
The Role of Insulin Signaling in Hippocampal-Related Diseases: A Focus on Alzheimer’s Disease



Abstract
1. Introduction
2. Insulin and Hippocampus: Memory as a Key Link
2.1. Evidence in Animal Studies
2.2. Evidence in Human Studies
2.3. Mechanisms by Which Insulin Affects Memory
3. The Source of Insulin in the Brain
3.1. External Insulin Reaches the Brain
3.2. Local Insulin Synthesis in the Brain
4. Insulin Signaling and Hippocampal Disease: AD Is a Key Point
4.1. The Role of the Hippocampus in AD
4.1.1. Hippocampal Neuroinflammation and AD
4.1.2. Hippocampal Ferroptosis and AD
4.1.3. Hippocampal Mitophagy and AD
4.1.4. Hippocampal Oxidative Stress and AD
4.2. T2DM and AD
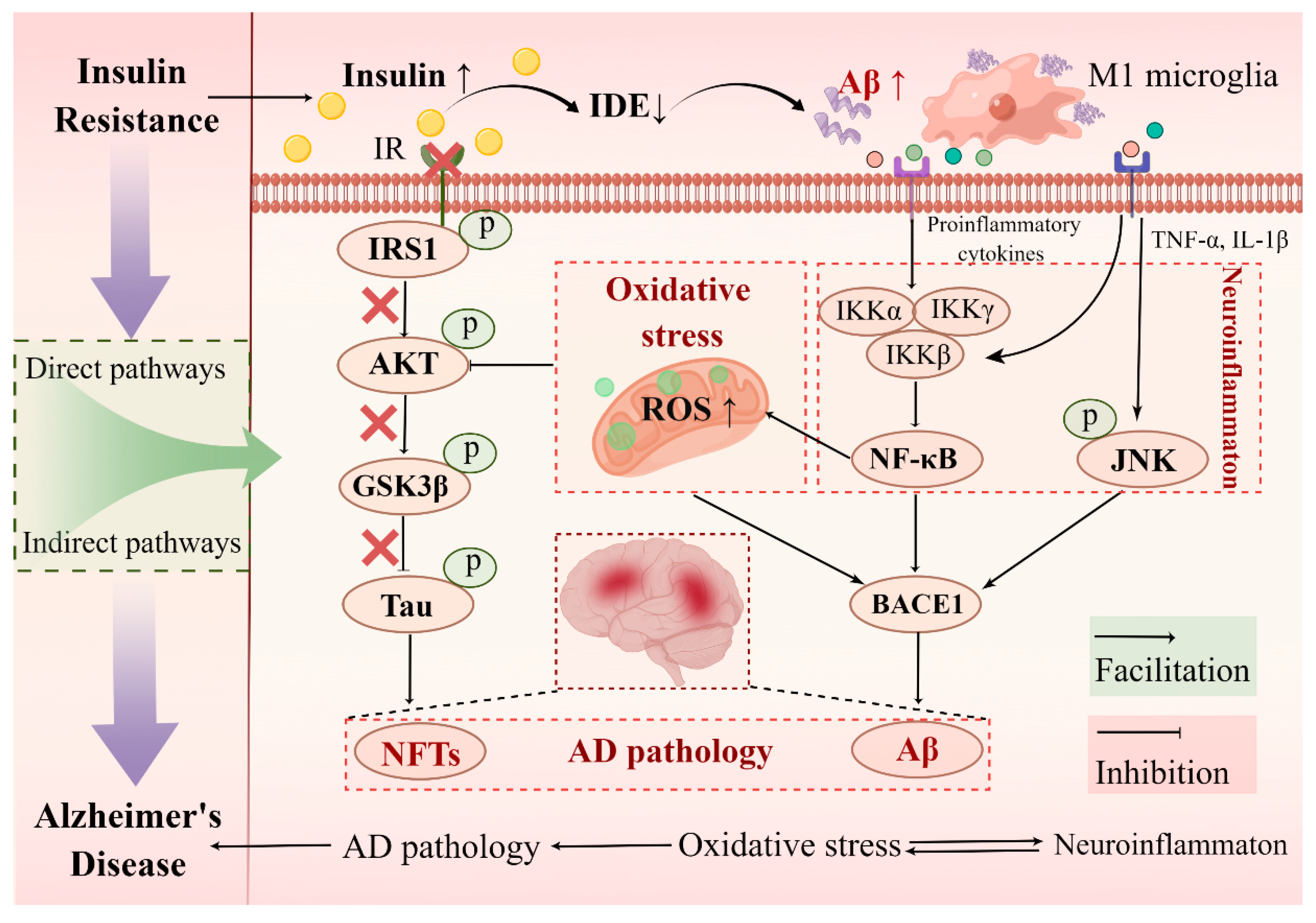
4.3. Molecular Link between Hippocampal Insulin Resistance and AD
4.3.1. Direct Pathways of Hippocampal Insulin Resistance Induced AD Pathology: Aβ Aggregation and Tau Hyperphosphorylation
4.3.2. Indirect Pathways of Hippocampal Insulin Resistance Induced AD Pathology: Neuroinflammation and Oxidative Stress
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Yaribeygi, H.; Farrokhi, F.R.; Butler, A.E.; Sahebkar, A. Insulin resistance: Review of the underlying molecular mechanisms. J. Cell. Physiol. 2019, 234, 8152–8161. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.E.; Arvanitakis, Z.; Macauley-Rambach, S.L.; Koenig, A.M.; Wang, H.Y.; Ahima, R.S.; Craft, S.; Gandy, S.; Buettner, C.; Stoeckel, L.E.; et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat. Rev. Neurol. 2018, 14, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Koepsell, H. Glucose transporters in brain in health and disease. Pflugers Arch. 2020, 472, 1299–1343. [Google Scholar] [CrossRef] [PubMed]
- Muta, K.; Morgan, D.A.; Rahmouni, K. The role of hypothalamic mTORC1 signaling in insulin regulation of food intake, body weight, and sympathetic nerve activity in male mice. Endocrinology 2015, 156, 1398–1407. [Google Scholar] [CrossRef] [PubMed]
- Nasca, C.; Dobbin, J.; Bigio, B.; Watson, K.; de Angelis, P.; Kautz, M.; Cochran, A.; Mathe, A.A.; Kocsis, J.H.; Lee, F.S.; et al. Insulin receptor substrate in brain-enriched exosomes in subjects with major depression: On the path of creation of biosignatures of central insulin resistance. Mol. Psychiatry 2021, 26, 5140–5149. [Google Scholar] [CrossRef] [PubMed]
- Soto, M.; Cai, W.; Konishi, M.; Kahn, C.R. Insulin signaling in the hippocampus and amygdala regulates metabolism and neurobehavior. Proc. Natl. Acad. Sci. USA 2019, 116, 6379–6384. [Google Scholar] [CrossRef] [PubMed]
- Park, K.H.; Noh, Y.; Choi, E.J.; Kim, H.; Chun, S.; Son, Y.D. Functional Connectivity of the Hippocampus in Early- and vs. Late-Onset Alzheimer’s Disease. J. Clin. Neurol. 2017, 13, 387–393. [Google Scholar] [CrossRef]
- Maruszak, A.; Thuret, S. Why looking at the whole hippocampus is not enough-a critical role for anteroposterior axis, subfield and activation analyses to enhance predictive value of hippocampal changes for Alzheimer’s disease diagnosis. Front. Cell. Neurosci. 2014, 8, 95. [Google Scholar] [CrossRef]
- Gralle, M.; Labrecque, S.; Salesse, C.; De Koninck, P. Spatial dynamics of the insulin receptor in living neurons. J. Neurochem. 2021, 156, 88–105. [Google Scholar] [CrossRef]
- Zimmet, P.; Shi, Z.; El-Osta, A.; Ji, L. Epidemic T2DM, early development and epigenetics: Implications of the Chinese Famine. Nat. Rev. Endocrinol. 2018, 14, 738–746. [Google Scholar] [CrossRef]
- Antal, B.; McMahon, L.P.; Sultan, S.F.; Lithen, A.; Wexler, D.J.; Dickerson, B.; Ratai, E.M.; Mujica-Parodi, L.R. Type 2 diabetes mellitus accelerates brain aging and cognitive decline: Complementary findings from UK Biobank and meta-analyses. Elife 2022, 11, e73138. [Google Scholar] [CrossRef] [PubMed]
- Talbot, K.; Wang, H.Y.; Kazi, H.; Han, L.Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef] [PubMed]
- Craft, S.; Raman, R.; Chow, T.W.; Rafii, M.S.; Sun, C.K.; Rissman, R.A.; Donohue, M.C.; Brewer, J.B.; Jenkins, C.; Harless, K.; et al. Safety, Efficacy, and Feasibility of Intranasal Insulin for the Treatment of Mild Cognitive Impairment and Alzheimer Disease Dementia: A Randomized Clinical Trial. JAMA Neurol. 2020, 77, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Rhea, E.M.; Humann, S.R.; Nirkhe, S.; Farr, S.A.; Morley, J.E.; Banks, W.A. Intranasal Insulin Transport is Preserved in Aged SAMP8 Mice and is Altered by Albumin and Insulin Receptor Inhibition. J. Alzheimers Dis. 2017, 57, 241–252. [Google Scholar] [CrossRef]
- Frazier, H.N.; Ghoweri, A.O.; Sudkamp, E.; Johnson, E.S.; Anderson, K.L.; Fox, G.; Vatthanaphone, K.; Xia, M.; Lin, R.L.; Hargis-Staggs, K.E.; et al. Long-Term Intranasal Insulin Aspart: A Profile of Gene Expression, Memory, and Insulin Receptors in Aged F344 Rats. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 1021–1030. [Google Scholar] [CrossRef]
- Apostolatos, A.; Song, S.; Acosta, S.; Peart, M.; Watson, J.E.; Bickford, P.; Cooper, D.R.; Patel, N.A. Insulin promotes neuronal survival via the alternatively spliced protein kinase CdeltaII isoform. J. Biol. Chem. 2012, 287, 9299–9310. [Google Scholar] [CrossRef]
- Peng, S.; Yang, J.; Wang, Y.; Fan, Y.; Tang, F.; Hou, C.; Yu, J.; Wang, X.; Jiang, G. Low-dose intranasal insulin improves cognitive function and suppresses the development of epilepsy. Brain Res. 2020, 1726, 146474. [Google Scholar] [CrossRef]
- Beirami, E.; Oryan, S.; Seyedhosseini Tamijani, S.M.; Ahmadiani, A.; Dargahi, L. Intranasal insulin treatment restores cognitive deficits and insulin signaling impairment induced by repeated methamphetamine exposure. J. Cell. Biochem. 2018, 119, 2345–2355. [Google Scholar] [CrossRef]
- Lv, H.; Tang, L.J.; Guo, C.S.; Jiang, Y.M.; Gao, C.; Wang, Y.F.; Jian, C.D. Intranasal insulin administration may be highly effective in improving cognitive function in mice with cognitive dysfunction by reversing brain insulin resistance. Cogn. Neurodynamics 2020, 14, 323–338. [Google Scholar] [CrossRef]
- Chen, Y.; Dai, C.L.; Wu, Z.; Iqbal, K.; Liu, F.; Zhang, B.; Gong, C.X. Intranasal Insulin Prevents Anesthesia-Induced Cognitive Impairment and Chronic Neurobehavioral Changes. Front. Aging Neurosci. 2017, 9, 136. [Google Scholar] [CrossRef]
- Rajasekar, N.; Nath, C.; Hanif, K.; Shukla, R. Intranasal insulin improves cerebral blood flow, Nrf-2 expression and BDNF in STZ (ICV)Q-induced memory impaired rats. Life Sci. 2017, 173, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.Y.; Zhang, X.; Li, S.S.; Wang, H.M.; Zhang, X.N.; Liu, L.J.; Xie, A.M. Intranasal insulin ameliorates cognitive impairment in a rat model of Parkinson’s disease through Akt/GSK3 beta signaling pathway. Life Sci. 2020, 259, 118159. [Google Scholar] [CrossRef] [PubMed]
- Dou, J.T.; Chen, M.; Dufour, F.; Alkon, D.L.; Zhao, W.Q. Insulin receptor signaling in long-term memory consolidation following spatial learning. Learn. Mem. 2005, 12, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Grillo, C.A.; Piroli, G.G.; Lawrence, R.C.; Wrighten, S.A.; Green, A.J.; Wilson, S.P.; Sakai, R.R.; Kelly, S.J.; Wilson, M.A.; Mott, D.D.; et al. Hippocampal Insulin Resistance Impairs Spatial Learning and Synaptic Plasticity. Diabetes 2015, 64, 3927–3936. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Fang, X.; Xu, F.; Liu, S.; Qian, Y.; Gong, X.; Zhao, X.; Ma, Z.; Xia, T.; Gu, X. Amelioration of Hippocampal Insulin Resistance Reduces Tau Hyperphosphorylation and Cognitive Decline Induced by Isoflurane in Mice. Front. Aging Neurosci. 2021, 13, 686506. [Google Scholar] [CrossRef]
- Zhang, P.A.; Sun, Q.; Li, Y.C.; Weng, R.X.; Wu, R.; Zhang, H.H.; Xu, G.Y. Overexpression of Purinergic P2X4 Receptors in Hippocampus Rescues Memory Impairment in Rats with Type 2 Diabetes. Neurosci. Bull. 2020, 36, 719–732. [Google Scholar] [CrossRef]
- Yermakov, L.M.; Griggs, R.B.; Drouet, D.E.; Sugimoto, C.; Williams, M.T.; Vorhees, C.V.; Susuki, K. Impairment of cognitive flexibility in type 2 diabetic db/db mice. Behav. Brain Res. 2019, 371, 111978. [Google Scholar] [CrossRef]
- Born, J.; Lange, T.; Kern, W.; McGregor, G.P.; Bickel, U.; Fehm, H.L. Sniffing neuropeptides: A transnasal approach to the human brain. Nat. Neurosci. 2002, 5, 514–516. [Google Scholar] [CrossRef]
- Benedict, C.; Hallschmid, M.; Hatke, A.; Schultes, B.; Fehm, H.L.; Born, J.; Kern, W. Intranasal insulin improves memory in humans. Psychoneuroendocrinology 2004, 29, 1326–1334. [Google Scholar] [CrossRef]
- Benedict, C.; Hallschmid, M.; Schultes, B.; Born, J.; Kern, W. Intranasal insulin to improve memory function in humans. Neuroendocrinology 2007, 86, 136–142. [Google Scholar] [CrossRef]
- Benedict, C.; Kern, W.; Schultes, B.; Born, J.; Hallschmid, M. Differential sensitivity of men and women to anorexigenic and memory-improving effects of intranasal insulin. J. Clin. Endocrinol. Metab. 2008, 93, 1339–1344. [Google Scholar] [CrossRef]
- Ritze, Y.; Kern, W.; Ebner, E.M.; Jahn, S.; Benedict, C.; Hallschmid, M. Metabolic and Cognitive Outcomes of Subchronic Once-Daily Intranasal Insulin Administration in Healthy Men. Front. Endocrinol. (Lausanne) 2018, 9, 663. [Google Scholar] [CrossRef] [PubMed]
- Claxton, A.; Baker, L.D.; Wilkinson, C.W.; Trittschuh, E.H.; Chapman, D.; Watson, G.S.; Cholerton, B.; Plymate, S.R.; Arbuckle, M.; Craft, S. Sex and ApoE genotype differences in treatment response to two doses of intranasal insulin in adults with mild cognitive impairment or Alzheimer’s disease. J. Alzheimers Dis. 2013, 35, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Kullmann, S.; Veit, R.; Peter, A.; Pohmann, R.; Scheffler, K.; Haring, H.U.; Fritsche, A.; Preissl, H.; Heni, M. Dose-Dependent Effects of Intranasal Insulin on Resting-State Brain Activity. J. Clin. Endocrinol. Metab. 2018, 103, 253–262. [Google Scholar] [CrossRef]
- Avgerinos, K.I.; Kalaitzidis, G.; Malli, A.; Kalaitzoglou, D.; Myserlis, P.G.; Lioutas, V.A. Intranasal insulin in Alzheimer’s dementia or mild cognitive impairment: A systematic review. J. Neurol. 2018, 265, 1497–1510. [Google Scholar] [CrossRef] [PubMed]
- Lioutas, V.A.; Alfaro-Martinez, F.; Bedoya, F.; Chung, C.C.; Pimentel, D.A.; Novak, V. Intranasal Insulin and Insulin-Like Growth Factor 1 as Neuroprotectants in Acute Ischemic Stroke. Transl. Stroke Res. 2015, 6, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Long, C.; Han, X.; Yang, Y.; Li, T.; Zhou, Q.; Chen, Q. Efficacy of intranasal insulin in improving cognition in mild cognitive impairment or dementia: A systematic review and meta-analysis. Front. Aging Neurosci. 2022, 14, 963933. [Google Scholar] [CrossRef]
- Claxton, A.; Baker, L.D.; Hanson, A.; Trittschuh, E.H.; Cholerton, B.; Morgan, A.; Callaghan, M.; Arbuckle, M.; Behl, C.; Craft, S. Long-Acting Intranasal Insulin Detemir Improves Cognition for Adults with Mild Cognitive Impairment or Early-Stage Alzheimer’s Disease Dementia. J. Alzheimers Dis. 2015, 44, 897–906. [Google Scholar] [CrossRef]
- Kellar, D.; Lockhart, S.N.; Aisen, P.; Raman, R.; Rissman, R.A.; Brewer, J.; Craft, S. Intranasal Insulin Reduces White Matter Hyperintensity Progression in Association with Improvements in Cognition and CSF Biomarker Profiles in Mild Cognitive Impairment and Alzheimer’s Disease. J. Prev. Alzheimers Dis. 2021, 8, 240–248. [Google Scholar] [CrossRef]
- Craft, S.; Claxton, A.; Baker, L.D.; Hanson, A.J.; Cholerton, B.; Trittschuh, E.H.; Dahl, D.; Caulder, E.; Neth, B.; Montine, T.J.; et al. Effects of Regular and Long-Acting Insulin on Cognition and Alzheimer’s Disease Biomarkers: A Pilot Clinical Trial. J. Alzheimers Dis. 2017, 57, 1325–1334. [Google Scholar] [CrossRef]
- Mendez, P.; Stefanelli, T.; Flores, C.E.; Muller, D.; Luscher, C. Homeostatic Plasticity in the Hippocampus Facilitates Memory Extinction. Cell Rep. 2018, 22, 1451–1461. [Google Scholar] [CrossRef] [PubMed]
- Skeberdis, V.A.; Lan, J.; Zheng, X.; Zukin, R.S.; Bennett, M.V. Insulin promotes rapid delivery of N-methyl-D- aspartate receptors to the cell surface by exocytosis. Proc. Natl. Acad. Sci. USA 2001, 98, 3561–3566. [Google Scholar] [CrossRef]
- Christie, J.M.; Wenthold, R.J.; Monaghan, D.T. Insulin causes a transient tyrosine phosphorylation of NR2A and NR2B NMDA receptor subunits in rat hippocampus. J. Neurochem. 1999, 72, 1523–1528. [Google Scholar] [CrossRef]
- Derkach, V.A.; Oh, M.C.; Guire, E.S.; Soderling, T.R. Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat. Rev. Neurosci. 2007, 8, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Passafaro, M.; Piech, V.; Sheng, M. Subunit-specific temporal and spatial patterns of AMPA receptor exocytosis in hippocampal neurons. Nat. Neurosci. 2001, 4, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Penick, E.C.; Edwards, J.G.; Kauer, J.A.; Ehlers, M.D. Recycling endosomes supply AMPA receptors for LTP. Science 2004, 305, 1972–1975. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; Huang, C.C.; Wu, M.Y.; Hsu, K.S. Insulin stimulates postsynaptic density-95 protein translation via the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway. J. Biol. Chem. 2005, 280, 18543–18550. [Google Scholar] [CrossRef]
- Yarube, I.; Ayo, J.; Magaji, R.; Umar, I. Insulin treatment increases brain nitric oxide and oxidative stress, but does not affect memory function in mice. Physiol. Behav. 2019, 211, 112640. [Google Scholar] [CrossRef]
- Watson, G.S.; Bernhardt, T.; Reger, M.A.; Cholerton, B.A.; Baker, L.D.; Peskind, E.R.; Asthana, S.; Plymate, S.R.; Frolich, L.; Craft, S. Insulin effects on CSF norepinephrine and cognition in Alzheimer’s disease. Neurobiol. Aging 2006, 27, 38–41. [Google Scholar] [CrossRef] [PubMed]
- Hammoud, H.; Netsyk, O.; Tafreshiha, A.S.; Korol, S.V.; Jin, Z.; Li, J.P.; Birnir, B. Insulin differentially modulates GABA signalling in hippocampal neurons and, in an age-dependent manner, normalizes GABA-activated currents in the tg-APPSwe mouse model of Alzheimer’s disease. Acta Physiol. 2021, 232, e13623. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, M.; Bourassa, P.; Tremblay, C.; Caron, V.; Sugere, C.; Emond, V.; Bennett, D.A.; Calon, F. Cerebrovascular insulin receptors are defective in Alzheimer’s disease. Brain 2022, awac309. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.; Pemberton, S.; Babin, A.; Abdulhameed, N.; Noonan, C.; Brown, M.B.; Banks, W.A.; Rhea, E.M. Insulin blood-brain barrier transport and interactions are greater following exercise in mice. J. Appl. Physiol. (1985) 2022, 132, 824–834. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Zabolotny, J.M.; Huang, H.; Lee, H.; Kim, Y.B. Insulin in the nervous system and the mind: Functions in metabolism, memory, and mood. Mol. Metab. 2016, 5, 589–601. [Google Scholar] [CrossRef] [PubMed]
- Rhea, E.M.; Rask-Madsen, C.; Banks, W.A. Insulin transport across the blood-brain barrier can occur independently of the insulin receptor. J. Physiol. 2018, 596, 4753–4765. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.M.; Aylor, K.W.; Barrett, E.J. Unravelling the regulation of insulin transport across the brain endothelial cell. Diabetologia 2017, 60, 1512–1521. [Google Scholar] [CrossRef]
- Garcia-Caceres, C.; Quarta, C.; Varela, L.; Gao, Y.; Gruber, T.; Legutko, B.; Jastroch, M.; Johansson, P.; Ninkovic, J.; Yi, C.X.; et al. Astrocytic Insulin Signaling Couples Brain Glucose Uptake with Nutrient Availability. Cell 2016, 166, 867–880. [Google Scholar] [CrossRef]
- Hersom, M.; Helms, H.C.; Schmalz, C.; Pedersen, T.A.; Buckley, S.T.; Brodin, B. The insulin receptor is expressed and functional in cultured blood-brain barrier endothelial cells but does not mediate insulin entry from blood to brain. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E531–E542. [Google Scholar] [CrossRef]
- Xaio, H.; Banks, W.A.; Niehoff, M.L.; Morley, J.E. Effect of LPS on the permeability of the blood-brain barrier to insulin. Brain Res. 2001, 896, 36–42. [Google Scholar] [CrossRef]
- Urayama, A.; Banks, W.A. Starvation and triglycerides reverse the obesity induced impairment of insulin transport at the blood-brain barrier. Endocrinology 2008, 149, 3592–3597. [Google Scholar] [CrossRef]
- Banks, W.A. The Blood-Brain Barrier Interface in Diabetes Mellitus: Dysfunctions, Mechanisms and Approaches to Treatment. Curr. Pharm. Des. 2020, 26, 1438–1447. [Google Scholar] [CrossRef] [PubMed]
- Baranowska-Bik, A.; Bik, W. Insulin and brain aging. Prz Menopauzalny 2017, 16, 44–46. [Google Scholar] [CrossRef] [PubMed]
- Baskin, D.G.; Brewitt, B.; Davidson, D.A.; Corp, E.; Paquette, T.; Figlewicz, D.P.; Lewellen, T.K.; Graham, M.K.; Woods, S.G.; Dorsa, D.M. Quantitative autoradiographic evidence for insulin receptors in the choroid plexus of the rat brain. Diabetes 1986, 35, 246–249. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dorn, A.; Rinne, A.; Bernstein, H.G.; Hahn, H.J.; Ziegler, M. Insulin and C-peptide in human brain neurons (insulin/C-peptide/brain peptides/immunohistochemistry/radioimmunoassay). J. Fur Hirnforsch. 1983, 24, 495–499. [Google Scholar]
- Schechter, R.; Holtzclaw, L.; Sadiq, F.; Kahn, A.; Devaskar, S. Insulin synthesis by isolated rabbit neurons. Endocrinology 1988, 123, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Jaspan, J.B.; Kastin, A.J. Selective, physiological transport of insulin across the blood-brain barrier: Novel demonstration by species-specific radioimmunoassays. Peptides 1997, 18, 1257–1262. [Google Scholar] [CrossRef]
- Molnar, G.; Farago, N.; Kocsis, A.K.; Rozsa, M.; Lovas, S.; Boldog, E.; Baldi, R.; Csajbok, E.; Gardi, J.; Puskas, L.G.; et al. GABAergic neurogliaform cells represent local sources of insulin in the cerebral cortex. J. Neurosci. 2014, 34, 1133–1137. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, T.; Toyoshima-Aoyama, F.; Yanagita, T.; Maruta, T.; Fujita, H.; Koshida, T.; Yonaha, T.; Wada, A.; Sawaguchi, A.; Murakami, M. New insights concerning insulin synthesis and its secretion in rat hippocampus and cerebral cortex: Amyloid-beta1-42-induced reduction of proinsulin level via glycogen synthase kinase-3beta. Cell. Signal. 2014, 26, 253–259. [Google Scholar] [CrossRef]
- Pitt, J.; Wilcox, K.C.; Tortelli, V.; Diniz, L.P.; Oliveira, M.S.; Dobbins, C.; Yu, X.W.; Nandamuri, S.; Gomes, F.C.A.; DiNunno, N.; et al. Neuroprotective astrocyte-derived insulin/insulin-like growth factor 1 stimulates endocytic processing and extracellular release of neuron-bound Abeta oligomers. Mol. Biol. Cell 2017, 28, 2623–2636. [Google Scholar] [CrossRef]
- Takano, K.; Koarashi, K.; Kawabe, K.; Itakura, M.; Nakajima, H.; Moriyama, M.; Nakamura, Y. Insulin expression in cultured astrocytes and the decrease by amyloid beta. Neurochem. Int. 2018, 119, 171–177. [Google Scholar] [CrossRef]
- Montero-Crespo, M.; Dominguez-Alvaro, M.; Alonso-Nanclares, L.; DeFelipe, J.; Blazquez-Llorca, L. Three-dimensional analysis of synaptic organization in the hippocampal CA1 field in Alzheimer’s disease. Brain 2021, 144, 553–573. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E.; Bohl, J. Staging of Alzheimer-related cortical destruction. Eur. Neurol. 1993, 33, 403–408. [Google Scholar] [CrossRef]
- Ly, P.T.T.; Wu, Y.L.; Zou, H.Y.; Wang, R.T.; Zhou, W.H.; Kinoshita, A.; Zhang, M.M.; Yang, Y.; Cai, F.; Woodgett, J.; et al. Inhibition of GSK3 beta-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J. Clin. Investig. 2013, 123, 224–235. [Google Scholar] [CrossRef]
- Hemonnot-Girard, A.L.; Valverde, A.J.; Hua, J.; Delaygue, C.; Linck, N.; Maurice, T.; Rassendren, F.; Hirbec, H. Analysis of CX3CR1 haplodeficiency in male and female APP(swe)/PSEN1(dE9) mice along Alzheimer disease progression. Brain Behav. Immun. 2021, 91, 404–417. [Google Scholar] [CrossRef] [PubMed]
- Bradburn, S.; Murgatroyd, C.; Ray, N. Neuroinflammation in mild cognitive impairment and Alzheimer’s disease: A meta-analysis. Ageing Res. Rev. 2019, 50, 1–8. [Google Scholar] [CrossRef]
- Popp, J.; Oikonomidi, A.; Tautvydaite, D.; Dayon, L.; Bacher, M.; Migliavacca, E.; Henry, H.; Kirkland, R.; Severin, I.; Wojcik, J.; et al. Markers of neuroinflammation associated with Alzheimer’s disease pathology in older adults. Brain Behav. Immun. 2017, 62, 203–211. [Google Scholar] [CrossRef]
- d’Errico, P.; Ziegler-Waldkirch, S.; Aires, V.; Hoffmann, P.; Mezo, C.; Erny, D.; Monasor, L.S.; Liebscher, S.; Ravi, V.M.; Joseph, K.; et al. Microglia contribute to the propagation of a beta into unaffected brain tissue. Nat. Neurosci. 2022, 25, 20–25. [Google Scholar] [CrossRef]
- Friker, L.L.; Scheiblich, H.; Hochheiser, I.V.; Brinkschulte, R.; Riedel, D.; Latz, E.; Geyer, M.; Heneka, M.T. Beta-Amyloid Clustering around ASC Fibrils Boosts Its Toxicity in Microglia. Cell Rep. 2020, 30, 3743–3754 e6. [Google Scholar] [CrossRef] [PubMed]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, S.C.; Lin, Y.; Oakley, D.; Roe, A.D.; DeVos, S.L.; Hanlon, D.; Hyman, B.T. The role of microglia in processing and spreading of bioactive tau seeds in Alzheimer’s disease. J. Neuroinflamm. 2018, 15, 269. [Google Scholar] [CrossRef] [PubMed]
- Pascoal, T.A.; Benedet, A.L.; Ashton, N.J.; Kang, M.S.; Therriault, J.; Chamoun, M.; Savard, M.; Lussier, F.Z.; Tissot, C.; Karikari, T.K.; et al. Microglial activation and tau propagate jointly across Braak stages. Nat. Med. 2021, 27, 1592–1597. [Google Scholar] [CrossRef]
- Antharam, V.; Collingwood, J.F.; Bullivant, J.P.; Davidson, M.R.; Chandra, S.; Mikhaylova, A.; Finnegan, M.E.; Batich, C.; Forder, J.R.; Dobson, J. High field magnetic resonance microscopy of the human hippocampus in Alzheimer’s disease: Quantitative imaging and correlation with iron. Neuroimage 2012, 59, 1249–1260. [Google Scholar] [CrossRef]
- Gao, L.L.; Jiang, Z.H.; Cai, Z.C.; Cai, M.; Zhang, Q.; Ma, Y.Y.; Li, G.L.; Zhao, F.Z.; Ma, Q. Brain iron deposition analysis using susceptibility weighted imaging and its association with body iron level in patients with mild cognitive impairment. Mol. Med. Rep. 2017, 16, 8209–8215. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bao, W.D.; Pang, P.; Zhou, X.T.; Hu, F.; Xiong, W.; Chen, K.; Wang, J.; Wang, F.; Xie, D.; Hu, Y.Z.; et al. Loss of ferroportin induces memory impairment by promoting ferroptosis in Alzheimer’s disease. Cell Death Differ. 2021, 28, 1548–1562. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Alonso, M.; Fernandez, B.; Navarro, A.; Junceda, S.; Astudillo, A.; Pereiro, R. Laser ablation ICP-MS for simultaneous quantitative imaging of iron and ferroportin in hippocampus of human brain tissues with Alzheimer’s disease. Talanta 2019, 197, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.N.; Qian, Z.M.; Wu, K.C.; Yung, W.H.; Ke, Y. Expression of Iron Transporters and Pathological Hallmarks of Parkinson’s and Alzheimer’s Diseases in the Brain of Young, Adult, and Aged Rats. Mol. Neurobiol. 2017, 54, 5213–5224. [Google Scholar] [CrossRef]
- Leskovjan, A.C.; Kretlow, A.; Lanzirotti, A.; Barrea, R.; Vogt, S.; Miller, L.M. Increased brain iron coincides with early plaque formation in a mouse model of Alzheimer’s disease. Neuroimage 2011, 55, 32–38. [Google Scholar] [CrossRef]
- Hambright, W.S.; Fonseca, R.S.; Chen, L.; Na, R.; Ran, Q. Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol. 2017, 12, 8–17. [Google Scholar] [CrossRef]
- McIntosh, A.; Mela, V.; Harty, C.; Minogue, A.M.; Costello, D.A.; Kerskens, C.; Lynch, M.A. Iron accumulation in microglia triggers a cascade of events that leads to altered metabolism and compromised function in APP/PS1 mice. Brain Pathol. 2019, 29, 606–621. [Google Scholar] [CrossRef]
- Zeineh, M.M.; Chen, Y.; Kitzler, H.H.; Hammond, R.; Vogel, H.; Rutt, B.K. Activated iron-containing microglia in the human hippocampus identified by magnetic resonance imaging in Alzheimer disease. Neurobiol. Aging 2015, 36, 2483–2500. [Google Scholar] [CrossRef]
- Kroner, A.; Greenhalgh, A.D.; Zarruk, J.G.; dos Santos, R.P.; Gaestel, M.; David, S. TNF and Increased Intracellular Iron Alter Macrophage Polarization to a Detrimental M1 Phenotype in the Injured Spinal Cord. Neuron 2014, 83, 1098–1116. [Google Scholar] [CrossRef]
- Hirai, K.; Aliev, G.; Nunomura, A.; Fujioka, H.; Russell, R.L.; Atwood, C.S.; Johnson, A.B.; Kress, Y.; Vinters, H.V.; Tabaton, M.; et al. Mitochondrial abnormalities in Alzheimer’s disease. J. Neurosci. 2001, 21, 3017–3023. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yin, J.; Ma, X.; Zhao, F.; Siedlak, S.L.; Wang, Z.; Torres, S.; Fujioka, H.; Xu, Y.; Perry, G.; et al. Inhibition of mitochondrial fragmentation protects against Alzheimer’s disease in rodent model. Hum. Mol. Genet. 2017, 26, 4118–4131. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A.; Yang, D.S. Autophagy failure in Alzheimer’s disease-locating the primary defect. Neurobiol. Dis. 2011, 43, 38–45. [Google Scholar] [CrossRef] [PubMed]
- de la Cueva, M.; Antequera, D.; Ordonez-Gutierrez, L.; Wandosell, F.; Camins, A.; Carro, E.; Bartolome, F. Amyloid-beta impairs mitochondrial dynamics and autophagy in Alzheimer’s disease experimental models. Sci. Rep. 2022, 12, 10092. [Google Scholar] [CrossRef]
- Reddy, P.H.; Yin, X.; Manczak, M.; Kumar, S.; Pradeepkiran, J.A.; Vijayan, M.; Reddy, A.P. Mutant APP and amyloid beta-induced defective autophagy, mitophagy, mitochondrial structural and functional changes and synaptic damage in hippocampal neurons from Alzheimer’s disease. Hum. Mol. Genet. 2018, 27, 2502–2516. [Google Scholar] [CrossRef]
- Miller, M.B.; Huang, A.Y.; Kim, J.; Zhou, Z.; Kirkham, S.L.; Maury, E.A.; Ziegenfuss, J.S.; Reed, H.C.; Neil, J.E.; Rento, L.; et al. Somatic genomic changes in single Alzheimer’s disease neurons. Nature 2022, 604, 714–722. [Google Scholar] [CrossRef]
- Mandal, P.K.; Shukla, D.; Tripathi, M.; Ersland, L. Cognitive Improvement with Glutathione Supplement in Alzheimer’s Disease: A Way Forward. J. Alzheimer’s Dis. 2019, 68, 531–535. [Google Scholar] [CrossRef]
- Tan, J.; Li, Q.X.; Evin, G. Effects of Mild and Severe Oxidative Stress on BACE1 Expression and APP Amyloidogenic Processing. Methods Mol. Biol. 2016, 1303, 101–116. [Google Scholar]
- Butterfield, D.A.; Sultana, R. Methionine-35 of abeta(1-42): Importance for oxidative stress in Alzheimer disease. J. Amino Acids 2011, 2011, 198430. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Xiao, Y.; Miao, R.; Zhao, J.; Zhang, W.; Huang, G.; Ma, F. The characteristic of cognitive function in Type 2 diabetes mellitus. Diabetes Res. Clin. Pract. 2015, 109, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.W.; Zhang, J.Q.; Liu, C.; Wei, P.; Zhang, X.; Yuan, Q.Y.; Yin, X.T.; Wei, L.Q.; Cui, J.G.; Wang, J. Memory dysfunction in type 2 diabetes mellitus correlates with reduced hippocampal CA1 and subiculum volumes. Chin. Med. J. 2015, 128, 465–471. [Google Scholar] [CrossRef]
- Huang, R.R.; Jia, B.H.; Xie, L.; Ma, S.H.; Yin, J.J.; Sun, Z.B.; Le, H.B.; Xu, W.C.; Huang, J.Z.; Luo, D.X. Spatial working memory impairment in primary onset middle-age type 2 diabetes mellitus: An ethology and BOLD-fMRI study. J. Magn. Reson. Imaging 2016, 43, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Soares, E.; Prediger, R.D.; Nunes, S.; Castro, A.A.; Viana, S.D.; Lemos, C.; De Souza, C.M.; Agostinho, P.; Cunha, R.A.; Carvalho, E.; et al. Spatial memory impairments in a prediabetic rat model. Neuroscience 2013, 250, 565–577. [Google Scholar] [CrossRef]
- Singh, A.; Bodakhe, S.H. Resveratrol attenuates behavioural impairment associated with learning and memory in rats with diabetes induced by a high-fat diet and streptozotocin. Br. J. Pharmacol. 2022, 179, 4673–4691. [Google Scholar] [CrossRef]
- Morris, J.K.; Vidoni, E.D.; Honea, R.A.; Burns, J.M.; Alzheimer’s Disease Neuroimaging, I. Impaired glycemia increases disease progression in mild cognitive impairment. Neurobiol. Aging 2014, 35, 585–589. [Google Scholar] [CrossRef]
- Potenza, M.A.; Sgarra, L.; Desantis, V.; Nacci, C.; Montagnani, M. Diabetes and Alzheimer’s Disease: Might Mitochondrial Dysfunction Help Deciphering the Common Path? Antioxidants 2021, 10, 1257. [Google Scholar] [CrossRef]
- Wakabayashi, T.; Yamaguchi, K.; Matsui, K.; Sano, T.; Kubota, T.; Hashimoto, T.; Mano, A.; Yamada, K.; Matsuo, Y.; Kubota, N.; et al. Differential effects of diet- and genetically-induced brain insulin resistance on amyloid pathology in a mouse model of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 15. [Google Scholar] [CrossRef]
- Schmitz, L.; Kuglin, R.; Bae-Gartz, I.; Janoschek, R.; Appel, S.; Mesaros, A.; Jakovcevski, I.; Vohlen, C.; Handwerk, M.; Ensenauer, R.; et al. Hippocampal insulin resistance links maternal obesity with impaired neuronal plasticity in adult offspring. Psychoneuroendocrinology 2018, 89, 46–52. [Google Scholar] [CrossRef]
- Spinelli, M.; Fusco, S.; Mainardi, M.; Scala, F.; Natale, F.; Lapenta, R.; Mattera, A.; Rinaudo, M.; Li Puma, D.D.; Ripoli, C.; et al. Brain insulin resistance impairs hippocampal synaptic plasticity and memory by increasing GluA1 palmitoylation through FoxO3a. Nat. Commun. 2017, 8, 2009. [Google Scholar] [CrossRef] [PubMed]
- Denver, P.; English, A.; McClean, P.L. Inflammation, insulin signaling and cognitive function in aged APP/PS1 mice. Brain Behav. Immun. 2018, 70, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Willette, A.A.; Modanlo, N.; Kapogiannis, D.; Alzheimer’s Disease Neuroimaging Initiative. Insulin resistance predicts medial temporal hypermetabolism in mild cognitive impairment conversion to Alzheimer disease. Diabetes 2015, 64, 1933–1940. [Google Scholar] [CrossRef]
- Cui, Y.; Tang, T.Y.; Lu, C.Q.; Ju, S. Insulin Resistance and Cognitive Impairment: Evidence From Neuroimaging. J. Magn. Reson. Imaging 2022, 56, 1621–1649. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.T.; Happonen, K.E.; Burrola, P.G.; O’Connor, C.; Hah, N.; Huang, L.; Nimmerjahn, A.; Lemke, G. Microglia use TAM receptors to detect and engulf amyloid beta plaques. Nat. Immunol. 2021, 22, 586–594. [Google Scholar] [CrossRef]
- Maianti, J.P.; Tan, G.A.; Vetere, A.; Welsh, A.J.; Wagner, B.K.; Seeliger, M.A.; Liu, D.R. Substrate-selective inhibitors that reprogram the activity of insulin-degrading enzyme. Nat. Chem. Biol. 2019, 15, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, B.R.; Panda, P.K.; Liang, W.; Tang, W.J.; Ahuja, R.; Ramamoorthy, A. Degradation of Alzheimer’s Amyloid-beta by a Catalytically Inactive Insulin-Degrading Enzyme. J. Mol. Biol. 2021, 433, 166993. [Google Scholar] [CrossRef] [PubMed]
- Starks, E.J.; O’Grady, J.P.; Hoscheidt, S.M.; Racine, A.M.; Carlsson, C.M.; Zetterberg, H.; Blennow, K.; Okonkwo, O.C.; Puglielli, L.; Asthana, S.; et al. Insulin Resistance is Associated with Higher Cerebrospinal Fluid Tau Levels in Asymptomatic APOE epsilon 4 Carriers. J. Alzheimers Dis. 2015, 46, 525–533. [Google Scholar] [CrossRef]
- Yang, W.; Liu, Y.; Xu, Q.Q.; Xian, Y.F.; Lin, Z.X. Sulforaphene Ameliorates Neuroinflammation and Hyperphosphorylated Tau Protein via Regulating the PI3K/Akt/GSK-3beta Pathway in Experimental Models of Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2020, 2020, 4754195. [Google Scholar] [CrossRef]
- Zhao, W.H.; Xu, W.M. Glutaredoxin 2 (GRX2) deficiency exacerbates high fat diet (HFD)-induced insulin resistance, inflammation and mitochondrial dysfunction in brain injury: A mechanism involving GSK-3 beta. Biomed. Pharmacother. 2019, 118, 108940. [Google Scholar]
- Liao, Y.F.; Wang, B.J.; Cheng, H.T.; Kuo, L.H.; Wolfe, M.S. Tumor necrosis factor-alpha, interleukin-1 beta, and interferon-gamma stimulate gamma-secretase-mediated cleavage of amyloid precursor protein through a JNK-dependent MAPK pathway. J. Biol. Chem. 2004, 279, 49523–49532. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.S.; Muhammad, T.; Ikram, M.; Kim, M.O. Dietary Supplementation of the Antioxidant Curcumin Halts Systemic LPS-Induced Neuroinflammation-Associated Neurodegeneration and Memory/Synaptic Impairment via the JNK/NF-kappa B/Akt Signaling Pathway in Adult Rats. Oxid. Med. Cell. Longev. 2019, 2019, 7860650. [Google Scholar] [CrossRef] [PubMed]
- Ruegsegger, G.N.; Vanderboom, P.M.; Dasari, S.; Klaus, K.A.; Kabiraj, P.; McCarthy, C.B.; Lucchinetti, C.F.; Nair, K.S. Exercise and metformin counteract altered mitochondrial function in the insulin-resistant brain. JCI Insight 2019, 4, e130681. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Hemmelgarn, B.T.; Chuang, C.C.; Best, T.M. The Role of Oxidative Stress-Induced Epigenetic Alterations in Amyloid-beta Production in Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2015, 2015, 604658. [Google Scholar] [CrossRef]
- Rains, J.L.; Jain, S.K. Oxidative stress, insulin signaling, and diabetes. Free Radic. Biol. Med. 2011, 50, 567–575. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Dose of Insulin | Time of Intranasal Administration | Animal Models | Memory Detection Method | Effects on Memory | References |
|---|---|---|---|---|---|
| Low level (0.0715 IU) | once a day, 5 days a week, 12 weeks | 18-month-old male F344 rats | Morris water maze test | No obvious effects | [15] |
| Low level (0.24 IU) | once a day, 4 consecutive weeks | male C57BL6 mice | Radial arm water maze test | Improvement | [16] |
| Low level (0.1 IU and 0.5 IU) | once a day, 4 consecutive weeks | kainic acid-induced chronic epileptic mice | Morris water maze test | Improvement | [17] |
| Low level (0.5 IU) | once a day, 7 consecutive days | Wistar with methamphetamine for 10 days | Y-maze test, Novel object recognition test | Improvement | [18] |
| High level (1 IU) | twice a day, 14 consecutive days | C57BL/6J mice treated with an I.C.V. injection of STZ | Morris water maze test | Improvement | [19] |
| High level (1.75 IU) | once a day, 3 consecutive days | 3xTg-AD mice anesthetized with propofol/sevoflurane for 3 h | Morris water maze test, novel object recognition test | Improvement | [20] |
| High level (2 IU) | once a day, 14 consecutive days | rats were injected with STZ (3 mg/kg, ICV) bilaterally twice, on days 1 and 3 | Morris water maze test | Improvement | [21] |
| High level (2 IU) | once a day, 6consecutive weeks | Wistar rats were injected with 6-OHDA (12 μg/4 μL) into the unilateral medial forebrain bundle | T-maze rewarded alternation test | Improvement | [22] |
| Objective (MCI or Mild to Moderate AD) | Dose and Duration of Intranasal Insulin Administration | Memory Detection Method | Effects on Memory | References |
|---|---|---|---|---|
| 289 adults | 40 IU/day, 12 months | adas-cog-12 score | No benefits | [13] |
| 60 adults | 40 IU/day, 21 days | verbal working memory, visuospatial working memory | Improvement | [38] |
| 104 adults | 40 IU/day, 4 months | delayed story recall, the dementia severity rating scale | Improvement | [33] |
| 49 adults | 20 IU/day, 12 months | Alzheimer’s disease assessment scale-cognition, Alzheimer’s disease cooperative study-activities of daily living scale, a memory composite | Improvement | [39] |
| 36 adults | 40 IU/day, 4 months | global cognition (Alzheimer’s disease assessment scale-cognition) | Improvement | [40] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Q.; Wang, Z.; Cao, J.; Dong, Y.; Chen, Y. The Role of Insulin Signaling in Hippocampal-Related Diseases: A Focus on Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 14417. https://doi.org/10.3390/ijms232214417
Liu Q, Wang Z, Cao J, Dong Y, Chen Y. The Role of Insulin Signaling in Hippocampal-Related Diseases: A Focus on Alzheimer’s Disease. International Journal of Molecular Sciences. 2022; 23(22):14417. https://doi.org/10.3390/ijms232214417
Chicago/Turabian StyleLiu, Qi, Zixu Wang, Jing Cao, Yulan Dong, and Yaoxing Chen. 2022. "The Role of Insulin Signaling in Hippocampal-Related Diseases: A Focus on Alzheimer’s Disease" International Journal of Molecular Sciences 23, no. 22: 14417. https://doi.org/10.3390/ijms232214417
APA StyleLiu, Q., Wang, Z., Cao, J., Dong, Y., & Chen, Y. (2022). The Role of Insulin Signaling in Hippocampal-Related Diseases: A Focus on Alzheimer’s Disease. International Journal of Molecular Sciences, 23(22), 14417. https://doi.org/10.3390/ijms232214417