
Joining Forces for Cancer Treatment: From “TCR versus CAR” to “TCR and CAR”

 , and
, and
Abstract
1. Introduction
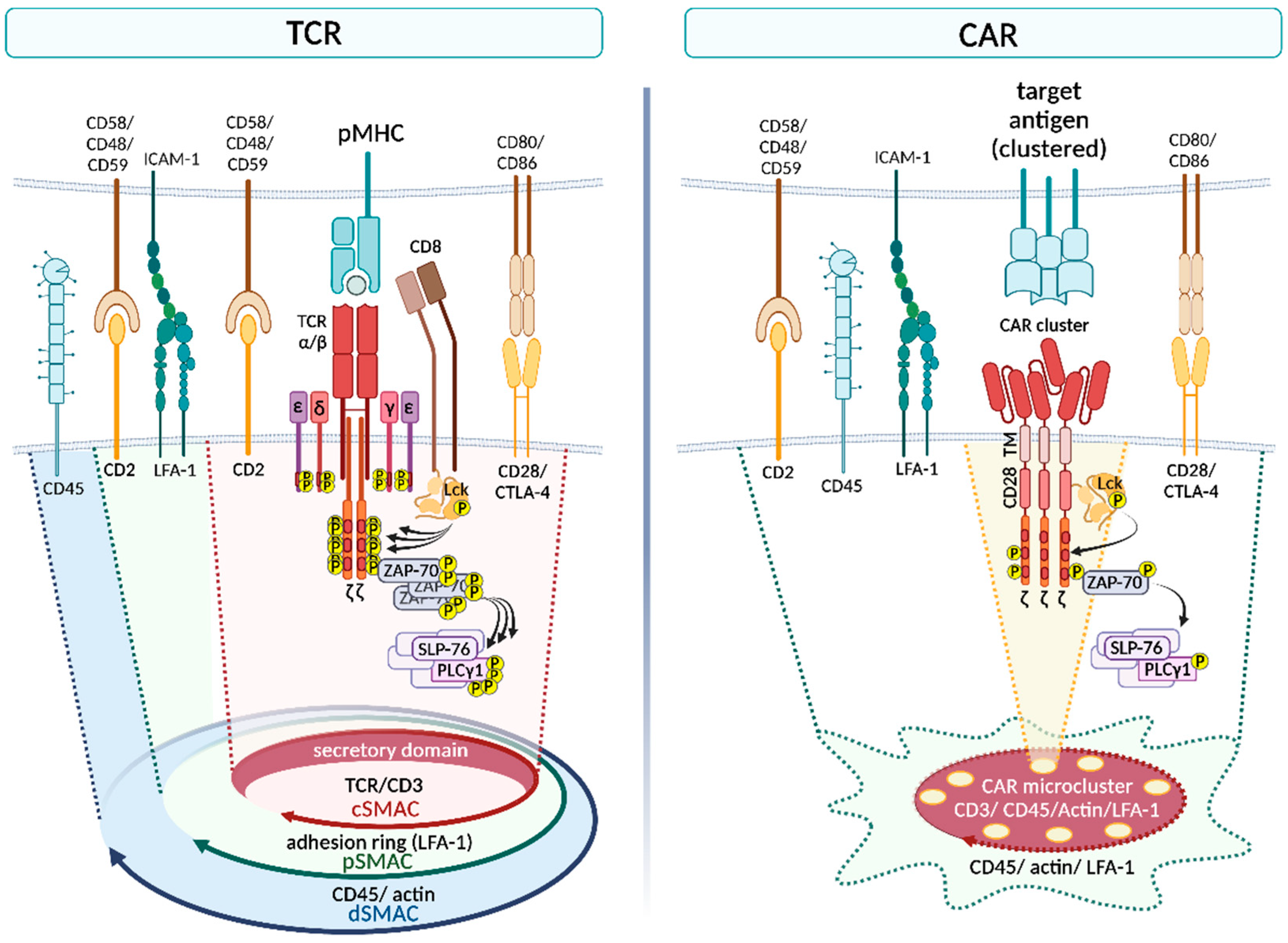
2. Part I—TCR versus CAR
2.1. TCR versus CAR: Structure
2.2. TCR versus CAR: Activation upon Stimulation
2.2.1. Antigen Engagement for Initiation of Activation
2.2.2. Formation of the Immunological Synapse
2.3. TCR versus CAR: Signaling Cascade
2.3.1. Proximal Signaling
2.3.2. Downstream Signaling and Outcome
2.3.3. Calcium/NFAT Pathway
2.3.4. Ras/ERK/AP-1 and NF-κB Pathway
2.3.5. PI3K/AKT/mTOR Pathway
2.3.6. Endosomal Trafficking and Lysosomal Degradation
3. Part II—A Change in Perspective: From “TCR versus CAR” to “TCR and CAR”
3.1. Combination of an Endogenous TCR and a CAR
3.2. Combination of a Transgenic TCR and a CAR
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Rosenberg, S.A.; Spiess, P.; Lafreniere, R. A New Approach to the Adoptive Immunotherapy of Cancer with Tumor-Infiltrating Lymphocytes. Science 1986, 233, 1318–1321. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable Complete Responses in Heavily Pretreated Patients with Metastatic Melanoma Using Transfer Immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef] [PubMed]
- Zacharakis, N.; Huq, L.M. Breast Cancers Are Immunogenic: Immunologic Analyses and a Phase II Pilot Clinical Trial Using Mutation-Reactive Autologous Lymphocytes. J. Clin. Oncol. 2022, 40, 1741–1754. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.; Seipp, C.A.; et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N. Engl. J. Med. 1988, 319, 1676–1680. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Sun, J.; Chen, K.; Ma, P.; Lei, Q.; Xing, S.; Cao, Z.; Sun, S.; Yu, Z.; Liu, Y.; et al. Perspectives of tumor-infiltrating lymphocyte treatment in solid tumors. BMC Med. 2021, 19, 140. [Google Scholar] [CrossRef]
- Kumar, A.; Watkins, R.; Vilgelm, A.E. Cell Therapy With TILs: Training and Taming T Cells to Fight Cancer. Front. Immunol. 2021, 12, 690499. [Google Scholar] [CrossRef]
- Wei, F.; Cheng, X.X.; Xue, J.Z.; Xue, S.A. Emerging Strategies in TCR-Engineered T Cells. Front. Immunol. 2022, 13, 850358. [Google Scholar] [CrossRef]
- Sengsayadeth, S.; Savani, B.N.; Oluwole, O.; Dholaria, B. Overview of approved CAR-T therapies, ongoing clinical trials, and its impact on clinical practice. eJHaem 2022, 3, 6–10. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Morgan, R.A.; Chinnasamy, N.; Abate-Daga, D.; Gros, A.; Robbins, P.F.; Zheng, Z.; Dudley, M.E.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immunother. 2013, 36, 133–151. [Google Scholar] [CrossRef]
- Morgan, R.A.; Dudley, M.E.; Wunderlich, J.R.; Hughes, M.S.; Yang, J.C.; Sherry, R.M.; Royal, R.E.; Topalian, S.L.; Kammula, U.S.; Restifo, N.P.; et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 2006, 314, 126–129. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, L. The Emerging World of TCR-T Cell Trials Against Cancer: A Systematic Review. Technol. Cancer Res. Treat. 2019, 18, 1533033819831068. [Google Scholar] [CrossRef]
- Rapoport, A.P.; Stadtmauer, E.A.; Binder-Scholl, G.K.; Goloubeva, O.; Vogl, D.T.; Lacey, S.F.; Badros, A.Z.; Garfall, A.; Weiss, B.; Finklestein, J.; et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat. Med. 2015, 21, 914–921. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Druta, M.; Liebner, D.A.; Schuetze, S.; Somaiah, N.; Tine, B.A.V.; Tap, W.D.; Pulham, T.; Chagin, K.; Norry, E.; et al. Pilot study of NY-ESO-1c259 T cells in advanced myxoid/round cell liposarcoma. J. Clin. Oncol. 2018, 36 (Suppl. 15), 3005. [Google Scholar] [CrossRef]
- Robbins, P.F.; Kassim, S.H.; Tran, T.L.N.; Crystal, J.S.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Dudley, M.E.; Wunderlich, J.R.; Sherry, R.M.; et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive receptor: Long-term follow-up and correlates with response. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 1019–1027. [Google Scholar] [CrossRef]
- Robbins, P.F.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; Dudley, M.E.; Wunderlich, J.R.; Nahvi, A.V.; Helman, L.J.; Mackall, C.L.; et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 917–924. [Google Scholar] [CrossRef]
- Shafer, P.; Kelly, L.M.; Hoyos, V. Cancer Therapy With TCR-Engineered T Cells: Current Strategies, Challenges, and Prospects. Front. Immunol. 2022, 13, 835762. [Google Scholar] [CrossRef]
- Huang, J.; Brameshuber, M.; Zeng, X.; Xie, J.; Li, Q.-J.; Chien, Y.-H.; Valitutti, S.; Davis, M.M. A single peptide-major histocompatibility complex ligand triggers digital cytokine secretion in CD4(+) T cells. Immunity 2013, 39, 846–857. [Google Scholar] [CrossRef]
- Sykulev, Y.; Joo, M.; Vturina, I.; Tsomides, T.J.; Eisen, H.N. Evidence that a Single Peptide–MHC Complex on a Target Cell Can Elicit a Cytolytic T Cell Response. Immunity 1996, 4, 565–571. [Google Scholar] [CrossRef]
- Anikeeva, N.; Panteleev, S.; Mazzanti, N.W.; Terai, M.; Sato, T.; Sykulev, Y. Efficient killing of tumor cells by CAR-T cells requires greater number of engaged CARs than TCRs. J. Biol. Chem. 2021, 297, 101033. [Google Scholar] [CrossRef]
- Dong, R.; Libby, K.A.; Blaeschke, F.; Fuchs, W.; Marson, A.; Vale, R.D.; Su, X. Rewired signaling network in T cells expressing the chimeric antigen receptor (CAR). EMBO J. 2020, 39, e104730. [Google Scholar] [CrossRef]
- Wu, L.; Brzostek, J.; Sankaran, S.; Wei, Q.; Yap, J.; Tan, T.Y.Y.; Lai, J.; MacAry, P.A.; Gascoigne, N.R.J. Targeting CAR to the Peptide-MHC Complex Reveals Distinct Signaling Compared to That of TCR in a Jurkat T Cell Model. Cancers 2021, 13, 867. [Google Scholar] [CrossRef]
- Lindner, S.E.; Johnson, S.M.; Brown, C.E.; Wang, L.D. Chimeric antigen receptor signaling: Functional consequences and design implications. Sci. Adv. 2020, 6, eaaz3223. [Google Scholar] [CrossRef]
- Watanabe, K.; Kuramitsu, S.; Posey, A.D.; June, C.H. Expanding the Therapeutic Window for CAR T Cell Therapy in Solid Tumors: The Knowns and Unknowns of CAR T Cell Biology. Front. Immunol. 2018, 9, 2486. [Google Scholar] [CrossRef]
- Salter, A.I.; Rajan, A. Comparative analysis of TCR and CAR signaling informs CAR designs with superior antigen sensitivity and in vivo function. Sci. Signal. 2021, 14, eabe2606. [Google Scholar] [CrossRef]
- Kersh, E.N.; Shaw, A.S.; Allen, P.M. Fidelity of T Cell Activation Through Multistep T Cell Receptor ζ Phosphorylation. Science 1998, 281, 572–575. [Google Scholar] [CrossRef]
- Burton, J.; Siller-Farfán, J.; Pettmann, J.; Salzer, B.; Kutuzov, M.; van der Merwe, A.; Dushek, O. Inefficient exploitation of accessory receptors reduces the sensitivity of chimeric antigen receptors. bioRxiv 2021. [Google Scholar] [CrossRef]
- Gudipati, V.; Rydzek, J.; Doel-Perez, I.; Goncalves, V.D.R.; Scharf, L.; Konigsberger, S.; Lobner, E.; Kunert, R.; Einsele, H.; Stockinger, H.; et al. Inefficient CAR-proximal signaling blunts antigen sensitivity. Nat. Immunol. 2020, 21, 848–856. [Google Scholar] [CrossRef]
- Davenport, A.J.; Cross, R.S.; Watson, K.A.; Liao, Y.; Shi, W.; Prince, H.M.; Beavis, P.A.; Trapani, J.A.; Kershaw, M.H.; Ritchie, D.S.; et al. Chimeric antigen receptor T cells form nonclassical and potent immune synapses driving rapid cytotoxicity. Proc. Natl. Acad. Sci. USA 2018, 115, E2068–E2076. [Google Scholar] [CrossRef]
- Wachsmann, T.L.A.; Wouters, A.K.; Remst, D.F.G.; Hagedoorn, R.S.; Meeuwsen, M.H.; van Diest, E.; Leusen, J.; Kuball, J.; Falkenburg, J.H.F.; Heemskerk, M.H.M. Comparing CAR and TCR engineered T cell performance as a function of tumor cell exposure. Oncoimmunology 2022, 11, 2033528. [Google Scholar] [CrossRef]
- Thauland, T.J.; Koguchi, Y.; Wetzel, S.A.; Dustin, M.L.; Parker, D.C. Th1 and Th2 cells form morphologically distinct immunological synapses. J. Immunol. 2008, 181, 393–399. [Google Scholar] [CrossRef]
- Brossard, C.; Feuillet, V.; Schmitt, A.; Randriamampita, C.; Romao, M.; Raposo, G.; Trautmann, A. Multifocal structure of the T cell—Dendritic cell synapse. Eur. J. Immunol. 2005, 35, 1741–1753. [Google Scholar] [CrossRef]
- Freiberg, B.A.; Kupfer, H.; Maslanik, W.; Delli, J.; Kappler, J.; Zaller, D.M.; Kupfer, A. Staging and resetting T cell activation in SMACs. Nat. Immunol. 2002, 3, 911–917. [Google Scholar] [CrossRef]
- Evnouchidou, I.; Caillens, V.; Koumantou, D.; Saveanu, L. The role of endocytic trafficking in antigen T cell receptor activation. Biomed. J. 2022, 45, 310–320. [Google Scholar] [CrossRef]
- Monjas, A.; Alcover, A.; Alarcón, B. Engaged and bystander T cell receptors are down-modulated by different endocytotic pathways. J. Biol. Chem. 2004, 279, 55376–55384. [Google Scholar] [CrossRef]
- Li, W.; Qiu, S.; Chen, J.; Jiang, S.; Chen, W.; Jiang, J.; Wang, F.; Si, W.; Shu, Y.; Wei, P.; et al. Chimeric Antigen Receptor Designed to Prevent Ubiquitination and Downregulation Showed Durable Antitumor Efficacy. Immunity 2020, 53, 456–470.e456. [Google Scholar] [CrossRef]
- Olson, M.L.; Vander Mause, E.R.; Radhakrishnan, S.V.; Brody, J.D.; Rapoport, A.P.; Welm, A.L.; Atanackovic, D.; Luetkens, T. Low-Affinity CAR T Cells Exhibit Reduced Trogocytosis, Preventing Fratricide and Antigen-Negative Tumor Escape While Preserving Anti-Tumor Activity. Leukemia 2022, 36, 1943–1946. [Google Scholar] [CrossRef]
- Kettleborough, C.A.; Saldanha, J.; Heath, V.J.; Morrison, C.J.; Bendig, M.M. Humanization of a mouse monoclonal antibody by CDR–grafting: The importance of framework residues on loop conformation. Protein Eng. Des. Sel. 1991, 4, 773–783. [Google Scholar] [CrossRef]
- Gaciarz, A.; Ruddock, L.W. Complementarity determining regions and frameworks contribute to the disulfide bond independent folding of intrinsically stable scFv. PLoS ONE 2017, 12, e0189964. [Google Scholar] [CrossRef]
- Xiang, J.; Sha, Y.; Jia, Z.; Prasad, L.; Delbaere, L.T. Framework residues 71 and 93 of the chimeric B72.3 antibody are major determinants of the conformation of heavy-chain hypervariable loops. J. Mol. Biol. 1995, 253, 385–390. [Google Scholar] [CrossRef]
- Rammensee, H.-G.; Falk, K.; Rötzschke, O. MHC molecules as peptide receptors. Curr. Opin. Immunol. 1993, 5, 35–44. [Google Scholar] [CrossRef]
- Davis, M.M.; Bjorkman, P.J. T-cell antigen receptor genes and T-cell recognition. Nature 1988, 334, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, M.G.; Stanfield, R.L.; Wilson, I.A. How TCRs bind MHCs, peptides, and coreceptors. Annu. Rev. Immunol. 2006, 24, 419–466. [Google Scholar] [CrossRef] [PubMed]
- Klein, J. The major histocompatibility complex of the mouse. Science 1979, 203, 516–521. [Google Scholar] [CrossRef]
- Crux, N.B.; Elahi, S. Human Leukocyte Antigen (HLA) and Immune Regulation: How Do Classical and Non-Classical HLA Alleles Modulate Immune Response to Human Immunodeficiency Virus and Hepatitis C Virus Infections? Front. Immunol. 2017, 8, 832. [Google Scholar] [CrossRef]
- Janeway, C.A.; Travers, P.; Walport, M.; Shlomchik, M.J. Immunobiology: The Immune System in Health and Disease, 5th ed.; Garland Science: New York, NY, USA, 2001. [Google Scholar]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef]
- Branella, G.M.; Spencer, H.T. Natural Receptor- and Ligand-Based Chimeric Antigen Receptors: Strategies Using Natural Ligands and Receptors for Targeted Cell Killing. Cells 2021, 11, 21. [Google Scholar] [CrossRef]
- Wong, W.K.; Leem, J.; Deane, C.M. Comparative Analysis of the CDR Loops of Antigen Receptors. Front. Immunol. 2019, 10, 2454. [Google Scholar] [CrossRef]
- Rock, E.P.; Sibbald, P.R.; Davis, M.M.; Chien, Y.H. CDR3 length in antigen-specific immune receptors. J. Exp. Med. 1994, 179, 323–328. [Google Scholar] [CrossRef]
- Blevins, S.J.; Pierce, B.G.; Singh, N.K.; Riley, T.P.; Wang, Y.; Spear, T.T.; Nishimura, M.I.; Weng, Z.; Baker, B.M. How structural adaptability exists alongside HLA-A2 bias in the human αβ TCR repertoire. Proc. Natl. Acad. Sci. USA 2016, 113, E1276–E1285. [Google Scholar] [CrossRef]
- Yarmarkovich, M.; Marshall, Q.F.; Warrington, J.M.; Premaratne, R.; Farrel, A.; Groff, D.; Li, W.; di Marco, M.; Runbeck, E.; Truong, H.; et al. Cross-HLA targeting of intracellular oncoproteins with peptide-centric CARs. Nature 2021, 599, 477–484. [Google Scholar] [CrossRef]
- Zhang, G.; Wang, L.; Cui, H.; Wang, X.; Zhang, G.; Ma, J.; Han, H.; He, W.; Wang, W.; Zhao, Y.; et al. Anti-melanoma activity of T cells redirected with a TCR-like chimeric antigen receptor. Sci. Rep. 2014, 4, 3571. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Xu, Y.; Xiang, J.; Long, L.; Green, S.; Yang, Z.; Zimdahl, B.; Lu, J.; Cheng, N.; Horan, L.H.; et al. Targeting Alpha-Fetoprotein (AFP)-MHC Complex with CAR T-Cell Therapy for Liver Cancer. Clin. Cancer Res. 2017, 23, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Rafiq, S.; Purdon, T.J.; Daniyan, A.F.; Koneru, M.; Dao, T.; Liu, C.; Scheinberg, D.A.; Brentjens, R.J. Optimized T-cell receptor-mimic chimeric antigen receptor T cells directed toward the intracellular Wilms Tumor 1 antigen. Leukemia 2017, 31, 1788–1797. [Google Scholar] [CrossRef] [PubMed]
- Schuberth, P.C.; Jakka, G.; Jensen, S.M.; Wadle, A.; Gautschi, F.; Haley, D.; Haile, S.; Mischo, A.; Held, G.; Thiel, M.; et al. Effector memory and central memory NY-ESO-1-specific re-directed T cells for treatment of multiple myeloma. Gene Ther. 2013, 20, 386–395. [Google Scholar] [CrossRef]
- Baeuerle, P.A.; Ding, J.; Patel, E.; Thorausch, N.; Horton, H.; Gierut, J.; Scarfo, I.; Choudhary, R.; Kiner, O.; Krishnamurthy, J.; et al. Synthetic TRuC receptors engaging the complete T cell receptor for potent anti-tumor response. Nat. Commun. 2019, 10, 2087. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, G.; Wang, J.; Zheng, Z.-Y.; Jia, L.; Rui, W.; Huang, D.; Zhou, Z.-X.; Zhou, L.; Wu, X.; et al. Chimeric STAR receptors using TCR machinery mediate robust responses against solid tumors. Sci. Transl. Med. 2021, 13, eabb5191. [Google Scholar] [CrossRef]
- Mansilla-Soto, J.; Eyquem, J.; Haubner, S.; Hamieh, M.; Feucht, J.; Paillon, N.; Zucchetti, A.E.; Li, Z.; Sjostrand, M.; Lindenbergh, P.L.; et al. HLA-independent T cell receptors for targeting tumors with low antigen density. Nat. Med. 2022, 28, 345–352. [Google Scholar] [CrossRef]
- Davari, K.; Holland, T.; Prassmayer, L.; Longinotti, G.; Ganley, K.P.; Pechilis, L.J.; Diaconu, I.; Nambiar, P.R.; Magee, M.S.; Schendel, D.J.; et al. Development of a CD8 co-receptor independent T-cell receptor specific for tumor-associated antigen MAGE-A4 for next generation T-cell-based immunotherapy. J. Immunother. Cancer 2021, 9, e002035. [Google Scholar] [CrossRef]
- Mehrotra, S.; Al-Khami, A.A.; Klarquist, J.; Husain, S.; Naga, O.; Eby, J.M.; Murali, A.K.; Lyons, G.E.; Li, M.; Spivey, N.D.; et al. A coreceptor-independent transgenic human TCR mediates anti-tumor and anti-self immunity in mice. J. Immunol. 2012, 189, 1627–1638. [Google Scholar] [CrossRef]
- Williams, C.M.; Schonnesen, A.A.; Zhang, S.-Q.; Ma, K.-Y.; He, C.; Yamamoto, T.; Eckhardt, S.G.; Klebanoff, C.A.; Jiang, N. Normalized Synergy Predicts That CD8 Co-Receptor Contribution to T Cell Receptor (TCR) and pMHC Binding Decreases As TCR Affinity Increases in Human Viral-Specific T Cells. Front. Immunol. 2017, 8, 894. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, M.; King, J.W.; Xue, S.-A.; Voisine, C.; Holler, A.; Wright, G.P.; Waxman, J.; Morris, E.; Stauss, H.J. CD3 limits the efficacy of TCR gene therapy in vivo. Blood 2011, 118, 3528–3537. [Google Scholar] [CrossRef] [PubMed]
- Schamel, W.W.A. The stoichiometry of the T cell antigen receptor and its implications for the signal transduction mechanism. Signal Transduct. 2007, 7, 311–319. [Google Scholar] [CrossRef]
- Wucherpfennig, K.W.; Gagnon, E.; Call, M.J.; Huseby, E.S.; Call, M.E. Structural biology of the T-cell receptor: Insights into receptor assembly, ligand recognition, and initiation of signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a005140. [Google Scholar] [CrossRef] [PubMed]
- Bridgeman, J.S.; Hawkins, R.E.; Bagley, S.; Blaylock, M.; Holland, M.; Gilham, D.E. The optimal antigen response of chimeric antigen receptors harboring the CD3zeta transmembrane domain is dependent upon incorporation of the receptor into the endogenous TCR/CD3 complex. J. Immunol. 2010, 184, 6938–6949. [Google Scholar] [CrossRef]
- James, J.R. Tuning ITAM multiplicity on T cell receptors can control potency and selectivity to ligand density. Sci. Signal. 2018, 11, eaan1088. [Google Scholar] [CrossRef]
- Feucht, J.; Sun, J.; Eyquem, J.; Ho, Y.J.; Zhao, Z.; Leibold, J.; Dobrin, A.; Cabriolu, A.; Hamieh, M.; Sadelain, M. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat. Med. 2019, 25, 82–88. [Google Scholar] [CrossRef]
- Duan, Y.; Chen, J.; Meng, X.; Liu, L.; Shang, K.; Wu, X.; Wang, Y.; Huang, Z.; Liu, H.; Huang, Y.; et al. Balancing activation and costimulation of CAR tunes signaling dynamics and enhances therapeutic potency. bioRxiv 2022. [Google Scholar] [CrossRef]
- Wu, W.; Zhou, Q.; Masubuchi, T.; Shi, X.; Li, H.; Xu, X.; Huang, M.; Meng, L.; He, X.; Zhu, H.; et al. Multiple Signaling Roles of CD3ε and Its Application in CAR-T Cell Therapy. Cell 2020, 182, 855–871.e823. [Google Scholar] [CrossRef]
- Till, B.G.; Jensen, M.C.; Wang, J.; Chen, E.Y.; Wood, B.L.; Greisman, H.A.; Qian, X.; James, S.E.; Raubitschek, A.; Forman, S.J.; et al. Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood 2008, 112, 2261–2271. [Google Scholar] [CrossRef]
- Maher, J.; Brentjens, R.J.; Gunset, G.; Rivière, I.; Sadelain, M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRζ /CD28 receptor. Nat. Biotechnol. 2002, 20, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Krause, A.; Guo, H.F.; Latouche, J.B.; Tan, C.; Cheung, N.K.; Sadelain, M. Antigen-dependent CD28 signaling selectively enhances survival and proliferation in genetically modified activated human primary T lymphocytes. J. Exp. Med. 1998, 188, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Kagoya, Y.; Tanaka, S.; Guo, T.; Anczurowski, M.; Wang, C.H.; Saso, K.; Butler, M.O.; Minden, M.D.; Hirano, N. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat. Med. 2018, 24, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, M.; Abken, H. CAR T Cells Releasing IL-18 Convert to T-Bet(high) FoxO1(low) Effectors that Exhibit Augmented Activity against Advanced Solid Tumors. Cell Rep. 2017, 21, 3205–3219. [Google Scholar] [CrossRef]
- Avanzi, M.P.; Yeku, O.; Li, X.; Wijewarnasuriya, D.P.; van Leeuwen, D.G.; Cheung, K.; Park, H.; Purdon, T.J.; Daniyan, A.F.; Spitzer, M.H.; et al. Engineered Tumor-Targeted T Cells Mediate Enhanced Anti-Tumor Efficacy Both Directly and through Activation of the Endogenous Immune System. Cell Rep. 2018, 23, 2130–2141. [Google Scholar] [CrossRef]
- Gutcher, I.; Becher, B. APC-derived cytokines and T cell polarization in autoimmune inflammation. J. Clin. Investig. 2007, 117, 1119–1127. [Google Scholar] [CrossRef]
- Feucht, J.; Sadelain, M. Function and evolution of the prototypic CD28ζ and 4-1BBζ chimeric antigen receptors. Immuno Oncol. Technol. 2020, 8, 2–11. [Google Scholar] [CrossRef]
- Strebhardt, K.; Ullrich, A. Paul Ehrlich’s magic bullet concept: 100 years of progress. Nat. Rev. Cancer 2008, 8, 473–480. [Google Scholar] [CrossRef]
- Alabanza, L.; Pegues, M.; Geldres, C.; Shi, V.; Wiltzius, J.J.W.; Sievers, S.A.; Yang, S.; Kochenderfer, J.N. Function of Novel Anti-CD19 Chimeric Antigen Receptors with Human Variable Regions Is Affected by Hinge and Transmembrane Domains. Mol. Ther. 2017, 25, 2452–2465. [Google Scholar] [CrossRef]
- Watanabe, N.; Bajgain, P.; Sukumaran, S.; Ansari, S.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Leen, A.M.; Vera, J.F. Fine-tuning the CAR spacer improves T-cell potency. Oncoimmunology 2016, 5, e1253656. [Google Scholar] [CrossRef] [PubMed]
- Guest, R.D.; Hawkins, R.E.; Kirillova, N.; Cheadle, E.J.; Arnold, J.; O’Neill, A.; Irlam, J.; Chester, K.A.; Kemshead, J.T.; Shaw, D.M.; et al. The Role of Extracellular Spacer Regions in the Optimal Design of Chimeric Immune Receptors: Evaluation of Four Different scFvs and Antigens. J. Immunother. 2005, 28, 203–211. [Google Scholar] [CrossRef]
- Hudecek, M.; Lupo-Stanghellini, M.-T.; Kosasih, P.L.; Sommermeyer, D.; Jensen, M.C.; Rader, C.; Riddell, S.R. Receptor Affinity and Extracellular Domain Modifications Affect Tumor Recognition by ROR1-Specific Chimeric Antigen Receptor T Cells. Clin. Cancer Res. 2013, 19, 3153–3164. [Google Scholar] [CrossRef]
- Kunkele, A.; Johnson, A.J.; Rolczynski, L.S.; Chang, C.A.; Hoglund, V.; Kelly-Spratt, K.S.; Jensen, M.C. Functional Tuning of CARs Reveals Signaling Threshold above Which CD8+ CTL Antitumor Potency Is Attenuated due to Cell Fas-FasL-Dependent AICD. Cancer Immunol. Res. 2015, 3, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Textor, A.; Grunewald, L.; Anders, K.; Klaus, A.; Schwiebert, S.; Winkler, A.; Stecklum, M.; Rolff, J.; Henssen, A.G.; Hopken, U.E.; et al. CD28 Co-Stimulus Achieves Superior CAR T Cell Effector Function against Solid Tumors Than 4-1BB Co-Stimulus. Cancers 2021, 13, 1050. [Google Scholar] [CrossRef] [PubMed]
- Drent, E.; Poels, R.; Ruiter, R.; van de Donk, N.; Zweegman, S.; Yuan, H.; de Bruijn, J.; Sadelain, M.; Lokhorst, H.M.; Groen, R.W.J.; et al. Combined CD28 and 4-1BB Costimulation Potentiates Affinity-tuned Chimeric Antigen Receptor-engineered T Cells. Clin. Cancer Res. 2019, 25, 4014–4025. [Google Scholar] [CrossRef]
- Artyomov, M.N.; Lis, M.; Devadas, S.; Davis, M.M.; Chakraborty, A.K. CD4 and CD8 binding to MHC molecules primarily acts to enhance Lck delivery. Proc. Natl. Acad. Sci. USA 2010, 107, 16916–16921. [Google Scholar] [CrossRef]
- Palacios, E.H.; Weiss, A. Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene 2004, 23, 7990–8000. [Google Scholar] [CrossRef]
- Brameshuber, M.; Kellner, F.; Rossboth, B.K.; Ta, H.; Alge, K.; Sevcsik, E.; Göhring, J.; Axmann, M.; Baumgart, F.; Gascoigne, N.R.J.; et al. Monomeric TCRs drive T cell antigen recognition. Nat. Immunol. 2018, 19, 487–496. [Google Scholar] [CrossRef]
- Pageon, S.V.; Tabarin, T.; Yamamoto, Y.; Ma, Y.; Nicovich, P.R.; Bridgeman, J.S.; Cohnen, A.; Benzing, C.; Gao, Y.; Crowther, M.D.; et al. Functional role of T-cell receptor nanoclusters in signal initiation and antigen discrimination. Proc. Natl. Acad. Sci. USA 2016, 113, E5454–E5463. [Google Scholar] [CrossRef]
- Kumar, R.; Ferez, M.; Swamy, M.; Arechaga, I.; Rejas, M.T.; Valpuesta, J.M.; Schamel, W.W.A.; Alarcon, B.; van Santen, H.M. Increased Sensitivity of Antigen-Experienced T Cells through the Enrichment of Oligomeric T Cell Receptor Complexes. Immunity 2011, 35, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Burroughs, N.J.; Lazic, Z.; van der Merwe, P.A. Ligand Detection and Discrimination by Spatial Relocalization: A Kinase-Phosphatase Segregation Model of TCR Activation. Biophys. J. 2006, 91, 1619–1629. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.J.; van der Merwe, P.A. The kinetic-segregation model: TCR triggering and beyond. Nat. Immunol. 2006, 7, 803–809. [Google Scholar] [CrossRef]
- James, J.R.; Vale, R.D. Biophysical Mechanism of T Cell Receptor Triggering in a Reconstituted System. Nature 2012, 487, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.T.; Takeuchi, K.; Sun, Z.-Y.J.; Touma, M.; Castro, C.E.; Fahmy, A.; Lang, M.J.; Wagner, G.; Reinherz, E.L. The αβ T Cell Receptor Is an Anisotropic Mechanosensor. J. Biol. Chem. 2009, 284, 31028–31037. [Google Scholar] [CrossRef] [PubMed]
- Judokusumo, E.; Tabdanov, E.; Kumari, S.; Dustin, M.L.; Kam, L.C. Mechanosensing in T Lymphocyte Activation. Biophys. J. 2012, 102, L5–L7. [Google Scholar] [CrossRef] [PubMed]
- Das, D.K.; Feng, Y.; Mallis, R.J.; Li, X.; Keskin, D.B.; Hussey, R.E.; Brady, S.K.; Wang, J.-H.; Wagner, G.; Reinherz, E.L.; et al. Force-dependent transition in the T-cell receptor β-subunit allosterically regulates peptide discrimination and pMHC bond lifetime. Proc. Natl. Acad. Sci. USA 2015, 112, 1517–1522. [Google Scholar] [CrossRef]
- Feng, Y.; Brazin, K.N.; Kobayashi, E.; Mallis, R.J.; Reinherz, E.L.; Lang, M.J. Mechanosensing drives acuity of αβ T-cell recognition. Proc. Natl. Acad. Sci. USA 2017, 114, E8204–E8213. [Google Scholar] [CrossRef]
- Yousefi, O.S.; Günther, M.; Hörner, M.; Chalupsky, J.; Wess, M.; Brandl, S.M.; Smith, R.W.; Fleck, C.; Kunkel, T.; Zurbriggen, M.D.; et al. Optogenetic control shows that kinetic proofreading regulates the activity of the T cell receptor. eLife 2019, 8, e42475. [Google Scholar] [CrossRef]
- Chang, Z.L.; Lorenzini, M.H.; Chen, X.; Tran, U.; Bangayan, N.J.; Chen, Y.Y. Rewiring T-cell responses to soluble factors with chimeric antigen receptors. Nat. Chem. Biol. 2018, 14, 317–324. [Google Scholar] [CrossRef]
- Tischer, D.K.; Weiner, O.D. Light-based tuning of ligand half-life supports kinetic proofreading model of T cell signaling. eLife 2019, 8, e42498. [Google Scholar] [CrossRef] [PubMed]
- Harris, D.T.; Hager, M.V.; Smith, S.N.; Cai, Q.; Stone, J.D.; Kruger, P.; Lever, M.; Dushek, O.; Schmitt, T.M.; Greenberg, P.D.; et al. Comparison of T Cell Activities Mediated by Human TCRs and CARs That Use the Same Recognition Domains. J. Immunol. 2018, 200, 1088–1100. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, X.; Zhou, Z.; Liu, Y.; Yu, L.; Jia, L.; Yang, J.; Li, J.; Yu, H.; Li, W.; et al. A novel adoptive synthetic TCR and antigen receptor (STAR) T-Cell therapy for B-Cell acute lymphoblastic leukemia. Am. J. Hematol. 2022, 97, 992–1004. [Google Scholar] [CrossRef]
- Stone, J.D.; Harris, D.T.; Soto, C.M.; Chervin, A.S.; Aggen, D.H.; Roy, E.J.; Kranz, D.M. A novel T cell receptor single-chain signaling complex mediates antigen-specific T cell activity and tumor control. Cancer Immunol. Immunother. CII 2014, 63, 1163–1176. [Google Scholar] [CrossRef] [PubMed]
- Morch, A.M.; Balint, S.; Santos, A.M.; Davis, S.J.; Dustin, M.L. Coreceptors and TCR Signaling—The Strong and the Weak of It. Front. Cell Dev. Biol. 2020, 8, 597627. [Google Scholar] [CrossRef]
- Potter, T.A.; Grebe, K.; Freiberg, B.; Kupfer, A. Formation of supramolecular activation clusters on fresh ex vivo CD8+ T cells after engagement of the T cell antigen receptor and CD8 by antigen-presenting cells. Proc. Natl. Acad. Sci. USA 2001, 98, 12624–12629. [Google Scholar] [CrossRef] [PubMed]
- Stinchcombe, J.C.; Bossi, G.; Booth, S.; Griffiths, G.M. The Immunological Synapse of CTL Contains a Secretory Domain and Membrane Bridges. Immunity 2001, 15, 751–761. [Google Scholar] [CrossRef]
- Reichardt, P.; Dornbach, B.; Rong, S.; Beissert, S.; Gueler, F.; Loser, K.; Gunzer, M. Naive B cells generate regulatory T cells in the presence of a mature immunologic synapse. Blood 2007, 110, 1519–1529. [Google Scholar] [CrossRef]
- Thauland, T.J.; Parker, D.C. Diversity in immunological synapse structure. Immunology 2010, 131, 466–472. [Google Scholar] [CrossRef]
- Balamuth, F.; Leitenberg, D.; Unternaehrer, J.; Mellman, I.; Bottomly, K. Distinct Patterns of Membrane Microdomain Partitioning in Th1 and Th2 Cells. Immunity 2001, 15, 729–738. [Google Scholar] [CrossRef]
- Jackman, R.P.; Balamuth, F.; Bottomly, K. CTLA-4 Differentially Regulates the Immunological Synapse in CD4 T Cell Subsets. J. Immunol. 2007, 178, 5543–5551. [Google Scholar] [CrossRef] [PubMed]
- Hailman, E.; Burack, W.R.; Shaw, A.S.; Dustin, M.L.; Allen, P.M. Immature CD4+CD8+ Thymocytes Form a Multifocal Immunological Synapse with Sustained Tyrosine Phosphorylation. Immunity 2002, 16, 839–848. [Google Scholar] [CrossRef]
- Grakoui, A.; Bromley, S.K.; Sumen, C.; Davis, M.M.; Shaw, A.S.; Allen, P.M.; Dustin, M.L. The Immunological Synapse: A Molecular Machine Controlling T Cell Activation. Science 1999, 285, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-H.; Holdorf, A.D.; Dustin, M.L.; Chan, A.C.; Allen, P.M.; Shaw, A.S. T cell receptor signaling precedes immunological synapse formation. Science 2002, 295, 1539–1542. [Google Scholar] [CrossRef]
- Yokosuka, T.; Kobayashi, W.; Sakata-Sogawa, K.; Takamatsu, M.; Hashimoto-Tane, A.; Dustin, M.L.; Tokunaga, M.; Saito, T. Spatiotemporal Regulation of T Cell Costimulation by TCR-CD28 Microclusters and Protein Kinase C θ Translocation. Immunity 2008, 29, 589–601. [Google Scholar] [CrossRef]
- Yokosuka, T.; Sakata-Sogawa, K.; Kobayashi, W.; Hiroshima, M.; Hashimoto-Tane, A.; Tokunaga, M.; Dustin, M.L.; Saito, T. Newly generated T cell receptor microclusters initiate and sustain T cell activation by recruitment of Zap70 and SLP-76. Nat. Immunol. 2005, 6, 1253–1262. [Google Scholar] [CrossRef]
- Campi, G.; Varma, R.; Dustin, M.L. Actin and agonist MHC-peptide complex-dependent T cell receptor microclusters as scaffolds for signaling. J. Exp. Med. 2005, 202, 1031–1036. [Google Scholar] [CrossRef]
- Varma, R.; Campi, G.; Yokosuka, T.; Saito, T.; Dustin, M.L. T Cell Receptor-Proximal Signals Are Sustained in Peripheral Microclusters and Terminated in the Central Supramolecular Activation Cluster. Immunity 2006, 25, 117–127. [Google Scholar] [CrossRef]
- Ilani, T.; Vasiliver-Shamis, G.; Vardhana, S.; Bretscher, A.; Dustin, M.L. T cell antigen receptor signaling and immunological synapse stability require myosin IIA. Nat. Immunol. 2009, 10, 531–539. [Google Scholar] [CrossRef]
- Cemerski, S.; Das, J.; Giurisato, E.; Markiewicz, M.A.; Allen, P.M.; Chakraborty, A.K.; Shaw, A.S. The balance between T cell receptor signaling and degradation at the center of the immunological synapse is determined by antigen quality. Immunity 2008, 29, 414–422. [Google Scholar] [CrossRef]
- Alarcón, B.; Mestre, D.; Martínez-Martín, N. The immunological synapse: A cause or consequence of T-cell receptor triggering? Immunology 2011, 133, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Vardhana, S.; Choudhuri, K.; Varma, R.; Dustin, M.L. Essential Role of Ubiquitin and TSG101 Protein in Formation and Function of the Central Supramolecular Activation Cluster. Immunity 2010, 32, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Stinchcombe, J.C.; Majorovits, E.; Bossi, G.; Fuller, S.; Griffiths, G.M. Centrosome polarization delivers secretory granules to the immunological synapse. Nature 2006, 443, 462–465. [Google Scholar] [CrossRef]
- Xiong, W.; Chen, Y.; Kang, X.; Chen, Z.; Zheng, P.; Hsu, Y.-H.; Jang, J.H.; Qin, L.; Liu, H.; Dotti, G.; et al. Immunological Synapse Predicts Effectiveness of Chimeric Antigen Receptor Cells. Mol. Ther. 2018, 26, 963–975. [Google Scholar] [CrossRef]
- Ramello, M.C.; Benzaïd, I.; Kuenzi, B.M.; Lienlaf-Moreno, M.; Kandell, W.M.; Santiago, D.N.; Pabón-Saldaña, M.; Darville, L.; Fang, B.; Rix, U.; et al. An immunoproteomic approach to characterize the CAR interactome and signalosome. Sci. Signal. 2019, 12, eaap9777. [Google Scholar] [CrossRef]
- Larson, R.C.; Kann, M.C.; Bailey, S.R. CAR T cell killing requires the IFNγR pathway in solid but not liquid tumours. Nature 2022, 604, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Wei, Q.; Brzostek, J.; Gascoigne, N.R.J. Signaling from T cell receptors (TCRs) and chimeric antigen receptors (CARs) on T cells. Cell. Mol. Immunol. 2020, 17, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Nika, K.; Soldani, C.; Salek, M.; Paster, W.; Gray, A.; Etzensperger, R.; Fugger, L.; Polzella, P.; Cerundolo, V.; Dushek, O.; et al. Constitutively Active Lck Kinase in T Cells Drives Antigen Receptor Signal Transduction. Immunity 2010, 32, 766–777. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, L.I.R.; Ebert, P.J.R.; Krummel, M.F.; Weiss, A.; Davis, M.M. Dynamics of p56lck translocation to the T cell immunological synapse following agonist and antagonist stimulation. Immunity 2002, 17, 809–822. [Google Scholar] [CrossRef]
- Kim, P.W.; Sun, Z.-Y.J.; Blacklow, S.C.; Wagner, G.; Eck, M.J. A zinc clasp structure tethers Lck to T cell coreceptors CD4 and CD8. Science 2003, 301, 1725–1728. [Google Scholar] [CrossRef]
- Reth, M. Antigen receptor tail clue. Nature 1989, 338, 383–384. [Google Scholar] [CrossRef] [PubMed]
- Smith-Garvin, J.E.; Koretzky, G.A.; Jordan, M.S. T cell activation. Annu. Rev. Immunol. 2009, 27, 591–619. [Google Scholar] [CrossRef] [PubMed]
- Samelson, L.E. Signal transduction mediated by the T cell antigen receptor: The role of adapter proteins. Annu. Rev. Immunol. 2002, 20, 371–394. [Google Scholar] [CrossRef]
- Lo, W.-L.; Shah, N.H.; Ahsan, N.; Horkova, V.; Stepanek, O.; Salomon, A.R.; Kuriyan, J.; Weiss, A. Lck promotes Zap70-dependent LAT phosphorylation by bridging Zap70 to LAT. Nat. Immunol. 2018, 19, 733–741. [Google Scholar] [CrossRef]
- Zhang, W.; Trible, R.P.; Zhu, M.; Liu, S.K.; McGlade, C.J.; Samelson, L.E. Association of Grb2, Gads, and phospholipase C-gamma 1 with phosphorylated LAT tyrosine residues. Effect of LAT tyrosine mutations on T cell angigen receptor-mediated signaling. J. Biol. Chem. 2000, 275, 23355–23361. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.K.; Fang, N.; Koretzky, G.A.; McGlade, C.J. The hematopoietic-specific adaptor protein gads functions in T-cell signaling via interactions with the SLP-76 and LAT adaptors. Curr. Biol. 1999, 9, 67–75. [Google Scholar] [CrossRef]
- Su, X.; Ditlev, J.A.; Hui, E.; Xing, W.; Banjade, S.; Okrut, J.; King, D.S.; Taunton, J.; Rosen, M.K.; Vale, R.D. Phase separation of signaling molecules promotes T cell receptor signal transduction. Science 2016, 352, 595–599. [Google Scholar] [CrossRef]
- Huang, W.Y.C.; Yan, Q.; Lin, W.-C.; Chung, J.K.; Hansen, S.D.; Christensen, S.M.; Tu, H.-L.; Kuriyan, J.; Groves, J.T. Phosphotyrosine-mediated LAT assembly on membranes drives kinetic bifurcation in recruitment dynamics of the Ras activator SOS. Proc. Natl. Acad. Sci. USA 2016, 113, 8218–8223. [Google Scholar] [CrossRef]
- Shim, E.K.; Jung, S.H.; Lee, J.R. Role of two adaptor molecules SLP-76 and LAT in the PI3K signaling pathway in activated T cells. J. Immunol. 2011, 186, 2926–2935. [Google Scholar] [CrossRef]
- Salter, A.I.; Ivey, R.G.; Kennedy, J.J.; Voillet, V.; Rajan, A.; Alderman, E.J.; Voytovich, U.J.; Lin, C.; Sommermeyer, D.; Liu, L.; et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci. Signal. 2018, 11, eaat6753. [Google Scholar] [CrossRef]
- Sun, C.; Shou, P.; Du, H.; Hirabayashi, K.; Chen, Y.; Herring, L.E.; Ahn, S.; Xu, Y.; Suzuki, K.; Li, G.; et al. THEMIS-SHP1 Recruitment by 4-1BB Tunes LCK-Mediated Priming of Chimeric Antigen Receptor-Redirected T Cells. Cancer Cell 2020, 37, 216–225.e216. [Google Scholar] [CrossRef] [PubMed]
- Askonas, B.A. T-cell differentiation and effector functions. Immunology 1988, 64 (Suppl. 1), 51–52. [Google Scholar]
- Iezzi, G.; Karjalainen, K.; Lanzavecchia, A. The duration of antigenic stimulation determines the fate of naive and effector T cells. Immunity 1998, 8, 89–95. [Google Scholar] [CrossRef]
- Beal, A.M.; Anikeeva, N.; Varma, R.; Cameron, T.O.; Vasiliver-Shamis, G.; Norris, P.J.; Dustin, M.L.; Sykulev, Y. Kinetics of early T cell receptor signaling regulate the pathway of lytic granule delivery to the secretory domain. Immunity 2009, 31, 632–642. [Google Scholar] [CrossRef]
- Pores-Fernando, A.T.; Zweifach, A. Calcium influx and signaling in cytotoxic T-lymphocyte lytic granule exocytosis. Immunol. Rev. 2009, 231, 160–173. [Google Scholar] [CrossRef]
- Joseph, N.; Reicher, B.; Barda-Saad, M. The calcium feedback loop and T cell activation: How cytoskeleton networks control intracellular calcium flux. Biochim. Biophys. Acta Biomembr. 2014, 1838, 557–568. [Google Scholar] [CrossRef]
- Karttunen, J.; Shastri, N. Measurement of ligand-induced activation in single viable T cells using the lacZ reporter gene. Proc. Natl. Acad. Sci. USA 1991, 88, 3972–3976. [Google Scholar] [CrossRef]
- Lewis, R.S. Calcium signaling mechanisms in T lymphocytes. Annu. Rev. Immunol. 2001, 19, 497–521. [Google Scholar] [CrossRef]
- Hogan, P.G.; Chen, L.; Nardone, J.; Rao, A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003, 17, 2205–2232. [Google Scholar] [CrossRef]
- Macian, F. NFAT proteins: Key regulators of T-cell development and function. Nat. Rev. Immunol. 2005, 5, 472–484. [Google Scholar] [CrossRef]
- Shao, M.; Teng, X.; Guo, X.; Zhang, H.; Huang, Y.; Cui, J.; Si, X.; Ding, L.; Wang, X.; Li, X.; et al. Inhibition of Calcium Signaling Prevents Exhaustion and Enhances Anti-Leukemia Efficacy of CAR-T Cells via SOCE-Calcineurin-NFAT and Glycolysis Pathways. Adv. Sci. 2022, 9, e2103508. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.C. Regulation and Function of the RasGRP Family of Ras Activators in Blood Cells. Genes Cancer 2011, 2, 320–334. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. ERK1/2 MAP kinases: Structure, function, and regulation. Pharmacol. Res. 2012, 66, 105–143. [Google Scholar] [CrossRef] [PubMed]
- Roche, M.I.; Ramadas, R.A.; Medoff, B.D. The Role of CARMA1 in T Cells. Crit. Rev. Immunol. 2013, 33, 219–243. [Google Scholar] [CrossRef]
- Schulze-Luehrmann, J.; Ghosh, S. Antigen-Receptor Signaling to Nuclear Factor κB. Immunity 2006, 25, 701–715. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Yang, J. Molecular basis of lysophosphatidic acid-induced NF-κB activation. Cell. Signal. 2010, 22, 1799–1803. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-kappaB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef]
- Zhao, X.; Yang, J.; Zhang, X.; Lu, X.-A.; Xiong, M.; Zhang, J.; Zhou, X.; Qi, F.; He, T.; Ding, Y.; et al. Efficacy and Safety of CD28- or 4-1BB-Based CD19 CAR-T Cells in B Cell Acute Lymphoblastic Leukemia. Mol. Ther. Oncolytics 2020, 18, 272–281. [Google Scholar] [CrossRef]
- Ying, Z.; He, T.; Wang, X.; Zheng, W.; Lin, N.; Tu, M.; Xie, Y.; Ping, L.; Zhang, C.; Liu, W.; et al. Parallel Comparison of 4-1BB or CD28 Co-stimulated CD19-Targeted CAR-T Cells for B Cell Non-Hodgkin’s Lymphoma. Mol. Ther. Oncolytics 2019, 15, 60–68. [Google Scholar] [CrossRef]
- Philipson, B.I.; O’Connor, R.S.; May, M.J.; June, C.H.; Albelda, S.M.; Milone, M.C. 4-1BB costimulation promotes CAR T cell survival through noncanonical NF-κB signaling. Sci. Signal. 2020, 13, eaay8248. [Google Scholar] [CrossRef]
- Gibbins, J.M.; Briddon, S.; Shutes, A.; van Vugt, M.J.; van de Winkel, J.G.; Saito, T.; Watson, S.P. The p85 subunit of phosphatidylinositol 3-kinase associates with the Fc receptor gamma-chain and linker for activitor of T cells (LAT) in platelets stimulated by collagen and convulxin. J. Biol. Chem. 1998, 273, 34437–34443. [Google Scholar] [CrossRef] [PubMed]
- Shim, E.K.; Moon, C.S.; Lee, G.Y.; Ha, Y.J.; Chae, S.-K.; Lee, J.R. Association of the Src homology 2 domain-containing leukocyte phosphoprotein of 76 kD (SLP-76) with the p85 subunit of phosphoinositide 3-kinase. FEBS Lett. 2004, 575, 35–40. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. Regulation of mTORC1 and its impact on gene expression at a glance. J. Cell Sci. 2013, 126, 1713–1719. [Google Scholar] [CrossRef] [PubMed]
- Frauwirth, K.A.; Riley, J.L.; Harris, M.H.; Parry, R.V.; Rathmell, J.C.; Plas, D.R.; Elstrom, R.L.; June, C.H.; Thompson, C.B. The CD28 Signaling Pathway Regulates Glucose Metabolism. Immunity 2002, 16, 769–777. [Google Scholar] [CrossRef]
- Kofler, D.M.; Chmielewski, M.; Rappl, G.; Hombach, A.; Riet, T.; Schmidt, A.; Hombach, A.A.; Wendtner, C.-M.; Abken, H. CD28 Costimulation Impairs the Efficacy of a Redirected T-cell Antitumor Attack in the Presence of Regulatory T cells Which Can Be Overcome by Preventing Lck Activation. Mol. Ther. 2011, 19, 760–767. [Google Scholar] [CrossRef]
- Alzubi, J.; Dettmer-Monaco, V.; Kuehle, J.; Thorausch, N.; Seidl, M.; Taromi, S.; Schamel, W.; Zeiser, R.; Abken, H.; Cathomen, T.; et al. PSMA-Directed CAR T Cells Combined with Low-Dose Docetaxel Treatment Induce Tumor Regression in a Prostate Cancer Xenograft Model. Mol. Ther. Oncolytics 2020, 18, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Doherty, G.J.; McMahon, H.T. Mechanisms of endocytosis. Annu. Rev. Biochem. 2009, 78, 857–902. [Google Scholar] [CrossRef]
- Schmid, E.M.; McMahon, H.T. Integrating molecular and network biology to decode endocytosis. Nature 2007, 448, 883–888. [Google Scholar] [CrossRef]
- Sigismund, S.; Argenzio, E.; Tosoni, D.; Cavallaro, E.; Polo, S.; Di Fiore, P.P. Clathrin-Mediated Internalization Is Essential for Sustained EGFR Signaling but Dispensable for Degradation. Dev. Cell 2008, 15, 209–219. [Google Scholar] [CrossRef]
- Boucrot, E.; Ferreira, A.P.A.; Almeida-Souza, L.; Debard, S.; Vallis, Y.; Howard, G.; Bertot, L.; Sauvonnet, N.; McMahon, H.T. Endophilin marks and controls a clathrin-independent endocytic pathway. Nature 2015, 517, 460–465. [Google Scholar] [CrossRef]
- Szymczak, A.L.; Vignali, D.A.A. Plasticity and Rigidity in Adaptor Protein-2-Mediated Internalization of the TCR:CD3 Complex. J. Immunol. 2005, 174, 4153–4160. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, J.; Hou, X.; Wegener, A.M.; Geisler, C. CD3 gamma contains a phosphoserine-dependent di-leucine motif involved in down-regulation of the T cell receptor. EMBO J. 1994, 13, 2156–2166. [Google Scholar] [CrossRef] [PubMed]
- von Essen, M.; Menné, C.; Nielsen, B.L.; Lauritsen, J.P.H.; Dietrich, J.; Andersen, P.S.; Karjalainen, K.; Ødum, N.; Geisler, C. The CD3 gamma leucine-based receptor-sorting motif is required for efficient ligand-mediated TCR down-regulation. J. Immunol. 2002, 168, 4519–4523. [Google Scholar] [CrossRef]
- San José, E.; Borroto, A.; Niedergang, F.; Alcover, A.; Alarcón, B. Triggering the TCR complex causes the downregulation of nonengaged receptors by a signal transduction-dependent mechanism. Immunity 2000, 12, 161–170. [Google Scholar] [CrossRef]
- Alcover, A.; Alarcón, B.; Di Bartolo, V. Cell Biology of T Cell Receptor Expression and Regulation. Annu. Rev. Immunol. 2018, 36, 103–125. [Google Scholar] [CrossRef]
- Martínez-Martín, N.; Fernández-Arenas, E.; Cemerski, S.; Delgado, P.; Turner, M.; Heuser, J.; Irvine, D.J.; Huang, B.; Bustelo, X.R.; Shaw, A.; et al. T cell receptor internalization from the immunological synapse is mediated by TC21 and RhoG GTPase-dependent phagocytosis. Immunity 2011, 35, 208–222. [Google Scholar] [CrossRef] [PubMed]
- Joly, E.; Hudrisier, D. What is trogocytosis and what is its purpose? Nat. Immunol. 2003, 4, 815. [Google Scholar] [CrossRef] [PubMed]
- Yudushkin, I.A.; Vale, R.D. Imaging T-cell receptor activation reveals accumulation of tyrosine-phosphorylated CD3ζ in the endosomal compartment. Proc. Natl. Acad. Sci. USA 2010, 107, 22128–22133. [Google Scholar] [CrossRef]
- Willinger, T.; Staron, M.; Ferguson, S.M.; De Camilli, P.; Flavell, R.A. Dynamin 2-dependent endocytosis sustains T-cell receptor signaling and drives metabolic reprogramming in T lymphocytes. Proc. Natl. Acad. Sci. USA 2015, 112, 4423–4428. [Google Scholar] [CrossRef]
- Onnis, A.; Baldari, C.T. Orchestration of Immunological Synapse Assembly by Vesicular Trafficking. Front. Cell Dev. Biol. 2019, 7, 110. [Google Scholar] [CrossRef]
- Stenger, D.; Stief, T.A.; Käuferle, T.; Willier, S.; Rataj, F.; Schober, K.; Vick, B.; Lotfi, R.; Wagner, B.; Grunewald, T.; et al. Endogenous TCR promotes in vivo persistence of CD19-CAR-T cells compared to a CRISPR/Cas9-mediated TCR knockout CAR. Blood 2020, 136, 1407–1418. [Google Scholar] [CrossRef] [PubMed]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gonen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Pule, M.A.; Savoldo, B.; Myers, G.D.; Rossig, C.; Russell, H.V.; Dotti, G.; Huls, M.H.; Liu, E.; Gee, A.P.; Mei, Z.; et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: Persistence and antitumor activity in individuals with neuroblastoma. Nat. Med. 2008, 14, 1264–1270. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Tashiro, H.; Omer, B.; Lapteva, N.; Ando, J.; Ngo, M.; Mehta, B.; Dotti, G.; Kinchington, P.R.; Leen, A.M.; et al. Vaccination Targeting Native Receptors to Enhance the Function and Proliferation of Chimeric Antigen Receptor (CAR)-Modified T Cells. Clin. Cancer Res. 2017, 23, 3499–3509. [Google Scholar] [CrossRef]
- Lapteva, N.; Gilbert, M.; Diaconu, I.; Rollins, L.A.; Al-Sabbagh, M.; Naik, S.; Krance, R.A.; Tripic, T.; Hiregange, M.; Raghavan, D.; et al. T-Cell Receptor Stimulation Enhances the Expansion and Function of CD19 Chimeric Antigen Receptor–Expressing T Cells. Clin. Cancer Res. 2019, 25, 7340–7350. [Google Scholar] [CrossRef]
- Omer, B.; Castillo, P.A.; Tashiro, H.; Shum, T.; Huynh, M.T.A.; Cardenas, M.; Tanaka, M.; Lewis, A.; Sauer, T.; Parihar, R.; et al. Chimeric Antigen Receptor Signaling Domains Differentially Regulate Proliferation and Native T Cell Receptor Function in Virus-Specific T Cells. Front. Med. 2018, 5, 343. [Google Scholar] [CrossRef]
- Louis, C.U.; Savoldo, B.; Dotti, G.; Pule, M.; Yvon, E.; Myers, G.D.; Rossig, C.; Russell, H.V.; Diouf, O.; Liu, E.; et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 2011, 118, 6050–6056. [Google Scholar] [CrossRef]
- Cruz, C.R.; Micklethwaite, K.P.; Savoldo, B.; Ramos, C.A.; Lam, S.; Ku, S.; Diouf, O.; Liu, E.; Barrett, A.J.; Ito, S.; et al. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: A phase 1 study. Blood 2013, 122, 2965–2973. [Google Scholar] [CrossRef]
- Vardhana, S.A.; Hwee, M.A.; Berisa, M.; Wells, D.K.; Yost, K.E.; King, B.; Smith, M.; Herrera, P.S.; Chang, H.Y.; Satpathy, A.T.; et al. Impaired mitochondrial oxidative phosphorylation limits the self-renewal of T cells exposed to persistent antigen. Nat. Immunol. 2020, 21, 1022–1033. [Google Scholar] [CrossRef]
- Weber, E.W.; Parker, K.R.; Sotillo, E.; Lynn, R.C.; Anbunathan, H.; Lattin, J.; Good, Z.; Belk, J.A.; Daniel, B.; Klysz, D.; et al. Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Science 2021, 372, eaba1786. [Google Scholar] [CrossRef]
- van der Stegen, S.J.; Hamieh, M.; Sadelain, M. The pharmacology of second-generation chimeric antigen receptors. Nat. Rev. Drug Discov. 2015, 14, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Burr, J.S.; Savage, N.D.; Messah, G.E.; Kimzey, S.L.; Shaw, A.S.; Arch, R.H.; Green, J.M. Cutting edge: Distinct motifs within CD28 regulate T cell proliferation and induction of Bcl-XL. J. Immunol. 2001, 166, 5331–5335. [Google Scholar] [CrossRef] [PubMed]
- Kerstan, A.; Armbruster, N.; Leverkus, M.; Hunig, T. Cyclosporin A abolishes CD28-mediated resistance to CD95-induced apoptosis via superinduction of caspase-3. J. Immunol. 2006, 177, 7689–7697. [Google Scholar] [CrossRef]
- Zhong, X.S.; Matsushita, M.; Plotkin, J.; Riviere, I.; Sadelain, M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol. Ther. 2010, 18, 413–420. [Google Scholar] [CrossRef]
- Yang, Y.; Kohler, M.E.; Chien, C.D.; Sauter, C.T.; Jacoby, E.; Yan, C.; Hu, Y.; Wanhainen, K.; Qin, H.; Fry, T.J. TCR engagement negatively affects CD8 but not CD4 CAR T cell expansion and leukemic clearance. Sci. Transl. Med. 2017, 9, eaag1209. [Google Scholar] [CrossRef]
- Evgin, L.; Kottke, T. Oncolytic virus-mediated expansion of dual-specific CAR T cells improves efficacy against solid tumors in mice. Sci. Transl. Med. 2022, 14, eabn2231. [Google Scholar] [CrossRef]
- Slaney, C.Y.; von Scheidt, B.; Davenport, A.J.; Beavis, P.A.; Westwood, J.A.; Mardiana, S.; Tscharke, D.C.; Ellis, S.; Prince, H.M.; Trapani, J.A.; et al. Dual-specific Chimeric Antigen Receptor T Cells and an Indirect Vaccine Eradicate a Variety of Large Solid Tumors in an Immunocompetent, Self-antigen Setting. Clin. Cancer Res. 2017, 23, 2478–2490. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F.; Lenig, D.; Förster, R.; Lipp, M.; Lanzavecchia, A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 1999, 401, 708–712. [Google Scholar] [CrossRef]
- Klebanoff, C.A.; Gattinoni, L.; Torabi-Parizi, P.; Kerstann, K.; Cardones, A.R.; Finkelstein, S.E.; Palmer, D.C.; Antony, P.A.; Hwang, S.T.; Rosenberg, S.A.; et al. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc. Natl. Acad. Sci. USA 2005, 102, 9571–9576. [Google Scholar] [CrossRef]
- Kueberuwa, G.; Gornall, H.; Alcantar-Orozco, E.M.; Bouvier, D.; Kapacee, Z.A.; Hawkins, R.E.; Gilham, D.E. CCR7(+) selected gene-modified T cells maintain a central memory phenotype and display enhanced persistence in peripheral blood in vivo. J. Immunother. Cancer 2017, 5, 14. [Google Scholar] [CrossRef]
- Uslu, U.; Schuler, G.; Dörrie, J.; Schaft, N. Combining a chimeric antigen receptor and a conventional T-cell receptor to generate T cells expressing two additional receptors (TETARs) for a multi-hit immunotherapy of melanoma. Exp. Dermatol. 2016, 25, 872–879. [Google Scholar] [CrossRef] [PubMed]
- Simon, B.; Harrer, D.C.; Schuler-Thurner, B.; Schuler, G.; Uslu, U. Arming T Cells with a gp100-Specific TCR and a CSPG4-Specific CAR Using Combined DNA- and RNA-Based Receptor Transfer. Cancers 2019, 11, 696. [Google Scholar] [CrossRef]
- Garber, K. Driving T-cell immunotherapy to solid tumors. Nat. Biotechnol. 2018, 36, 215–219. [Google Scholar] [CrossRef]
- Miyao, K.; Terakura, S.; Okuno, S.; Julamanee, J.; Watanabe, K.; Hamana, H.; Kishi, H.; Sakemura, R.; Koyama, D.; Goto, T.; et al. Introduction of Genetically Modified CD3ζ Improves Proliferation and Persistence of Antigen-Specific CTLs. Cancer Immunol. Res. 2018, 6, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Thakur, A.; Scholler, J.; Kubicka, E.; Bliemeister, E.T.; Schalk, D.L.; June, C.H.; Lum, L.G. Bispecific Antibody Armed Metabolically Enhanced Headless CAR T Cells. Front. Immunol. 2021, 12, 690437. [Google Scholar] [CrossRef] [PubMed]
- Omer, B.; Cardenas, M.G.; Pfeiffer, T.; Daum, R.; Huynh, M.; Sharma, S.; Nouraee, N.; Xie, C.; Tat, C.; Perconti, S.; et al. A Costimulatory CAR Improves TCR-based Cancer Immunotherapy. Cancer Immunol. Res. 2022, 10, 512–524. [Google Scholar] [CrossRef] [PubMed]
- Morris, E.C.; Neelapu, S.S.; Giavridis, T.; Sadelain, M. Cytokine release syndrome and associated neurotoxicity in cancer immunotherapy. Nat. Reviews. Immunol. 2022, 22, 85–96. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.S.; Hegde, M.; Robertson, C.; Ghazi, A.; Gerken, C.; Liu, E.; Dakhova, O.; Ashoori, A.; Corder, A. Human Epidermal Growth Factor Receptor 2 (HER2) –Specific Chimeric Antigen Receptor–Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J. Clin. Oncol. 2015, 33, 1688–1696. [Google Scholar] [CrossRef]
- Siegler, E.L.; Kenderian, S.S. Neurotoxicity and Cytokine Release Syndrome After Chimeric Antigen Receptor T Cell Therapy: Insights Into Mechanisms and Novel Therapies. Front. Immunol. 2020, 11, 1973. [Google Scholar] [CrossRef]
- Liu, Y.; Yan, X.; Zhang, F.; Zhang, X.; Tang, F.; Han, Z.; Li, Y. TCR-T Immunotherapy: The Challenges and Solutions. Front Oncol. 2021, 11, 794183. [Google Scholar] [CrossRef]
- Morton, L.T.; Reijmers, R.M.; Wouters, A.K.; Kweekel, C.; Remst, D.F.; Pothast, C.R.; Falkenburg, J.F.; Heemskerk, M.H. Simultaneous Deletion of Endogenous TCRαβ for TCR Gene Therapy Creates an Improved and Safe Cellular Therapeutic. Mol. Therapy. 2020, 28, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.R.; Jarosch, S.; Hammel, M.; Leube, J.; Grassmann, S.; Bernard, B.; Effenberger, M.; Andrä, I.; Chaudhry, M.Z.; Käuferle, T.; et al. Targeted T cell receptor gene editing provides predictable T cell product function for immunotherapy. Cell Rep. Med. 2021, 2, 100374. [Google Scholar] [CrossRef] [PubMed]
- Besser, M.J.; Shapira-Frommer, R.; Itzhaki, O.; Treves, A.J.; Zippel, D.B.; Levy, D.; Kubi, A.; Shoshani, N.; Zikich, D.; Ohayon, Y.; et al. Adoptive transfer of tumor-infiltrating lymphocytes in patients with metastatic melanoma: Intent-to-treat analysis and efficacy after failure to prior immunotherapies. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 4792–4800. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.A.; Heemskerk, B.; Powell, D.J.; Cohen, C.J.; Morgan, R.A.; Dudley, M.E.; Robbins, P.F.; Rosenberg, S.A. Gene transfer of tumor-reactive TCR confers both high avidity and tumor reactivity to nonreactive peripheral blood mononuclear cells and tumor-infiltrating lymphocytes. J. Immunol. 2006, 177, 6548–6559. [Google Scholar] [CrossRef]
- Andersen, R.; Donia, M.; Ellebaek, E.; Borch, T.H.; Kongsted, P.; Iversen, T.Z.; Hölmich, L.R.; Hendel, H.W.; Met, Ö.; Andersen, M.H.; et al. Long-Lasting Complete Responses in Patients with Metastatic Melanoma after Adoptive Cell Therapy with Tumor-Infiltrating Lymphocytes and an Attenuated IL2 Regimen. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 3734–3745. [Google Scholar] [CrossRef]
- Zhao, Z.; Condomines, M.; van der Stegen, S.J.C.; Perna, F.; Kloss, C.C.; Gunset, G.; Plotkin, J.; Sadelain, M. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell 2015, 28, 415–428. [Google Scholar] [CrossRef]


{kind=link}
| TCR | CAR | ||
|---|---|---|---|
| Structure | Receptor clustering | One pMHC potentially enough [18,19] | Clustering required [20,21,22] |
| ITAM number | 10 ITAMs provided by the CD3 complex [23] | Up to 3 ITAMs per CAR [23] | |
| Affinity/ Sensitivity | Lower affinity, higher sensitivity | Higher affinity [24], lower sensitivity [25] | |
| Phosph. of CD3 subunits | Phosph. of CD3 ζ, γ, δ, ε [26] | Phosph. of only CD3 ζ [25] | |
| Signaling | Phosph. of signaling molecules | Stronger phosph. of ZAP-70, ITAMs and PLCγ1 than in CAR [27,28] | Stronger phosph. of Lck and ERK than in TCR [29] |
| Recruitment of signaling molecules | More efficient recruitment of ZAP-70, CD2 and LFA-1 than in CAR [27,28] | Less dependent on LFA-1:ICAM-1 interaction and LAT [21,25,29] | |
| Upon increased antigen exposure | Maintain an earlier differentiation phenotype upon strong stimulation [30] | Higher levels of co-inhibitory molecules upon activation [30] | |
| IS structure | Classical “bull’s eye” structure [29] or multifocal structures formed by Th2 cells [31] or at the interface with DCs [32] | Non-classical, disorganized IS with multifocal pattern [29] | |
| Immunological Synapse | SMACs | Conventional IS consisting of cSMAC, pSMAC and dSMAC [33] | Merged cSMAC and pSMAC, no adhesion molecule ring [21,29] |
| Lck | One central Lck cluster [29] | Disorganized Lck patches [29] | |
| Duration | Usually slower/weaker effector function [29,30]; longer IS duration, slower off-rate from target [29] | Faster cytotoxic granule secretion and faster resolution of IS [29] | |
| Resting state | Constitutive internalization of TCR complex through clathrin-dependent endocytosis (CDE) [34] | Unknown | |
| Trafficking | Upon activation | Engaged TCRs: Clathrin- independent endocytosis (CIE) for internalization, recycling or lysosomal degradation [34,35] Bystander TCRs: CDE for internalization and recycling [34,35] | Engagement of antigens induced rapid lysosomal ubiquitination [36] High-affinity CAR T cells demonstrated enhanced trogocytosis [37] |
| Combination of Endogenous TCR and CAR | |||||
|---|---|---|---|---|---|
| CAR | TCR Specificity | TCR Stimulus | Study | Main Result | Citation |
| GD2-ζ | EBV- specific TCR | Patients with EBV pre-infection | NCT000 85930 | Prolonged survival and expansion compared to anti-CD3 antibody activation | Pule et al., 2008 [184] Louis et al., 2011 [188] |
| CD19-28ζ | EBV- specific TCR | EBV-transformed lymphoblastoid B cell lines | NCT008 40853 | Stimulation of native TCR increased CAR T cell expansion; T cells were donor-derived after allogeneic HSCT (no GVHD) | Cruz et al., 2013 [189] |
| GD2-28-OX40ζ | VZV- specific TCR | VZV peptide mix-loaded DCs | In vitro | Exhausted and dysfunctional CAR T cells recovered upon stimulation of native TCR | Tanaka et al., 2017 [185] |
| CD19-28ζ | HY- specific TCR | Male bone-marrow-derived cells (HY) | In vivo | Dual stimulation led to exhaustion and apoptosis in CD8+ (not in CD4+) CAR T cells | Yang et al., 2017 [196] |
| Her2-28ζ | gp100- specific TCR | Recombinant vaccinia virus encoding gp100 | In vivo | Increased expansion, persistence, tumor infiltration and functionality upon native TCR stimulation | Slaney et al., 2017 [198] |
| (1) GD2-ζ (2) GD2-28ζ (3) GD2-BBζ | VZV-/EBV- specific TCR | VZV or EBV peptide mix-loaded DCs | In vitro | TCR stimulation led to increased expansion and functionality in GD2-28ζ (but not in GD2-BBζ) CAR T cells | Omer et al., 2018 [187] |
| CD19-28ζ | EBV- specific TCR | Patients with EBV pre-infection | NCT008 40853 | Virus load-dependent increase in CAR T cell expansion | Lapteva et al., 2019 [186] |
| EGFRvIII-28-BBζ | Oncolytic VSV or reovirus | Oncolytic virus co-administered with CAR T cell | In vivo | Enhanced trafficking, infiltration and functionality; long-term effects through in vivo reactivation with TCR-directed oncolytic virus | Evgin et al., 2022 [197] |
| Combination of Transgenic TCR and CAR | |||||
|---|---|---|---|---|---|
| CAR | TCR Specificity | CAR/TCR Stimulus | Study | Main Result | Citation |
| CSPG4-28ζ (transient) | gp100 (transient) | Target cell line | In vitro | Functionally co- expressed, without reciprocal inhibition | Uslu et al., 2016 [202] |
| CSPG4-28ζ (transient) | gp100 (stable) | Target cell line | In vitro | Functionally co- expressed; reduced cytotoxicity compared to TCR T cells | Simon et al., 2019 [203] |
| BBζ (lacking scFv) | NY-ESO-1 | TCR-target- expressing cell line | In vitro/ in vivo | Increased proliferation and tumor regression upon single and repeated TCR stimulation | Miyao et al., 2018 [205] |
| (1) CD19-28 (2) CD19-BB (3) CD19-28-OX40 (lacking signaling domain) | Survivin | Target cell line | In vitro/ in vivo | Enhanced apoptosis with CD19-BB CAR; CD19-28-OX40 (not CD19-28) increased repeated killing and prolonged tumor control in vivo | Omer et al., 2022 [206] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teppert, K.; Wang, X.; Anders, K.; Evaristo, C.; Lock, D.; Künkele, A. Joining Forces for Cancer Treatment: From “TCR versus CAR” to “TCR and CAR”. Int. J. Mol. Sci. 2022, 23, 14563. https://doi.org/10.3390/ijms232314563
Teppert K, Wang X, Anders K, Evaristo C, Lock D, Künkele A. Joining Forces for Cancer Treatment: From “TCR versus CAR” to “TCR and CAR”. International Journal of Molecular Sciences. 2022; 23(23):14563. https://doi.org/10.3390/ijms232314563
Chicago/Turabian StyleTeppert, Karin, Xueting Wang, Kathleen Anders, César Evaristo, Dominik Lock, and Annette Künkele. 2022. "Joining Forces for Cancer Treatment: From “TCR versus CAR” to “TCR and CAR”" International Journal of Molecular Sciences 23, no. 23: 14563. https://doi.org/10.3390/ijms232314563
APA StyleTeppert, K., Wang, X., Anders, K., Evaristo, C., Lock, D., & Künkele, A. (2022). Joining Forces for Cancer Treatment: From “TCR versus CAR” to “TCR and CAR”. International Journal of Molecular Sciences, 23(23), 14563. https://doi.org/10.3390/ijms232314563