NK Cell Metabolism. J. Immunol. 2018, 200, 3934–3941. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Han, Y.; Guo, Q.; Zhang, M.; Cao, X. Cancer-Expanded Myeloid-Derived Suppressor Cells Induce Anergy of NK Cells through Membrane-Bound TGF-β1. J. Immunol. 2009, 182, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Bruno, A.; Mortara, L.; Baci, D.; Noonan, D.M.; Albini, A. Myeloid Derived Suppressor Cells Interactions with Natural Killer Cells and Pro-angiogenic Activities: Roles in Tumor Progression. Front. Immunol. 2019, 10, 771. [Google Scholar] [CrossRef]
- Stiff, A.; Trikha, P.; Mundy-Bosse, B.; McMichael, E.; Mace, T.A.; Benner, B.; Kendra, K.; Campbell, A.; Gautam, S.; Abood, D.; et al. Nitric oxide production by myeloid-derived suppressor cells plays a role in impairing Fc receptor–mediated natural killer cell function. Clin. Cancer Res. 2018, 24, 1891–1904. [Google Scholar] [CrossRef]
- Marzec, M.; Liu, X.; Kasprzycka, M.; Witkiewicz, A.; Raghunath, P.N.; El-Salem, M.; Robertson, E.; Odum, N.; Wasik, M.A. IL-2- and IL-15-induced activation of the rapamycin-sensitive mTORC1 pathway in malignant CD4+ T lymphocytes. Blood 2008, 111, 2181–2189. [Google Scholar] [CrossRef]
- Hay, N.; Sonenberg, N. Upstream and downstream of mTOR. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef]
- Marçais, A.; Cherfils-Vicini, J.; Viant, C.; Degouve, S.; Viel, S.; Fenis, A.; Rabilloud, J.; Mayol, K.; Tavares, A.; Bienvenu, J.; et al. The metabolic checkpoint kinase mTOR is essential for IL-15 signaling during the development and activation of NK cells. Nat. Immunol. 2014, 15, 749–757. [Google Scholar] [CrossRef]
- Ouyang, W.; Jacobson, N.G.; Bhattacharya, D.; Gorham, J.D.; Fenoglio, D.; Sha, W.C.; Murphy, T.L.; Murphy, K.M. The Ets transcription factor ERM is Th1-specific and induced by IL-12 through a Stat4-dependent pathway. Proc. Natl. Acad. Sci. USA 1999, 96, 3888–3893. [Google Scholar] [CrossRef]
- Lazarevic, V.; Glimcher, L.H.; Lord, G.M. T-bet: A bridge between innate and adaptive immunity. Nat. Rev. Immunol. 2013, 13, 777–789. [Google Scholar] [CrossRef]
- Zhang, S.; Kaplan, M.H. The p38 Mitogen-Activated Protein Kinase Is Required for IL-12-Induced IFN-γ Expression. J. Immunol. 2000, 165, 1374–1380. [Google Scholar] [CrossRef] [PubMed]
- Alazawi, W.; Pirmadjid, N.; Lahiri, R.; Bhattacharya, S. Inflammatory and immune responses to surgery and their clinical impact. Ann. Surg. 2016, 264, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Wei, M.; Becknell, B.; Trotta, R.; Liu, S.; Boyd, Z.; Jaung, M.S.; Blaser, B.W.; Sun, J.; Benson, D.M.; et al. Pro- and Antiinflammatory Cytokine Signaling: Reciprocal Antagonism Regulates Interferon-gamma Production by Human Natural Killer Cells. Immunity 2006, 24, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Trotta, R.; Col, J.D.; Yu, J.; Ciarlariello, D.; Thomas, B.; Zhang, X.; Allard, J.; Wei, M.; Mao, H.; Byrd, J.C.; et al. TGF-β Utilizes SMAD3 to Inhibit CD16-Mediated IFN-γ Production and Antibody-Dependent Cellular Cytotoxicity in Human NK Cells. J. Immunol. 2008, 181, 3784–3792. [Google Scholar] [CrossRef]
- Wang, T.; Li, B.Y.; Danielson, P.D.; Shah, P.C.; Rockwell, S.; Lechleider, R.J.; Martin, J.; Manganaro, T.; Donahoe, P.K. The immunophilin FKBP12 functions as a common inhibitor of the TGFβ family type I receptors. Cell 1996, 86, 435–444. [Google Scholar] [CrossRef]
- Sehgal, S.N. Sirolimus: Its discovery, biological properties, and mechanism of action. Transplant. Proc. 2003, 35 (Suppl. 3), 7S–14S. [Google Scholar] [CrossRef]
- Choudhry, M.A.; Sir, Ö.; Sayeed, M.M. TGF-β abrogates TCR-mediated signaling by upregulating tyrosine phosphatases in T cells. Shock 2001, 15, 193–199. [Google Scholar] [CrossRef]
- Park, I.-K.; Shultz, L.D.; Letterio, J.J.; Gorham, J.D. TGF-β1 Inhibits T-bet Induction by IFN-γ in Murine CD4 + T Cells through the Protein Tyrosine Phosphatase Src Homology Region 2 Domain-Containing Phosphatase-1. J. Immunol. 2005, 175, 5666–5674. [Google Scholar] [CrossRef]
- Viant, C.; Fenis, A.; Chicanne, G.; Payrastre, B.; Ugolini, S.; Vivier, E. SHP-1-mediated inhibitory signals promote responsiveness and anti-tumour functions of natural killer cells. Nat. Commun. 2014, 5, 5108. [Google Scholar] [CrossRef]
- Marçais, A.; Marotel, M.; Degouve, S.; Koenig, A.; Fauteux-Daniel, S.; Drouillard, A.; Schlums, H.; Viel, S.; Besson, L.; Allatif, O.; et al. High mTOR activity is a hallmark of reactive natural killer cells and amplifies early signaling through activating receptors. Elife 2017, 6, e26423. [Google Scholar] [CrossRef]
- Viel, S.; Besson, L.; Marotel, M.; Walzer, T.; Marçais, A. Regulation of mTOR metabolic fitness and effector functions by cytokines in natural killer cells. Cancers 2017, 9, 132. [Google Scholar] [CrossRef] [PubMed]
- Cong, J. Metabolism of Natural Killer Cells and Other Innate Lymphoid Cells. Front. Immunol. 2020, 11, 1989. [Google Scholar] [CrossRef] [PubMed]
- Market, M.; Tennakoon, G.; Ng, J.; Scaffidi, M.; Tanese de Souza, C.; Kennedy, M.A.; Auer, R.C.; Tanese de Souza, C.; Auer, R.C.; Market, M. A Method of Assessment of Human Natural Killer Cell Phenotype and Function in Whole Blood. Front. Immunol. 2020, 11, 963. [Google Scholar] [CrossRef] [PubMed]
- McGinnis, C.S.; Patterson, D.M.; Winkler, J.; Conrad, D.N.; Hein, M.Y.; Srivastava, V.; Hu, J.L.; Murrow, L.M.; Weissman, J.S.; Werb, Z.; et al. MULTI-seq: Sample multiplexing for single-cell RNA sequencing using lipid-tagged indices. Nat. Methods 2019, 16, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Dillekås, H.; Rogers, M.S.; Straume, O. Are 90% of deaths from cancer caused by metastases? Cancer Med. 2019, 8, 5574–5576. [Google Scholar] [CrossRef]
- Arruebo, M.; Vilaboa, N.; Sáez-Gutierrez, B.; Lambea, J.; Tres, A.; Valladares, M.; González-Fernández, Á. Assessment of the evolution of cancer treatment therapies. Cancers 2011, 3, 3279–3330. [Google Scholar] [CrossRef]
- Mukherjee, S. The Emperor of All Maladies: A Biography of Cancer. J. Postgrad. Med. Educ. Res. 2012, 46, 112. [Google Scholar] [CrossRef]
- Benish, M.; Bartal, I.; Goldfarb, Y.; Levi, B.; Avraham, R.; Raz, A.; Ben-Eliyahu, S. Perioperative use of β-blockers and COX-2 inhibitors may improve immune competence and reduce the risk of tumor metastasis. Ann. Surg. Oncol. 2008, 15, 2042–2052. [Google Scholar] [CrossRef]
- Goldfarb, Y.; Sorski, L.; Benish, M.; Levi, B.; Melamed, R.; Ben-Eliyahu, S. Improving postoperative immune status and resistance to cancer metastasis: A combined perioperative approach of immunostimulation and prevention of excessive surgical stress responses. Ann. Surg. 2011, 253, 798–810. [Google Scholar] [CrossRef]
- Tartter, P.I.; Steinberg, B.; Barron, D.M.; Martinelli, G. The Prognostic Significance of Natural Killer Cytotoxicity in Patients With Colorectal Cancer. Arch. Surg. 1987, 122, 1264–1268. [Google Scholar] [CrossRef]
- Schantz, S.P.; Brown, B.W.; Lira, E.; Taylor, D.L.; Beddingfield, N. Evidence for the role of natural immunity in the control of metastatic spread of head and neck cancer. Cancer Immunol. Immunother. 1987, 25, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, T.; Yamaguchi, Y. Autologous tumor killing activity as a prognostic factor in primary resected nonsmall cell carcinoma of the lung. Cancer 1997, 79, 474–481. [Google Scholar] [CrossRef]
- Strobl, H.; Knapp, W. TGF-β1 regulation of dendritic cells. Microbes Infect. 1999, 1, 1283–1290. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.P.; Choi, S.C.; Kiesler, P.; Gil-Krzewska, A.; Borrego, F.; Weck, J.; Krzewski, K.; Coligan, J.E. Complex regulation of human NKG2D-DAP10 cell surface expression: Opposing roles of the γ c cytokines and TGF-β1. Blood 2011, 118, 3019–3027. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Mao, F.Y.; Zhao, Y.L.; Lv, Y.P.; Teng, Y.S.; Duan, M.; Chen, W.; Cheng, P.; Wang, T.T.; Liang, Z.Y.; et al. Altered NKp30, NKp46, NKG2D, and DNAM-1 Expression on Circulating NK Cells Is Associated with Tumor Progression in Human Gastric Cancer. J. Immunol. Res. 2018. [Google Scholar] [CrossRef]
- Sun, C.; Fu, B.; Gao, Y.; Liao, X.; Sun, R.; Tian, Z.; Wei, H. TGF-β1 down-regulation of NKG2D/DAP10 and 2B4/SAP expression on human NK cells contributes to HBV persistence. PLoS Pathog. 2012, 8, e1002594. [Google Scholar] [CrossRef]
- Slattery, K.; Gardiner, C.M. NK Cell Metabolism and TGFβ—Implications for Immunotherapy. Front. Immunol. 2019, 10, 2915. [Google Scholar] [CrossRef]
- Donnelly, R.P.; Loftus, R.M.; Keating, S.E.; Liou, K.T.; Biron, C.A.; Gardiner, C.M.; Finlay, D.K. mTORC1-Dependent Metabolic Reprogramming Is a Prerequisite for NK Cell Effector Function. J. Immunol. 2014, 193, 4477–4484. [Google Scholar] [CrossRef]
- Slattery, K.; Woods, E.; Bittencourt, V. TGFβ drives NK cell metabolic dysfunction in human metastatic breast cancer. J. Immunother. Cancer 2021, 9, e002044. [Google Scholar] [CrossRef]
- Pakyari, M.; Farrokhi, A.; Maharlooei, M.K.; Ghahary, A. Critical Role of Transforming Growth Factor Beta in Different Phases of Wound Healing. Adv. Wound Care 2013, 2, 215–224. [Google Scholar] [CrossRef]
- Teixeira, A.F.; ten Dijke, P.; Zhu, H.-J. On-Target Anti-TGF-β Therapies Are Not Succeeding in Clinical Cancer Treatments: What Are Remaining Challenges? Front. Cell Dev. Biol. 2020, 8, 605. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Saulis, A.S.; Liu, W.R.; Roy, N.K.; Chao, J.D.; Ledbetter, S.; Mustoe, T.A. The temporal effects of anti-TGF-β1, 2, and 3 monoclonal antibody on wound healing and hypertrophic scar formation. J. Am. Coll. Surg. 2005, 201, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Yamakawa, S.; Hayashida, K. Advances in surgical applications of growth factors for wound healing. Burn. Trauma 2019, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.; Tai, L.H.; Falls, T.; De Souza, C.T.; Bell, J.C.; Carrier, M.; Atkins, H.; Boushey, R.; Auer, R.A. Surgical stress promotes the development of cancer metastases by a coagulation-dependent mechanism involving natural killer cells in a murine model. Ann. Surg. 2013, 258, 158–168. [Google Scholar] [CrossRef]
- Karolczak, K.; Watala, C. Blood Platelets as an Important but Underrated Circulating Source of TGFβ. Int. J. Mol. Sci. 2021, 22, 4492. [Google Scholar] [CrossRef]
- Ahamed, J.; Burg, N.; Yoshinaga, K.; Janczak, C.A.; Rifkin, D.B.; Coller, B.S. In vitro and in vivo evidence for shear-induced activation of latent transforming growth factor-β1. Blood 2008, 112, 3650–3660. [Google Scholar] [CrossRef]
- Blakytny, R.; Ludlow, A.; Martin, G.E.M.; Ireland, G.; Lund, L.R.; Ferguson, M.W.J.; Brunner, G. Latent TGF-?1 activation by platelets. J. Cell. Physiol. 2004, 199, 67–76. [Google Scholar] [CrossRef]
- Naesh, O.; Hindberg, I.; Friis, J.; Christiansen, C.; Pedersen, T.; Trap-Jensen, J.; Lund, J.O. Platelet activation in major surgical stress: Influence of combined epidural and general anaesthesia. Acta Anaesthesiol. Scand. 1994, 38, 820–825. [Google Scholar] [CrossRef]
- Guo, S.-W.; Du, Y.; Liu, X. Platelet-derived TGF-β1 mediates the down-modulation of NKG2D expression and may be responsible for impaired natural killer (NK) cytotoxicity in women with endometriosis. Hum. Reprod. 2016, 31, 1462–1474. [Google Scholar] [CrossRef]
- Cluxton, C.D.; Spillane, C.; O’Toole, S.A.; Sheils, O.; Gardiner, C.M.; O’Leary, J.J. Suppression of Natural Killer cell NKG2D and CD226 anti-tumour cascades by platelet cloaked cancer cells: Implications for the metastatic cascade. PLoS ONE 2019, 14, e0211538. [Google Scholar] [CrossRef]
- Koupenova, M.; Clancy, L.; Corkrey, H.A.; Freedman, J.E. Circulating Platelets as Mediators of Immunity, Inflammation, and Thrombosis. Circ. Res. 2018, 122, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Kopp, H.-G.G.; Placke, T.; Salih, H.R. Platelet-derived transforming growth factor-β down-regulates NKG2D thereby inhibiting natural killer cell antitumor reactivity. Cancer Res. 2009, 69, 7775–7783. [Google Scholar] [CrossRef]
- Lee, J.-C.; Lee, K.-M.; Kim, D.-W.; Heo, D.S. Elevated TGF-β1 Secretion and Down-Modulation of NKG2D Underlies Impaired NK Cytotoxicity in Cancer Patients. J. Immunol. 2004, 172, 7335–7340. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Liu, X.; Guo, S.-W. Platelets impair natural killer cell reactivity and function in endometriosis through multiple mechanisms. Hum. Reprod. 2017, 32, 794–810. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I. Myeloid-derived suppressor cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef]
- Tai, L.H.; Alkayyal, A.A.; Leslie, A.L.; Sahi, S.; Bennett, S.; Tanese de Souza, C.; Baxter, K.; Angka, L.; Xu, R.; Kennedy, M.A.; et al. Phosphodiesterase-5 inhibition reduces postoperative metastatic disease by targeting surgery-induced myeloid derived suppressor cell-dependent inhibition of Natural Killer cell cytotoxicity. Oncoimmunology 2018, 7, e1431082. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Wang, M.; Yin, T.; Zhao, Y.; Wei, X. Myeloid-Derived Suppressor Cells Promote Metastasis in Breast Cancer After the Stress of Operative Removal of the Primary Cancer. Front. Oncol. 2019, 9, 855. [Google Scholar] [CrossRef]
- Xu, P.; He, H.; Gu, Y.; Wang, Y.; Sun, Z.; Yang, L.; Miao, C. Surgical trauma contributes to progression of colon cancer by downregulating CXCL4 and recruiting MDSCs. Exp. Cell Res. 2018, 370, 692–698. [Google Scholar] [CrossRef]
- Wang, J.; Yang, L.; Yu, L.; Wang, Y.Y.; Chen, R.; Qian, J.; Hong, Z.P.; Su, X.S. Surgery-induced monocytic myeloid-derived suppressor cells expand regulatory T cells in lung cancer. Oncotarget 2017, 8, 17050–17058. [Google Scholar] [CrossRef]
- Yuan, L.; Xu, B.; Fan, H.; Yuan, P.; Zhao, P.; Suo, Z. Pre- and post-operative evaluation: Percentages of circulating myeloid-derived suppressor cells in rectal cancer patients. Neoplasma 2015, 62, 239–249. [Google Scholar] [CrossRef]
- Wang, P.F.; Song, S.Y.; Wang, T.J.; Ji, W.J.; Li, S.W.; Liu, N.; Yan, C.X. Prognostic role of pretreatment circulating MDSCs in patients with solid malignancies: A meta-analysis of 40 studies. Oncoimmunology 2018, 7, e1494113. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Shen, W.; Zhang, Y.; Liu, M.; Zhang, L.; Liu, Q.; Lu, H.h.; Bo, J. Accumulation of myeloid-derived suppressor cells (MDSCs) induced by low levels of IL-6 correlates with poor prognosis in bladder cancer. Oncotarget 2017, 8, 38378–38388. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Pang, Y.; Gara, S.K.; Achyut, B.R.; Heger, C.; Goldsmith, P.K.; Lonning, S.; Yang, L. Gr-1+CD11b+ cells are responsible for tumor promoting effect of TGF-β in breast cancer progression. Int. J. Cancer 2012, 131, 2584–2595. [Google Scholar] [CrossRef] [PubMed]
- Tumino, N.; Di Pace, A.L.; Besi, F.; Quatrini, L.; Vacca, P.; Moretta, L. Interaction Between MDSC and NK Cells in Solid and Hematological Malignancies: Impact on HSCT. Front. Immunol. 2021, 12, 638841. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.R.; Lee, W.; Cho, S.K.; Park, S.G. Characterization of multiple cytokine combinations and TGF-β on differentiation and functions of myeloid-derived suppressor cells. Int. J. Mol. Sci. 2018, 19, 869. [Google Scholar] [CrossRef]
- Li, W.; Tanikawa, T.; Kryczek, I.; Xia, H.; Li, G.; Wu, K.; Wei, S.; Zhao, L.; Vatan, L.; Wen, B.; et al. Aerobic Glycolysis Controls Myeloid-Derived Suppressor Cells and Tumor Immunity via a Specific CEBPB Isoform in Triple-Negative Breast Cancer. Cell Metab. 2018, 28, 87–103.e6. [Google Scholar] [CrossRef]
- Burga, R.; Yvon, E.; Chorvinsky, E.; Fernandes, R.; Cruz, C.R.; Bolalrd, C. Engineering the TGFβ receptor to Enhance the Therapeutic Potential of Natural Killer Cells as an Immunotherapy for Neuroblastoma. Clin. Cancer Res. 2019, 25, 4400–4412. [Google Scholar] [CrossRef]
- Neviani, P.; Wise, P.M.; Murtadha, M.; Liu, C.W.; Wu, C.-H.; Jong, A.Y.; Seeger, R.C.; Fabbri, M. Natural Killer-derived exosomal miR-186 inhibits neuroblastoma growth and immune escape mechanisms. Cancer Res. 2020, 79, 1151–1164. [Google Scholar] [CrossRef]
- Khongorzul, P.; Ling, C.J.; Khan, F.U.; Ihsan, A.U.; Zhang, J. Antibody-drug conjugates: A comprehensive review. Mol. Cancer Res. 2020, 18, 3–19. [Google Scholar] [CrossRef]
- McPherson, M.J.; Hobson, A.D. Pushing the Envelope: Advancement of ADCs Outside of Oncology. In Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2020. [Google Scholar]
- Gingrich, J. How the Next Generation Antibody Drug Conjugates Expands Beyond Cytotoxic Payloads for Cancer Therapy. ADC Rev. 2020. [Google Scholar] [CrossRef]
- Carr, E.J.; Dooley, J.; Garcia-Perez, J.E.; Lagou, V.; Lee, J.C.; Wouters, C.; Meyts, I.; Goris, A.; Boeckxstaens, G.; Linterman, M.A.; et al. The cellular composition of the human immune system is shaped by age and cohabitation. Nat. Immunol. 2016, 17, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Shen-Orr, S.S.; Furman, D.; Kidd, B.A.; Hadad, F.; Lovelace, P.; Huang, Y.W.; Rosenberg-Hasson, Y.; Mackey, S.; Grisar, F.A.G.; Pickman, Y.; et al. Defective Signaling in the JAK-STAT Pathway Tracks with Chronic Inflammation and Cardiovascular Risk in Aging Humans. Cell Syst. 2016, 3, 374–384.e4. [Google Scholar] [CrossRef] [PubMed]
- Brodin, P.; Davis, M.M. Human immune system variation. Nat. Rev. Immunol. 2017, 17, 21–29. [Google Scholar] [CrossRef]
- Scherer, S.D.; Bauer, J.; Schmaus, A.; Neumaier, C.; Herskind, C.; Veldwijk, M.R.; Wenz, F.; Sleeman, J.P. TGF-β1 is present at high levels in wound fluid from breast cancer patients immediately post-surgery, and is not increased by intraoperative radiation therapy (IORT). PLoS ONE 2016, 11, e0162221. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Honda, I.; Suzuki, H.; Murakami, M.; Matsukawa, S.; Hashimoto, Y. Interleukin-10 production during and after upper abdominal surgery. J. Clin. Anesth. 1998, 10, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Torrance, H.D.T.; Pearse, R.M.; O’Dwyer, M.J. Does major surgery induce immune suppression and increase the risk of postoperative infection? Curr. Opin. Anaesthesiol. 2016, 29, 376–383. [Google Scholar] [CrossRef]
- The History of Cancer. Available online: https://www.cancer.org/cancer/cancer-basics/history-of-cancer/cancer-treatment-surgery.html (accessed on 1 January 2020).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Vendor | Cat # | Clone |
|---|---|---|---|
| CD3 FITC (mouse) | Invitrogen | 11-0039-41 | HIT3a |
| CD56 BV421 (mouse) | BD biosciences | 562751 | NCAM16.2 |
| CD16 BV650 (mouse) | BD biosciences | 563692 | 3G8 |
| CD14 APC-Cy7 (mouse) | BD biosciences | 557831 | MφP9 |
| CD45 AF700 (mouse) | BD biosciences | 560566 | HI30 |
| Fixable viability dye BV510 | BD biosciences | 564406 | - |
| IFNγ APC (mouse) | Invitrogen | 17-7319-82 | 4S.B3 |
| CD25 PE-Cy7 (mouse) | BD biosciences | 557741 | M-A251 |
| CD122 PE (mouse) | BD biosciences | 554522 | Mik-β2 |
| CD132 APC (rat) | Biolegend | 338607 | TUGh4 |
| p-STAT5 PE-Cy7 (pY694) (mouse) | BD biosciences | 560117 | 47/Stat5 |
| CD212 BV786 (mouse) | BD biosciences | 744207 | 2.4E6 |
| p-STAT4 PE (pY693) (mouse) | BD biosciences | 558249 | 38/p-Stat4 |
| NKG2D BV650 (mouse) | BD biosciences | 563408 | 1D11 |
| NKG2A PE (mouse) | R&D Systems | FAB1059P-025 | 131411 |
| PE-Cy7 DNAM-1 (mouse) | BioLegend | 338315 | 11A8 |
| APC TIGIT (mouse) | BioLegend | 372705 | A15153G |
| PD-1 PerCP-Cy5.5 (mouse) | BioLegend | 329913 | EH12.2H7 |
| S6 PE (pS235/236) (mouse) | BD biosciences | 560433 | NF-548 |
| p38 MAPK APC (pThr180, Tyr 182) (mouse) | Invitrogen | 17-9078-42 | 4NIT4KK |
| Smad2(pS465/pS467)/Smad3(pS423/pS425) PE (mouse) | BD biosciences | 562586 | O72-670 |
| Mouse PE IgG2a | BioLegend | 400214 | MOPC-173 |
| Mouse APC IgG2A | BioLegend | 400219 | MOPC-173 |
| Mouse PerCP-Cy5.5 IgG1 | BioLegend | 400149 | MOPC-21 |
| Mouse APC IgG1 | Biolegend | 400119 | MOPC-21 |
| Mouse PE-Cy7 IgG1 | BD biosciences | 557872 | MOPC-21 |
| Mouse PE IgG2b | Invitrogen | 12-4732-41 | eBMG2b |
| Mouse BV650 IgG1 | BD biosciences | 563231 | X40 |
| Rat APC IgG2b | Biolegend | 400611 | RTK4530 |
| Mouse BV786 IgG1 | BD biosciences | 563330 | X40 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Market, M.; Tennakoon, G.; Scaffidi, M.; Cook, D.P.; Angka, L.; Ng, J.; Tanese de Souza, C.; Kennedy, M.A.; Vanderhyden, B.C.; Auer, R.C. Preventing Surgery-Induced NK Cell Dysfunction Using Anti-TGF-β Immunotherapeutics. Int. J. Mol. Sci. 2022, 23, 14608. https://doi.org/10.3390/ijms232314608
Market M, Tennakoon G, Scaffidi M, Cook DP, Angka L, Ng J, Tanese de Souza C, Kennedy MA, Vanderhyden BC, Auer RC. Preventing Surgery-Induced NK Cell Dysfunction Using Anti-TGF-β Immunotherapeutics. International Journal of Molecular Sciences. 2022; 23(23):14608. https://doi.org/10.3390/ijms232314608
Chicago/Turabian StyleMarket, Marisa, Gayashan Tennakoon, Marlena Scaffidi, David P. Cook, Leonard Angka, Juliana Ng, Christiano Tanese de Souza, Michael A. Kennedy, Barbara C. Vanderhyden, and Rebecca C. Auer. 2022. "Preventing Surgery-Induced NK Cell Dysfunction Using Anti-TGF-β Immunotherapeutics" International Journal of Molecular Sciences 23, no. 23: 14608. https://doi.org/10.3390/ijms232314608
APA StyleMarket, M., Tennakoon, G., Scaffidi, M., Cook, D. P., Angka, L., Ng, J., Tanese de Souza, C., Kennedy, M. A., Vanderhyden, B. C., & Auer, R. C. (2022). Preventing Surgery-Induced NK Cell Dysfunction Using Anti-TGF-β Immunotherapeutics. International Journal of Molecular Sciences, 23(23), 14608. https://doi.org/10.3390/ijms232314608