
DFT Study of the Direct Radical Scavenging Potency of Two Natural Catecholic Compounds


Abstract
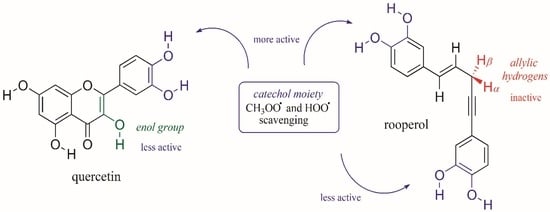
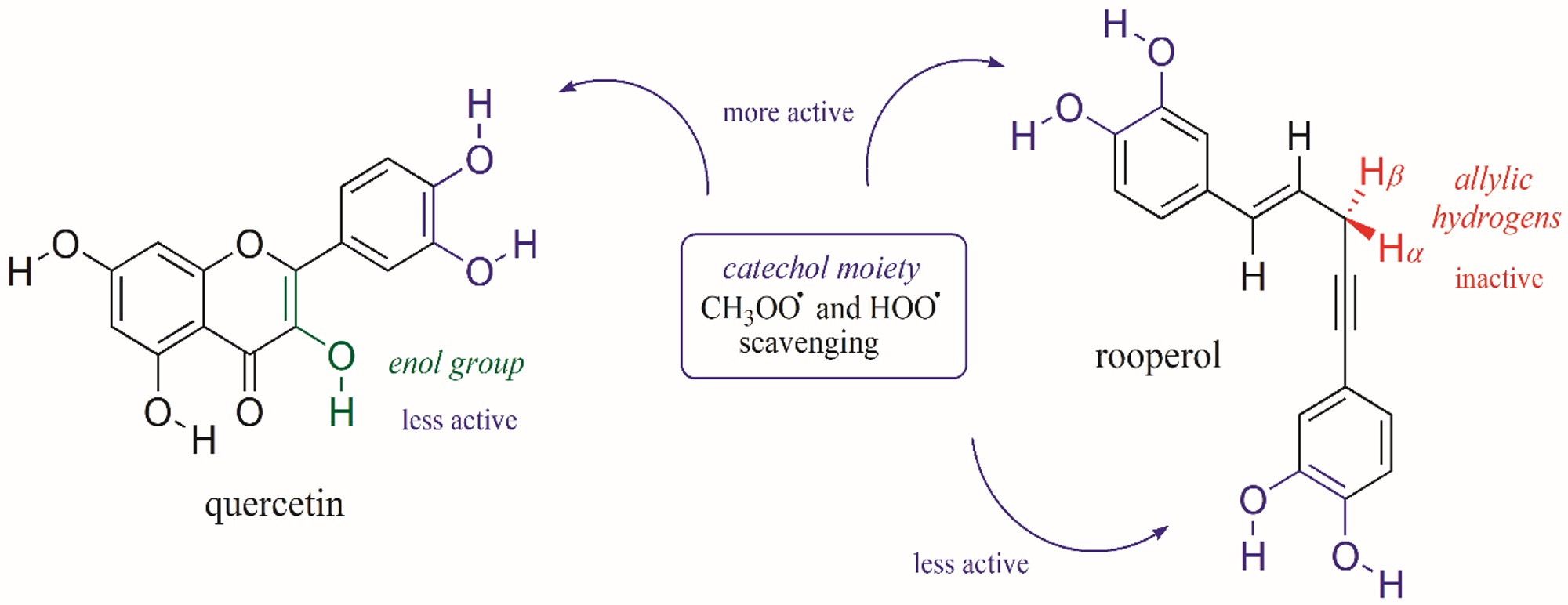
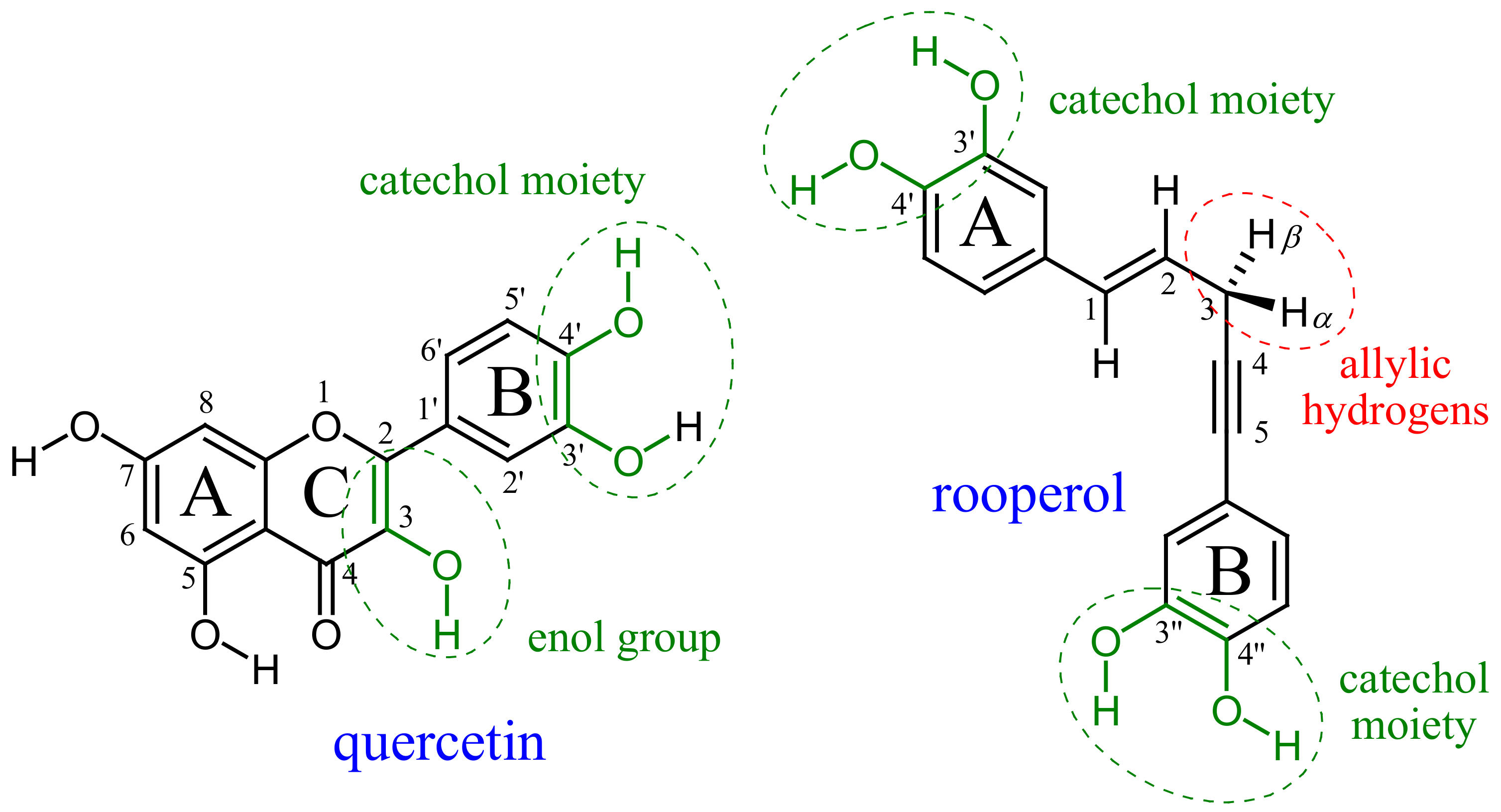
1. Introduction
2. Results and Discussion
2.1. PCET from Phenolic O–H Groups of Quercetin to CH3OO• and HOO• Radicals
2.2. PCET from Phenolic O–H and HAT from Allylic C–H Groups of Rooperol to HOO• and CH3OO• Radicals
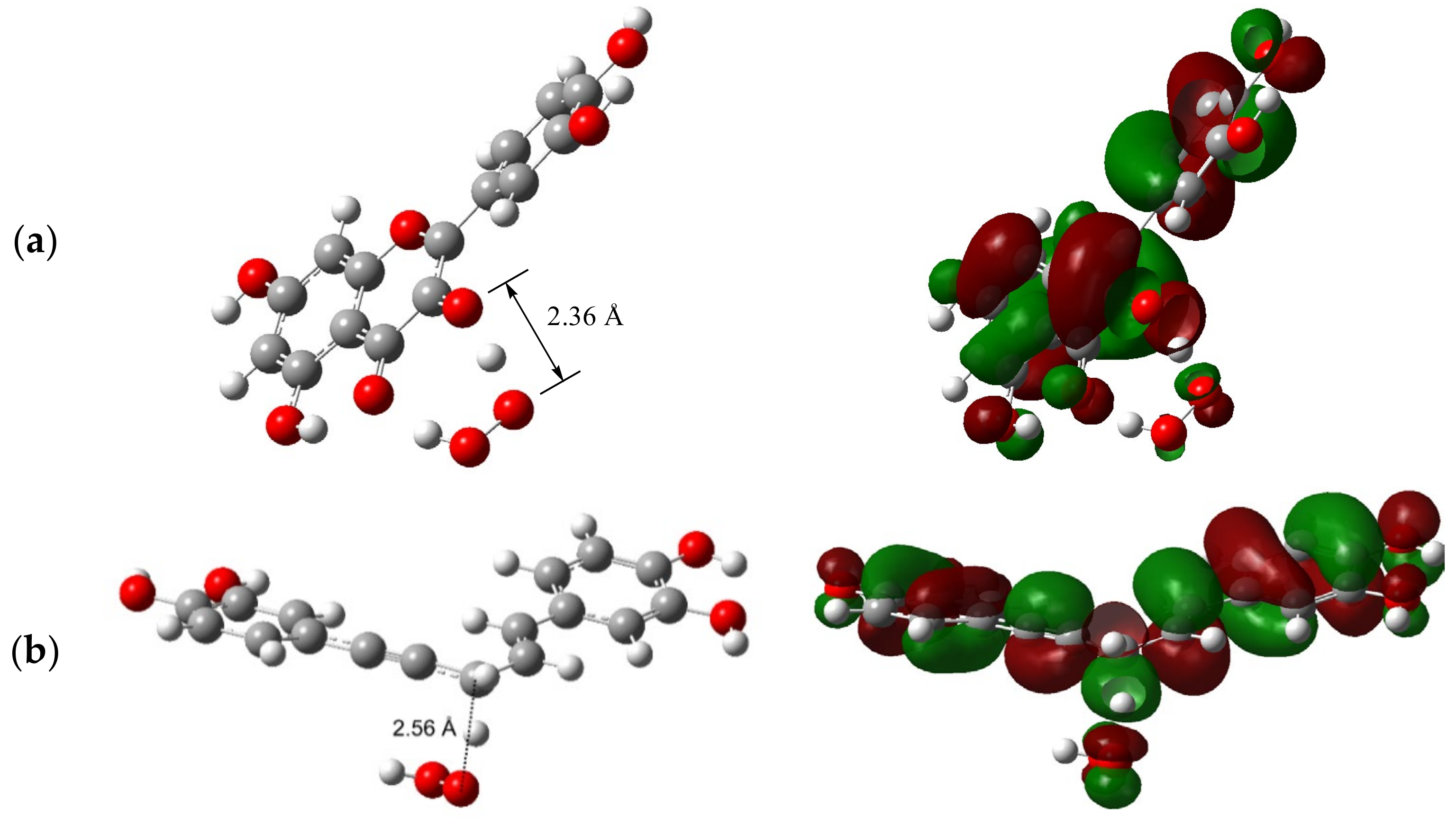
2.3. SOMO at TS of H-Atom Transfer from Phenolic O–H and Allylic C–H Groups
2.4. SET from Phenoxide Anions of Quercetin and Rooperol to HOO• and CH3OO• Radicals
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Dangles, O.; Dufour, C.; Tonnele, C.; Trouillas, P. The physical chemistry of polyphenols: Insights into the activity of polyphenols in humans at the molecular level. In Recent Advances in Polyphenol Research; Yoshida, K., Cheynier, V., Quiedau, S., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2017; pp. 1–35. [Google Scholar]
- Bors, W.; Heller, W.; Michel, C.; Saran, M. Flavonoids as antioxidants: Determination of radical-scavenging efficiencies. In Methods in Enzymology; Packer, L., Glazer, A.N., Eds.; Academic Press: San Diego, CA, USA, 1990; pp. 343–355. [Google Scholar]
- Bielski, B.H.J.; Arudi, R.L.; Sutherland, M.W. A study of the reactivity of HO2/O2− with unsaturated fatty acids. J. Biol. Chem. 1983, 258, 4759–4761. [Google Scholar] [CrossRef]
- Ulusoy, H.G.; Sanlier, N. A minireview of quercetin: From its metabolism to possible mechanisms of its biological activities. Crit. Rev. Food Sci. Nutr. 2020, 60, 3290–3303. [Google Scholar] [CrossRef] [PubMed]
- Laporta, O.; Perez-Fons, L.; Mallavia, R.; Caturla, N.; Micol, V. Isolation, characterization and antioxidant capacity assessment of the bioactive compounds derived from Hypoxis rooperi corm extract (African potato). Food Chem. 2007, 101, 1425–1437. [Google Scholar] [CrossRef]
- Kabanda, M.M. Antioxidant activity of rooperol investigated through Cu (I and II) chelation ability and the hydrogen transfer mechanism: A DFT study. Chem. Res. Toxicol. 2012, 25, 2153–2166. [Google Scholar] [CrossRef]
- Amorati, R.; Baschieri, A.; Cowden, A.; Valgimigli, L. The antioxidant activity of quercetin in water solution. Biomimetics 2017, 2, 9. [Google Scholar] [CrossRef]
- Zheng, Y.-Z.; Fu, Z.-M.; Deng, G.; Guo, R.; Chen, D.-F. Role of C–H bond in the antioxidant activities of rooperol and its derivatives: A DFT study. Phytochemistry 2020, 178, 112454. [Google Scholar] [CrossRef]
- Ingold, K.U.; Pratt, D.A. Advances in radical-trapping antioxidant chemistry in the 21st century: A kinetics and mechanisms perspective. Chem. Rev. 2014, 114, 9022–9046. [Google Scholar] [CrossRef]
- Di Meo, F.; Lemaur, V.; Cornil, J.; Lazzaroni, R.; Duroux, J.-L.; Olivier, Y.; Trouillas, P. Free radical scavenging by natural polyphenols: Atom versus electron transfer. J. Phys. Chem. A 2013, 117, 2082–2092. [Google Scholar] [CrossRef]
- Wright, J.S.; Johnson, E.R.; DiLabio, G.A. Predicting the activity of phenolic antioxidants: Theoretical method, analysis of substituent effects, and application to major families of antioxidants. J. Am. Chem. Soc. 2001, 123, 1173–1183. [Google Scholar] [CrossRef]
- Lucarini, M.; Pedrielli, P.; Pedulli, G.F.; Cabiddu, S.; Fattuoni, C. Bond dissociation energies of O–H bonds in substituted phenols from equilibration studies. J. Org. Chem. 1996, 61, 9259–9263. [Google Scholar] [CrossRef]
- Lucarini, M.; Pedulli, G.F. Free radical intermediates in the inhibition of the autoxidation reaction. Chem. Soc. Rev. 2010, 39, 2106–2119. [Google Scholar] [CrossRef] [PubMed]
- Leopoldini, M.; Russo, N.; Toscano, M. The molecular basis of working mechanisms of natural polyphenolic antioxidants. Food Chem. 2011, 125, 288–306. [Google Scholar] [CrossRef]
- Galano, A. Free radicals induced oxidative stress at a molecular level: The current status, challenges and perspectives of computational chemistry based protocols. J. Mex. Chem. Soc. 2015, 59, 231–262. [Google Scholar] [CrossRef]
- Mayer, J.M. Understanding hydrogen atom transfer: From bond strengths to Marcus theory. Acc. Chem. Res. 2011, 44, 36–46. [Google Scholar] [CrossRef]
- Tejero, I.; Gonzalez-Garcia, N.; Gonzalez-Lafont, A.; Lluch, J.M. Tunneling in green tea: Understanding the antioxidant activity of catechol-containing compounds. A variational transition-state theory study. J. Am. Chem. Soc. 2007, 129, 5846–5854. [Google Scholar] [CrossRef]
- Schreiner, P.R. Quantum mechanical tunneling is essential to understanding chemical reactivity. Trends Chem. 2020, 2, 980–989. [Google Scholar] [CrossRef]
- McMahon, R.J. Chemical reactions involving quantum tunneling. Science 2003, 299, 833–834. [Google Scholar] [CrossRef]
- Galano, A.; Alvarez-Idaboy, J.R. A computational methodology for accurate predictions of rate constants in solution: Application to the assessment of primary antioxidant activity. J. Comput. Chem. 2013, 34, 2430–2445. [Google Scholar] [CrossRef]
- Li, Z.; Moalin, M.; Zhang, M.; Vervoort, L.; Mommers, A.; Haenen, G.R.M.M. Delocalization of the unpaired electron in the quercetin radical: Comparison of experimental ESR data with DFT calculations. Int. J. Mol. Sci. 2020, 21, 2033. [Google Scholar] [CrossRef]
- Klippenstein, S.J.; Pande, V.S.; Truhlar, D.G. Chemical kinetics and mechanisms of complex systems: A perspective on recent theoretical advances. J. Am. Chem. Soc. 2014, 136, 528–546. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, I.; Shoji, Y.; Ohkubo, K.; Fukuzumi, S. Tunneling in the hydrogen-transfer reaction from a vitamin E analog to an inclusion complex of 2,2-diphenyl-1-picrylhydrazyl radical with β-cyclodextrin in an aqueous buffer solution at ambient temperature. Antioxidants 2021, 10, 1966. [Google Scholar] [CrossRef] [PubMed]
- Nagaoka, S.-i.; Inoue, M.; Nishioka, C.; Nishioku, Y.; Tsunoda, S.; Ohguchi, C.; Ohara, K.; Mukai, K.; Nagashima, U. Tunneling effect in antioxidant, prooxidant, and regeneration reactions of vitamin E. J. Phys. Chem. B 2000, 104, 856–862. [Google Scholar] [CrossRef]
- Dzib, E.; Cabellos, J.L.; Ortiz-Chi, F.; Pan, S.; Galano, A.; Merino, G. Eyringpy: A program for computing rate constants in the gas phase and in solution. Int. J. Quantum Chem. 2019, 119, e25686. [Google Scholar] [CrossRef]
- Sirjean, B.; Dames, E.; Wang, H.; Tsang, W. Tunneling in hydrogen-transfer isomerization of n-alkyl radicals. J. Phys. Chem. A 2012, 116, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Truong, T.N. Thermal rates of hydrogen exchange of methane with zeolite: A direct ab initio dynamics study on the importance of quantum tunneling effects. J. Phys. Chem. B 1997, 101, 2750–2752. [Google Scholar] [CrossRef]
- Butković, V.; Klasinc, L.; Bors, W. Kinetic study of flavonoid reactions with stable radicals. J. Agric. Food Chem. 2004, 52, 2816–2820. [Google Scholar] [CrossRef]
- Fukuhara, K.; Nakanishi, I.; Kansui, H.; Sugiyama, E.; Kimura, M.; Shimada, T.; Urano, S.; Yamaguchi, K.; Miyata, N. Enhanced radical-scavenging activity of a planar catechin analogue. J. Am. Chem. Soc. 2002, 124, 5952–5953. [Google Scholar] [CrossRef]
- Chiodo, S.G.; Leopoldini, M.; Russo, N.; Toscano, M. The inactivation of lipid peroxide radical by quercetin. A theoretical insight. Phys. Chem. Chem. Phys. 2010, 12, 7662–7670. [Google Scholar] [CrossRef]
- Marković, Z.S.; Dimitrić Marković, J.M.; Doličanin, Ć.B. Mechanistic pathways for the reaction of quercetin with hydroperoxy radical. Theor. Chem. Acc. 2010, 127, 69–80. [Google Scholar] [CrossRef]
- Amić, A.; Milenković, D.; Dimitrić Marković, J.M.; Marković, Z. Do equol’s C-ring hydrogens contribute to free radical scavenging? J. Serb. Soc. Comput. Mech. 2020, SI, 45–58. [Google Scholar] [CrossRef]
- Amić, A.; Milenković, D.; Marković, Z.; Cagardová, D.; Rodríguez-Guerra Pedregal, J.; Dimitrić Marković, J.M. Impact of the phenolic O–H vs. C-ring C−H bond cleavage on the antioxidant potency of dihydrokaempferol. New J. Chem. 2021, 45, 7977–7986. [Google Scholar] [CrossRef]
- Amić, A.; Mastiľák Cagardová, D. Mactanamide and lariciresinol as radical scavengers and Fe2+ ion chelators—A DFT study. Phytochemistry 2022, 204, 113442. [Google Scholar] [CrossRef] [PubMed]
- Mayer, J.M.; Hrovat, D.A.; Thomas, J.L.; Borden, W.T. Proton-coupled electron transfer versus hydrogen atom transfer in benzyl/toluene, methoxyl/methanol, and phenoxyl/phenol self-exchange reactions. J. Am. Chem. Soc. 2002, 124, 11142–11147. [Google Scholar] [CrossRef]
- Litwinienko, G.; Ingold, K.U. Abnormal solvent effects on hydrogen atom abstractions. 1. The reactions of phenols with 2,2-diphenyl-1-picrylhydrazyl (dpph•) in alcohols. J. Org. Chem. 2003, 68, 3433–3438. [Google Scholar] [CrossRef]
- Musialik, M.; Kuzmicz, R.; Pawlowski, T.S.; Litwinienko, G. Acidity of hydroxyl groups: An overlooked influence on antiradical properties of flavonoids. J. Org. Chem. 2009, 74, 2699–2709. [Google Scholar] [CrossRef]
- Alvarez-Diduk, R.; Ramirez-Silva, M.T.; Galano, A.; Merkoci, A. Deprotonation mechanism and acidity constants in aqueous solution of flavonols: A combined experimental and theoretical study. J. Phys. Chem. B 2013, 117, 12347–12359. [Google Scholar] [CrossRef]
- Mezzetti, A.; Protti, S.; Lapouge, C.; Cornard, J.-P. Protic equilibria as the key factor of quercetin emission in solution. Relevance to biochemical and analytical studies. Phys. Chem. Chem. Phys. 2011, 13, 6858–6864. [Google Scholar] [CrossRef]
- Lee-Hilz, Y.Y.; Boerboom, A.-M.J.F.; Westphal, A.H.; van Berkel, W.J.H.; Aarts, J.M.M.J.G.; Rietjens, I.M.C.M. Pro-oxidant activity of flavonoids induces EpRE-mediated gene expression. Chem. Res. Toxicol. 2006, 19, 1499–1505. [Google Scholar] [CrossRef]
- Cabelli, D.E.; Bielski, B.H.J. Kinetics and mechanism for the oxidation of ascorbic acid/ascorbate by HO2/O2− radicals. A pulse radiolysis and stopped-flow photolysis study. J. Phys. Chem. 1983, 87, 1809–1812. [Google Scholar] [CrossRef]
- Kim, D.-O.; Lee, C.Y. Comprehensive study on vitamin C equivalent antioxidant capacity (VCEAC) of various polyphenolics in scavenging a free radical and its structural relationship. Crit. Rev. Food Sci. Nutr. 2004, 44, 253–273. [Google Scholar] [CrossRef] [PubMed]
- Nair, V.D.P.; Dairam, A.; Agbonon, A.; Arnason, J.T.; Foster, B.C.; Kanfer, I. Investigation of the antioxidant activity of African potato (Hypoxis hemerocallidea). J. Agric. Food Chem. 2007, 55, 1707–1711. [Google Scholar] [CrossRef] [PubMed]
- Pérez-González, A.; Alvarez-Idaboy, J.R.; Galano, A. Dual antioxidant/pro-oxidant behavior of the tryptophan metabolite 3-hydroxyanthranilic acid: A theoretical investigation of reaction mechanisms and kinetics. New J. Chem. 2017, 41, 3829–3845. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Zhao, Y.; Schultz, N.E.; Truhlar, D.G. Design of density functionals by combining the method of constraint satisfaction withparametrization for thermochemistry, thermochemical kinetics, and noncovalent interactions. J. Chem. Theory Comput. 2006, 2, 364–382. [Google Scholar] [CrossRef] [PubMed]
- Mardirossian, N.; Head-Gordon, M. How accurate are the Minnesota density functionals for noncovalent interactions, isomerization energies, thermochemistry, and barrier heights involving molecules composed of main-group elements? J. Chem. Theory Comput. 2016, 12, 4303–4325. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuummodel of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Amić, A.; Lučić, B.; Stepanić, V.; Marković, Z.; Marković, S.; Dimitrić Marković, J.M.; Amić, D. Free radical scavenging potency of quercetin catecholic colonic metabolites: Thermodynamics of 2H+/2e− processes. Food Chem. 2017, 218, 144–151. [Google Scholar] [CrossRef]
- ACD/Percepta. ACD/Labs Release 2020.2.0. Available online: https://www.acdlabs.com/products/percepta/predictors/pka/ (accessed on 4 September 2021).
- Alvarez-Idaboy, J.R.; Mora-Diez, N.; Boyd, R.J.; Vivier-Bunge, A. On the importance of prereactive complexes in molecule-radical reactions: Hydrogen abstraction from aldehydes by OH. J. Am. Chem. Soc. 2001, 123, 2018–2024. [Google Scholar] [CrossRef]
- Marcus, R.A. Electron transfer reactions in chemistry. Theory and experiment. Pure Appl. Chem. 1997, 69, 13–29. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
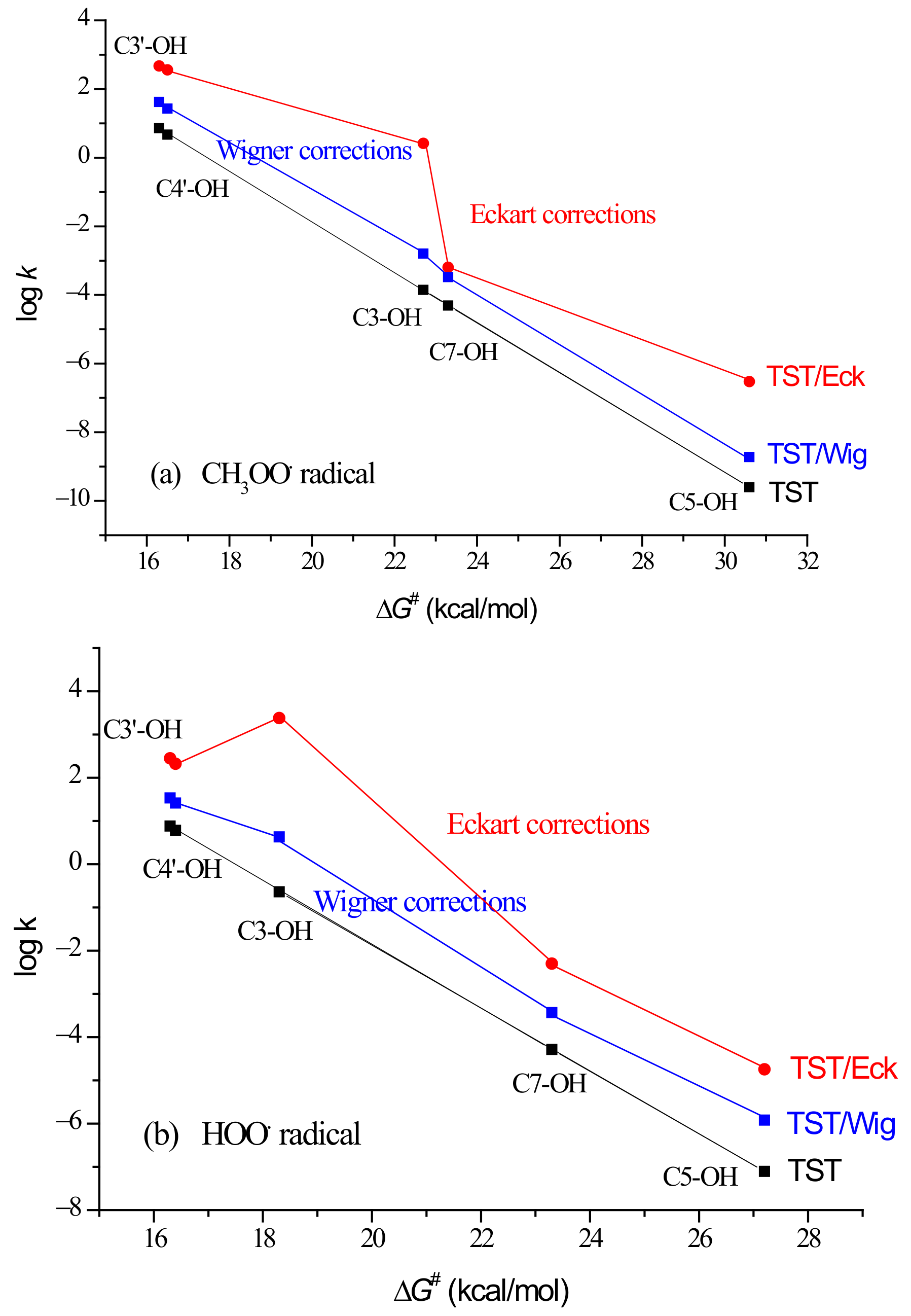
| Path | BDE | ΔrG | ν | ΔG≠ | kTST | κEck | kTST/Eck | ΓEck | κWig | kTST/Wig | ΓWig |
|---|---|---|---|---|---|---|---|---|---|---|---|
| CH3OO• | |||||||||||
| 3-OH | 85.41 | 0.9 | −3307 | 22.7 | 1.4 × 10−4 | 18350.5 | 2.6 × 100 | 0.3 | 11.6 | 1.6 × 10−3 | 0 |
| 5-OH | 99.41 | 14.9 | −2631 | 30.6 | 2.5 × 10−10 | 120.0 | 3.0 × 10−8 | 0 | 7.7 | 1.9 × 10−9 | 0 |
| 7-OH | 94.55 | 10.4 | −2456 | 23.3 | 4.9 × 10−5 | 13.2 | 6.4 × 10−4 | 0 | 6.9 | 3.3 × 10−4 | 0 |
| 3′-OH | 82.36 | −1.1 | −2227 | 16.3 | 7.2 × 100 | 65.8 | 4.7 × 102 | 56.4 | 5.8 | 4.2 × 101 | 60.0 |
| 4′-OH | 80.35 | −3.5 | −2259 | 16.5 | 4.7 × 100 | 77.1 | 3.6 × 102 | 43.2 | 6.0 | 2.8 × 101 | 40.0 |
| = 1.2 × 101 | = 8.3 × 102 | = 7.0 × 101 | |||||||||
| HOO• | |||||||||||
| 3-OH | 85.41 | −1.3 | −4274 | 18.3 | 2.3 × 10−1 | 10301.8 | 2.4 × 103 | 83.0 | 18.7 | 4.3 × 100 | 6.7 |
| 5-OH | 99.41 | 12.7 | −3894 | 27.2 | 7.8 × 10−8 | 233.0 | 1.8 × 10−5 | 0 | 15.7 | 1.2 × 10−6 | 0 |
| 7-OH | 94.55 | 8.1 | −2521 | 23.3 | 5.2 × 10−5 | 96.8 | 5.0 × 10−3 | 0 | 7.2 | 3.7 × 10−4 | 0 |
| 3′-OH | 82.36 | −3.4 | −1884 | 16.3 | 7.6 × 100 | 36.7 | 2.8 × 102 | 9.7 | 4.4 | 3.4 × 101 | 52.9 |
| 4′-OH | 80.35 | −5.8 | −1843 | 16.4 | 6.1 × 100 | 34.0 | 2.1 × 102 | 7.3 | 4.3 | 2.6 × 101 | 40.4 |
| = 1.4 × 101 | = 2.9 × 103 | = 6.4 × 101 | |||||||||
| Path | BDE | ΔrG | ν | ΔG≠ | kTST | κEck | kTST/Eck | ΓEck | κWig | kTST/Wig | ΓWig |
|---|---|---|---|---|---|---|---|---|---|---|---|
| (a) HOO• | |||||||||||
| phenolic reaction paths | |||||||||||
| 3′-OH | 80.48 | −4.8 | −1801 | 16.3 | 7.4 × 100 | 23.5 | 1.7 × 102 | 12.0 | 4.1 | 3.1 × 101 | 7.8 |
| 4′-OH | 77.04 | −8.0 | −1616 | 14.7 | 9.7 × 101 | 11.1 | 1.1 × 103 | 77.6 | 3.5 | 3.4 × 102 | 86.1 |
| 3″-OH | 81.81 | −3.0 | −1817 | 17.6 | 8.2 × 10−1 | 30.6 | 2.5 × 101 | 1.7 | 4.2 | 3.5 × 100 | 0.9 |
| 4″-OH | 79.63 | −5.3 | −1761 | 16.5 | 5.0 × 100 | 24.6 | 1.2 × 102 | 8.5 | 4.0 | 2.0 × 101 | 5.1 |
| = 1.1 × 102 | = 1.4 × 103 | 99.8 | = 3.9 × 102 | 99.9 | |||||||
| allylic reaction paths | |||||||||||
| allylic Hα | 72.21 | −14.1 | −1739 | 19.4 | 3.6 × 10−2 | 39.6 | 1.4 × 100 | 0.1 | 3.9 | 1.4 × 10−1 | 0.03 |
| allylic Hβ | 72.21 | −14.1 | −1730 | 19.3 | 4.3 × 10−2 | 37.0 | 1.6 × 100 | 0.1 | 3.9 | 1.7 × 10−1 | 0.04 |
| = 7.9 × 10−2 | = 3.0 × 100 | 0.2 | = 3.1 × 10−1 | 0.07 | |||||||
| vinylic reaction paths | |||||||||||
| vinylic H1 | 101.07 | 17.7 | −1881 | 32.8 | 5.9 × 10−12 | 36.8 | 2.2 × 10−10 | 0.0 | 4.4 | 2.6 × 10−11 | 0.00 |
| vinylic H2 | 109.70 | 23.8 | −1913 | 33.5 | 1.6 × 10−12 | 0.8 | 1.3 × 10−12 | 0.0 | 4.6 | 7.3 × 10−12 | 0.00 |
| = 7.5 × 10−12 | = 2.2 × 10−10 | 0.0 | = 3.3 × 10−11 | 0.00 | |||||||
| (b) CH3OO• | |||||||||||
| 3’-OH | 80.48 | −2.6 | −2085 | 17.1 | 1.7 × 100 | 45.0 | 7.8 × 101 | 16.0 | 5.2 | 9.1 × 100 | 9.5 |
| 4’-OH | 77.04 | −5.7 | −1866 | 15.8 | 1.7 × 101 | 18.4 | 3.2 × 102 | 65.4 | 4.4 | 7.6 × 101 | 79.3 |
| 3’’-OH | 81.81 | −0.7 | −2087 | 17.7 | 6.7 × 10−1 | 53.6 | 3.6 × 101 | 7.4 | 5.2 | 3.5 × 100 | 3.7 |
| 4’’-OH | 79.63 | −3.0 | −1992 | 17.2 | 1.5 × 100 | 36.9 | 5.5 × 101 | 11.2 | 4.8 | 7.2 × 100 | 7.5 |
| = 2.0 × 101 | = 4.9 × 102 | 100.0 | = 9.6 × 101 | 100.0 | |||||||
| Path | ΔrG | ΔG≠ | λ | kTST | kD | kapp | kMf | Γ |
|---|---|---|---|---|---|---|---|---|
| (a) scavenging of •OOH by quercetin phenoxide anions | ||||||||
| 3-O− | 3.0 | 5.9 | 16.9 | 3.2 × 108 | 8.2 × 109 | 3.1 × 108 | 5.2 × 105 | 90.3 |
| 5-O− | 13.4 | 13.5 | 15.1 | 8.0 × 102 | 8.3 × 109 | 8.0 × 102 | 1.3 × 100 | 0 |
| 7-O− | 18.5 | 18.8 | 14.3 | 1.0 × 10−1 | 8.2 × 109 | 1.0 × 10−1 | 1.7 × 10−4 | 0 |
| 3′-O− | 5.8 | 7.5 | 16.2 | 2.0 × 107 | 8.2 × 109 | 2.0 × 107 | 3.4 × 104 | 5.7 |
| 4′-O− | 6.3 | 7.7 | 15.6 | 1.4 × 107 | 8.2 × 109 | 1.4 × 107 | 2.4 × 104 | 4.0 |
| = | 5.8 × 105 | |||||||
| scavenging of CH3OO• by quercetin phenoxide anions | ||||||||
| 3-O− | 4.9 | 6.9 | 16.5 | 5.1 × 107 | 7.7 × 109 | 5.1 × 107 | 3.4 × 107 | 99.8 |
| 5-O− | 15.4 | 15.4 | 14.7 | 3.4 × 101 | 7.8 × 109 | 3.4 × 101 | 2.3 × 101 | 0 |
| 7-O− | 20.4 | 21.2 | 13.9 | 1.9 × 10−3 | 7.8 × 109 | 1.9 × 10−3 | 1.3 × 10−3 | 0 |
| 3′-O− | 7.8 | 8.8 | 15.8 | 2.3 × 106 | 7.7 × 109 | 2.3 × 106 | 1.5 × 106 | 0.1 |
| 4′-O− | 8.3 | 9.0 | 15.2 | 1.5 × 106 | 7.7 × 109 | 1.5 × 106 | 1.0 × 106 | 0.1 |
| = | 3.7 × 107 | |||||||
| (b) scavenging of •OOH by rooperol phenoxide anions | ||||||||
| 3′-O− | 2.5 | 5.4 | 16.4 | 6.3 × 108 | 8.1 × 109 | 5.8 × 108 | 7.3 × 103 | 9.8 |
| 4′-O− | −0.8 | 3.7 | 16.3 | 1.2 × 1010 | 8.2 × 109 | 4.9 × 109 | 6.1 × 104 | 82.6 |
| 3″-O− | 3.5 | 6.3 | 17.5 | 1.6 × 108 | 8.3 × 109 | 1.5 × 108 | 1.9 × 103 | 2.5 |
| 4″-O− | 3.4 | 5.9 | 15.9 | 3.1 × 108 | 8.3 × 109 | 3.0 × 108 | 3.8 × 103 | 5.1 |
| = | 7.4 × 104 | |||||||
| scavenging of CH3OO• by rooperol phenoxide anions | ||||||||
| 3′-O− | 4.5 | 6.5 | 16.0 | 1.0 × 108 | 7.7 × 109 | 1.0 × 108 | 5.0 × 105 | 4.4 |
| 4′-O− | 1.1 | 4.6 | 15.8 | 2.8 × 109 | 7.8 × 109 | 2.1 × 109 | 1.1 × 107 | 92.5 |
| 3″-O− | 5.4 | 7.4 | 17.0 | 2.4 × 107 | 7.8 × 109 | 2.4 × 107 | 1.2 × 105 | 1.0 |
| 4″-O− | 5.3 | 7.0 | 15.5 | 4.7 × 107 | 7.8 × 109 | 4.7 × 107 | 2.4 × 105 | 2.1 |
| = | 1.1 × 107 | |||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amić, A.; Mastiľák Cagardová, D. DFT Study of the Direct Radical Scavenging Potency of Two Natural Catecholic Compounds. Int. J. Mol. Sci. 2022, 23, 14497. https://doi.org/10.3390/ijms232214497
Amić A, Mastiľák Cagardová D. DFT Study of the Direct Radical Scavenging Potency of Two Natural Catecholic Compounds. International Journal of Molecular Sciences. 2022; 23(22):14497. https://doi.org/10.3390/ijms232214497
Chicago/Turabian StyleAmić, Ana, and Denisa Mastiľák Cagardová. 2022. "DFT Study of the Direct Radical Scavenging Potency of Two Natural Catecholic Compounds" International Journal of Molecular Sciences 23, no. 22: 14497. https://doi.org/10.3390/ijms232214497
APA StyleAmić, A., & Mastiľák Cagardová, D. (2022). DFT Study of the Direct Radical Scavenging Potency of Two Natural Catecholic Compounds. International Journal of Molecular Sciences, 23(22), 14497. https://doi.org/10.3390/ijms232214497