
The Impacts of Iron Overload and Ferroptosis on Intestinal Mucosal Homeostasis and Inflammation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Importance of Maintaining Intestinal Mucosal Homeostasis
2.1. The Role of the Intestinal Barrier
2.1.1. Intestinal Stem Cells (ISC)
2.1.2. Absorptive Enterocytes
2.1.3. Enteroendocrine Cells
2.1.4. Goblet Cells
2.1.5. Paneth Cells and Tuff Cells
2.2. The Role of Intestinal Microbiota
2.3. The Role of Intestinal Immune Cells
2.3.1. Macrophages and M Cells
2.3.2. T and B Cells
2.3.3. Dendritic Cells
3. Intestinal Iron Metabolism
4. Microbial Iron Absorption Mechanism
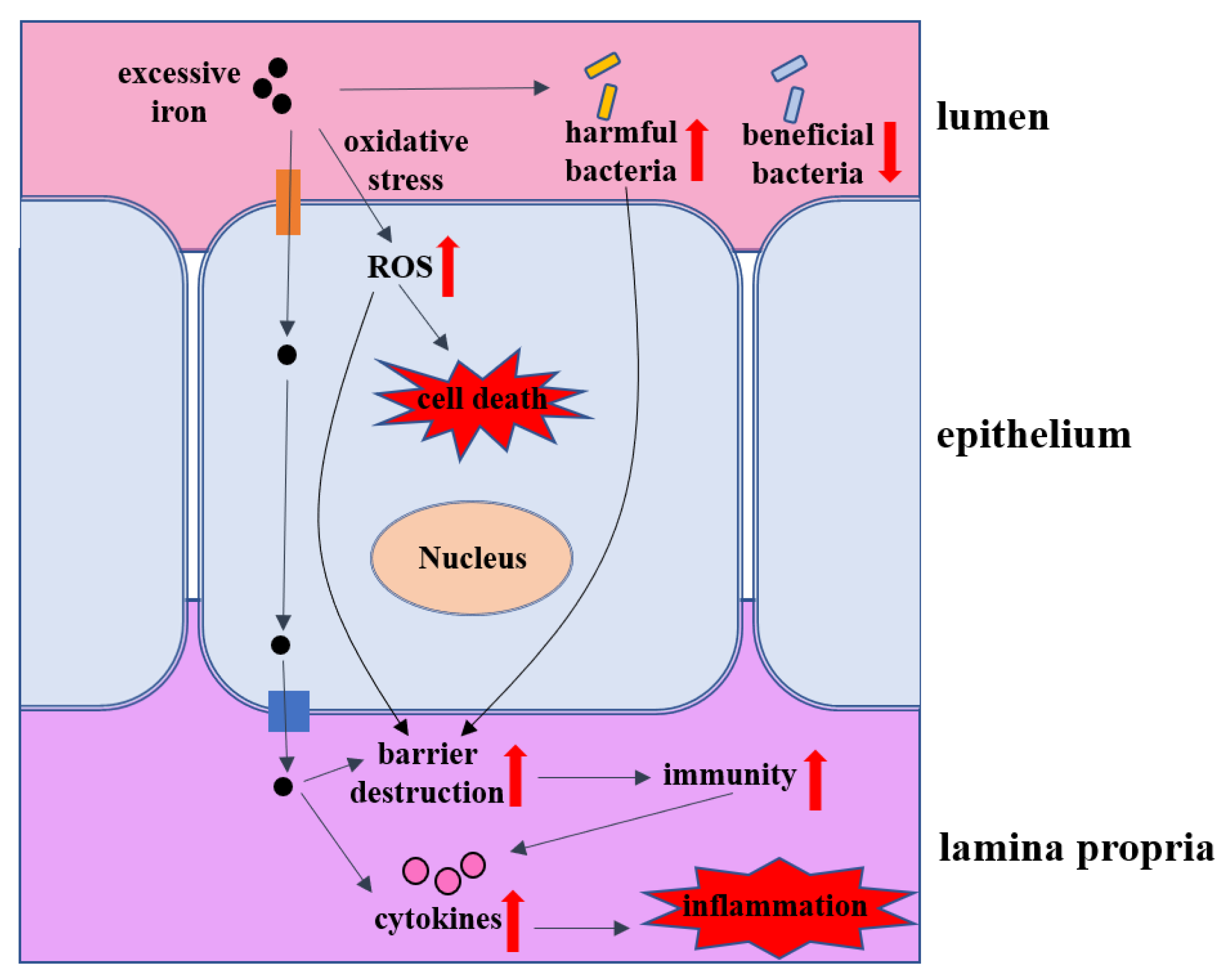
5. The Impacts of Iron Overload on Intestinal Homeostasis
5.1. Intestinal Barrier
5.2. Intestinal Inflammation
5.3. Intestinal Microbes
5.4. Intestinal Transport Proteins
5.5. Cell Death
6. Ferroptosis Machinery and Its Role in Intestinal Diseases
6.1. Major Metabolic Mechanisms of Ferroptosis
6.1.1. Amino acid Metabolism
6.1.2. Lipid Metabolism
6.1.3. Iron Metabolism
6.2. The Role of Ferroptosis in Intestinal Diseases
6.2.1. Inflammatory Bowel Disease (IBD)
6.2.2. Colorectal Cancer (CRC)
6.2.3. Intestinal Ischemia/Reperfusion (I/R) Injury
7. The Progress of Iron Overload and Ferroptosis-Targeting Therapeutic Strategies in Intestinal Diseases
7.1. Iron Chelators
7.2. Antioxidants
7.3. Anti-Inflammatory Treatments
8. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mowat, A.M.; Agace, W.W. Regional specialization within the intestinal immune system. Nat. Rev. Immunol. 2014, 14, 667–685. [Google Scholar] [CrossRef] [PubMed]
- Salvo Romero, E.; Alonso Cotoner, C.; Pardo Camacho, C.; Casado Bedmar, M.; Vicario, M. The intestinal barrier function and its involvement in digestive disease. Rev. Esp. Enferm. Dig. 2015, 107, 686–696. [Google Scholar] [CrossRef] [PubMed]
- Peterson, L.W.; Artis, D. Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 2014, 14, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Amar, J.; Chabo, C.; Waget, A.; Klopp, P.; Vachoux, C.; Bermudez-Humaran, L.G.; Smirnova, N.; Berge, M.; Sulpice, T.; Lahtinen, S.; et al. Intestinal mucosal adherence and translocation of commensal bacteria at the early onset of type 2 diabetes: Molecular mechanisms and probiotic treatment. EMBO Mol. Med. 2011, 3, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Sandler, N.G.; Koh, C.; Roque, A.; Eccleston, J.L.; Siegel, R.B.; Demino, M.; Kleiner, D.E.; Deeks, S.G.; Liang, T.J.; Heller, T.; et al. Host response to translocated microbial products predicts outcomes of patients with HBV or HCV infection. Gastroenterology 2011, 141, 1220–1230.e3. [Google Scholar] [CrossRef] [PubMed]
- Pickert, G.; Neufert, C.; Leppkes, M.; Zheng, Y.; Wittkopf, N.; Warntjen, M.; Lehr, H.A.; Hirth, S.; Weigmann, B.; Wirtz, S.; et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J. Exp. Med. 2009, 206, 1465–1472. [Google Scholar] [CrossRef]
- Wu, Y.; Tang, L.; Wang, B.; Sun, Q.; Zhao, P.; Li, W. The role of autophagy in maintaining intestinal mucosal barrier. J. Cell Physiol. 2019, 234, 19406–19419. [Google Scholar] [CrossRef]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to tango: Regulation of Mammalian iron metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef]
- Cornelissen, A.; Guo, L.; Sakamoto, A.; Virmani, R.; Finn, A.V. New insights into the role of iron in inflammation and atherosclerosis. EBioMedicine 2019, 47, 598–606. [Google Scholar] [CrossRef]
- Anderson, G.J.; Frazer, D.M. Current understanding of iron homeostasis. Am. J. Clin. Nutr. 2017, 106 (Suppl. 6), 1559S–1566S. [Google Scholar] [CrossRef]
- Lund, E.K.; Fairweather-Tait, S.J.; Wharf, S.G.; Johnson, I.T. Chronic exposure to high levels of dietary iron fortification increases lipid peroxidation in the mucosa of the rat large intestine. J. Nutr. 2001, 131, 2928–2931. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Lao, C.; Huang, C.; Xia, Y.; Ma, W.; Liu, W.; Chen, Z. Iron Overload Resulting from the Chronic Oral Administration of Ferric Citrate Impairs Intestinal Immune and Barrier in Mice. Biol. Trace Elem. Res. 2021, 199, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Mou, Y.; Wang, J.; Wu, J.; He, D.; Zhang, C.; Duan, C.; Li, B. Ferroptosis, a new form of cell death: Opportunities and challenges in cancer. J. Hematol. Oncol. 2019, 12, 34. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, C.; Zhao, Y.; Gao, G. Mitochondria regulation in ferroptosis. Eur. J. Cell Biol. 2020, 99, 151058. [Google Scholar] [CrossRef]
- Qiu, Y.; Cao, Y.; Cao, W.; Jia, Y.; Lu, N. The Application of Ferroptosis in Diseases. Pharmacol. Res. 2020, 159, 104919. [Google Scholar] [CrossRef]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Hassannia, B.; Vandenabeele, P.; Vanden Berghe, T. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell 2019, 35, 830–849. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Cao, J.Y.; Dixon, S.J. Mechanisms of ferroptosis. Cell Mol. Life Sci. 2016, 73, 2195–2209. [Google Scholar] [CrossRef]
- Hu, Z.; Zhang, H.; Yang, S.K.; Wu, X.; He, D.; Cao, K.; Zhang, W. Emerging Role of Ferroptosis in Acute Kidney Injury. Oxidative Med. Cell. Longev. 2019, 2019, 8010614. [Google Scholar] [CrossRef] [PubMed]
- Del Re, D.P.; Amgalan, D.; Linkermann, A.; Liu, Q.; Kitsis, R.N. Fundamental Mechanisms of Regulated Cell Death and Implications for Heart Disease. Physiol. Rev. 2019, 99, 1765–1817. [Google Scholar] [CrossRef]
- Li, Y.; Feng, D.; Wang, Z.; Zhao, Y.; Sun, R.; Tian, D.; Liu, D.; Zhang, F.; Ning, S.; Yao, J.; et al. Ischemia-induced ACSL4 activation contributes to ferroptosis-mediated tissue injury in intestinal ischemia/reperfusion. Cell Death Differ. 2019, 26, 2284–2299. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Yang, H.; Lin, R.; Jiang, K.; Wang, B.M. The role of ferroptosis in digestive system cancer. Oncol. Lett. 2019, 18, 2159–2164. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Zhang, T.; Wu, H. Emerging Pathological Engagement of Ferroptosis in Gut Diseases. Oxidative Med. Cell. Longev. 2021, 2021, 4246255. [Google Scholar] [CrossRef]
- Jakaria, M.; Belaidi, A.A.; Bush, A.I.; Ayton, S. Ferroptosis as a mechanism of neurodegeneration in Alzheimer’s disease. J. Neurochem. 2021, 159, 804–825. [Google Scholar] [CrossRef]
- Tsurusaki, S.; Tsuchiya, Y.; Koumura, T.; Nakasone, M.; Sakamoto, T.; Matsuoka, M.; Imai, H.; Yuet-Yin Kok, C.; Okochi, H.; Nakano, H.; et al. Hepatic ferroptosis plays an important role as the trigger for initiating inflammation in nonalcoholic steatohepatitis. Cell Death Dis. 2019, 10, 449. [Google Scholar] [CrossRef]
- Chen, J.; Li, X.; Ge, C.; Min, J.; Wang, F. The multifaceted role of ferroptosis in liver disease. Cell Death Differ. 2022, 29, 467–480. [Google Scholar] [CrossRef]
- Ajoolabady, A.; Aslkhodapasandhokmabad, H.; Libby, P.; Tuomilehto, J.; Lip, G.Y.H.; Penninger, J.M.; Richardson, D.R.; Tang, D.; Zhou, H.; Wang, S.; et al. Ferritinophagy and ferroptosis in the management of metabolic diseases. Trends Endocrinol. Metab. 2021, 32, 444–462. [Google Scholar] [CrossRef]
- Mazhar, M.; Din, A.U.; Ali, H.; Yang, G.; Ren, W.; Wang, L.; Fan, X.; Yang, S. Implication of ferroptosis in aging. Cell Death Discov. 2021, 7, 149. [Google Scholar] [CrossRef]
- Martens, E.C.; Neumann, M.; Desai, M.S. Interactions of commensal and pathogenic microorganisms with the intestinal mucosal barrier. Nat. Rev. Microbiol. 2018, 16, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Randall-Demllo, S.; Chieppa, M.; Eri, R. Intestinal epithelium and autophagy: Partners in gut homeostasis. Front. Immunol. 2013, 4, 301. [Google Scholar] [CrossRef] [PubMed]
- Lassen, K.G.; Xavier, R.J. Mechanisms and function of autophagy in intestinal disease. Autophagy 2018, 14, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Bach, S.P.; Renehan, A.G.; Potten, C.S. Stem cells: The intestinal stem cell as a paradigm. Carcinogenesis 2000, 21, 469–476. [Google Scholar] [CrossRef]
- van der Flier, L.G.; Clevers, H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu. Rev. Physiol. 2009, 71, 241–260. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Biehs, B.; Warming, S.; Leong, K.G.; Rangell, L.; Klein, O.D.; de Sauvage, F.J. A reserve stem cell population in small intestine renders Lgr5-positive cells dispensable. Nature 2011, 478, 255–259. [Google Scholar] [CrossRef]
- Barker, N.; van Es, J.H.; Kuipers, J.; Kujala, P.; van den Born, M.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J.; et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef]
- Sangiorgi, E.; Capecchi, M.R. Bmi1 is expressed in vivo in intestinal stem cells. Nat. Genet. 2008, 40, 915–920. [Google Scholar] [CrossRef]
- Kurokawa, K.; Hayakawa, Y.; Koike, K. Plasticity of Intestinal Epithelium: Stem Cell Niches and Regulatory Signals. Int. J. Mol. Sci. 2020, 22, 357. [Google Scholar] [CrossRef]
- Metcalfe, C.; Kljavin, N.M.; Ybarra, R.; de Sauvage, F.J. Lgr5+ stem cells are indispensable for radiation-induced intestinal regeneration. Cell Stem Cell 2014, 14, 149–159. [Google Scholar] [CrossRef]
- Marshman, E.; Booth, C.; Potten, C.S. The intestinal epithelial stem cell. Bioessays 2002, 24, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Tourkochristou, E.; Triantos, C.; Mouzaki, A. The Influence of Nutritional Factors on Immunological Outcomes. Front. Immunol. 2021, 12, 665968. [Google Scholar] [CrossRef] [PubMed]
- Steed, E.; Balda, M.S.; Matter, K. Dynamics and functions of tight junctions. Trends Cell Biol. 2010, 20, 142–149. [Google Scholar] [CrossRef]
- Turner, J.R. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 2009, 9, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Arrieta, M.C.; Madsen, K.; Doyle, J.; Meddings, J. Reducing small intestinal permeability attenuates colitis in the IL10 gene-deficient mouse. Gut 2009, 58, 41–48. [Google Scholar] [CrossRef]
- Gribble, F.M.; Reimann, F. Function and mechanisms of enteroendocrine cells and gut hormones in metabolism. Nat. Rev. Endocrinol. 2019, 15, 226–237. [Google Scholar] [CrossRef]
- Spreckley, E.; Murphy, K.G. The L-Cell in Nutritional Sensing and the Regulation of Appetite. Front. Nutr. 2015, 2, 23. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.E.; Larsson, J.M.; Hansson, G.C. The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host-microbial interactions. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4659–4665. [Google Scholar] [CrossRef]
- Rogers, D.F. Airway goblet cells: Responsive and adaptable front-line defenders. Eur. Respir. J. 1994, 7, 1690–1706. [Google Scholar] [CrossRef]
- Chang, S.K.; Dohrman, A.F.; Basbaum, C.B.; Ho, S.B.; Tsuda, T.; Toribara, N.W.; Gum, J.R.; Kim, Y.S. Localization of mucin (MUC2 and MUC3) messenger RNA and peptide expression in human normal intestine and colon cancer. Gastroenterology 1994, 107, 28–36. [Google Scholar] [CrossRef]
- Jumblatt, M.M.; McKenzie, R.W.; Jumblatt, J.E. MUC5AC mucin is a component of the human precorneal tear film. Investig. Ophthalmol. Vis. Sci. 1999, 40, 43–49. [Google Scholar]
- Van der Sluis, M.; De Koning, B.A.; De Bruijn, A.C.; Velcich, A.; Meijerink, J.P.; Van Goudoever, J.B.; Buller, H.A.; Dekker, J.; Van Seuningen, I.; Renes, I.B.; et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology 2006, 131, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Propheter, D.C.; Chara, A.L.; Harris, T.A.; Ruhn, K.A.; Hooper, L.V. Resistin-like molecule beta is a bactericidal protein that promotes spatial segregation of the microbiota and the colonic epithelium. Proc. Natl. Acad. Sci. USA 2017, 114, 11027–11033. [Google Scholar] [CrossRef]
- Herbert, D.R.; Yang, J.Q.; Hogan, S.P.; Groschwitz, K.; Khodoun, M.; Munitz, A.; Orekov, T.; Perkins, C.; Wang, Q.; Brombacher, F.; et al. Intestinal epithelial cell secretion of RELM-beta protects against gastrointestinal worm infection. J. Exp. Med. 2009, 206, 2947–2957. [Google Scholar] [CrossRef]
- Morampudi, V.; Dalwadi, U.; Bhinder, G.; Sham, H.P.; Gill, S.K.; Chan, J.; Bergstrom, K.S.; Huang, T.; Ma, C.; Jacobson, K.; et al. The goblet cell-derived mediator RELM-beta drives spontaneous colitis in Muc2-deficient mice by promoting commensal microbial dysbiosis. Mucosal Immunol. 2016, 9, 1218–1233. [Google Scholar] [CrossRef]
- Bergstrom, J.H.; Birchenough, G.M.; Katona, G.; Schroeder, B.O.; Schutte, A.; Ermund, A.; Johansson, M.E.; Hansson, G.C. Gram-positive bacteria are held at a distance in the colon mucus by the lectin-like protein ZG16. Proc. Natl. Acad. Sci. USA 2016, 113, 13833–13838. [Google Scholar] [CrossRef]
- McDole, J.R.; Wheeler, L.W.; McDonald, K.G.; Wang, B.; Konjufca, V.; Knoop, K.A.; Newberry, R.D.; Miller, M.J. Goblet cells deliver luminal antigen to CD103+ dendritic cells in the small intestine. Nature 2012, 483, 345–349. [Google Scholar] [CrossRef]
- Schulz, O.; Jaensson, E.; Persson, E.K.; Liu, X.; Worbs, T.; Agace, W.W.; Pabst, O. Intestinal CD103+, but not CX3CR1+, antigen sampling cells migrate in lymph and serve classical dendritic cell functions. J. Exp. Med. 2009, 206, 3101–3114. [Google Scholar] [CrossRef]
- Knoop, K.A.; Newberry, R.D. Goblet cells: Multifaceted players in immunity at mucosal surfaces. Mucosal Immunol. 2018, 11, 1551–1557. [Google Scholar] [CrossRef]
- Wang, S.L.; Shao, B.Z.; Zhao, S.B.; Fang, J.; Gu, L.; Miao, C.Y.; Li, Z.S.; Bai, Y. Impact of Paneth Cell Autophagy on Inflammatory Bowel Disease. Front. Immunol. 2018, 9, 693. [Google Scholar] [CrossRef]
- Wehkamp, J.; Stange, E.F. Paneth’s disease. J. Crohns Colitis 2010, 4, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, O.H.; Katajisto, P.; Lamming, D.W.; Gultekin, Y.; Bauer-Rowe, K.E.; Sengupta, S.; Birsoy, K.; Dursun, A.; Yilmaz, V.O.; Selig, M.; et al. mTORC1 in the Paneth cell niche couples intestinal stem-cell function to calorie intake. Nature 2012, 486, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; van Es, J.H.; Snippert, H.J.; Stange, D.E.; Vries, R.G.; van den Born, M.; Barker, N.; Shroyer, N.F.; van de Wetering, M.; Clevers, H. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 2011, 469, 415–418. [Google Scholar] [CrossRef] [PubMed]
- van Es, J.H.; Wiebrands, K.; Lopez-Iglesias, C.; van de Wetering, M.; Zeinstra, L.; van den Born, M.; Korving, J.; Sasaki, N.; Peters, P.J.; van Oudenaarden, A.; et al. Enteroendocrine and tuft cells support Lgr5 stem cells on Paneth cell depletion. Proc. Natl. Acad. Sci. USA 2019, 116, 26599–26605. [Google Scholar] [CrossRef]
- Gill, S.R.; Pop, M.; Deboy, R.T.; Eckburg, P.B.; Turnbaugh, P.J.; Samuel, B.S.; Gordon, J.I.; Relman, D.A.; Fraser-Liggett, C.M.; Nelson, K.E. Metagenomic analysis of the human distal gut microbiome. Science 2006, 312, 1355–1359. [Google Scholar] [CrossRef]
- Chung, H.; Pamp, S.J.; Hill, J.A.; Surana, N.K.; Edelman, S.M.; Troy, E.B.; Reading, N.C.; Villablanca, E.J.; Wang, S.; Mora, J.R.; et al. Gut immune maturation depends on colonization with a host-specific microbiota. Cell 2012, 149, 1578–1593. [Google Scholar] [CrossRef]
- Noureldein, M.H.; Eid, A.A. Gut microbiota and mTOR signaling: Insight on a new pathophysiological interaction. Microb. Pathog. 2018, 118, 98–104. [Google Scholar] [CrossRef]
- Round, J.L.; Mazmanian, S.K. The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 2009, 9, 313–323. [Google Scholar] [CrossRef]
- Koenig, J.E.; Spor, A.; Scalfone, N.; Fricker, A.D.; Stombaugh, J.; Knight, R.; Angenent, L.T.; Ley, R.E. Succession of microbial consortia in the developing infant gut microbiome. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4578–4585. [Google Scholar] [CrossRef]
- Mathis, D.; Benoist, C. The influence of the microbiota on type-1 diabetes: On the threshold of a leap forward in our understanding. Immunol. Rev. 2012, 245, 239–249. [Google Scholar] [CrossRef]
- Duerkop, B.A.; Vaishnava, S.; Hooper, L.V. Immune responses to the microbiota at the intestinal mucosal surface. Immunity 2009, 31, 368–376. [Google Scholar] [CrossRef]
- Gardet, A.; Xavier, R.J. Common alleles that influence autophagy and the risk for inflammatory bowel disease. Curr. Opin. Immunol. 2012, 24, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Hunter, M.M.; Wang, A.; Parhar, K.S.; Johnston, M.J.; Van Rooijen, N.; Beck, P.L.; McKay, D.M. In vitro-derived alternatively activated macrophages reduce colonic inflammation in mice. Gastroenterology 2010, 138, 1395–1405. [Google Scholar] [CrossRef] [PubMed]
- Qualls, J.E.; Kaplan, A.M.; van Rooijen, N.; Cohen, D.A. Suppression of experimental colitis by intestinal mononuclear phagocytes. J. Leukoc. Biol. 2006, 80, 802–815. [Google Scholar] [CrossRef] [PubMed]
- Bain, C.C.; Scott, C.L.; Uronen-Hansson, H.; Gudjonsson, S.; Jansson, O.; Grip, O.; Guilliams, M.; Malissen, B.; Agace, W.W.; Mowat, A.M. Resident and pro-inflammatory macrophages in the colon represent alternative context-dependent fates of the same Ly6Chi monocyte precursors. Mucosal Immunol. 2013, 6, 498–510. [Google Scholar] [CrossRef]
- Rugtveit, J.; Nilsen, E.M.; Bakka, A.; Carlsen, H.; Brandtzaeg, P.; Scott, H. Cytokine profiles differ in newly recruited and resident subsets of mucosal macrophages from inflammatory bowel disease. Gastroenterology 1997, 112, 1493–1505. [Google Scholar] [CrossRef]
- Thiesen, S.; Janciauskiene, S.; Uronen-Hansson, H.; Agace, W.; Hogerkorp, C.M.; Spee, P.; Hakansson, K.; Grip, O. CD14(hi)HLA-DR(dim) macrophages, with a resemblance to classical blood monocytes, dominate inflamed mucosa in Crohn’s disease. J. Leukoc. Biol. 2014, 95, 531–541. [Google Scholar] [CrossRef]
- Perminow, G.; Reikvam, D.H.; Lyckander, L.G.; Brandtzaeg, P.; Vatn, M.H.; Carlsen, H.S. Increased number and activation of colonic macrophages in pediatric patients with untreated Crohn’s disease. Inflamm. Bowel Dis. 2009, 15, 1368–1378. [Google Scholar] [CrossRef]
- Kucharzik, T.; Lugering, N.; Rautenberg, K.; Lugering, A.; Schmidt, M.A.; Stoll, R.; Domschke, W. Role of M cells in intestinal barrier function. Ann. N. Y. Acad. Sci. 2000, 915, 171–183. [Google Scholar] [CrossRef]
- Saez, A.; Gomez-Bris, R.; Herrero-Fernandez, B.; Mingorance, C.; Rius, C.; Gonzalez-Granado, J.M. Innate Lymphoid Cells in Intestinal Homeostasis and Inflammatory Bowel Disease. Int. J. Mol. Sci. 2021, 22, 7618. [Google Scholar] [CrossRef]
- Cupedo, T.; Crellin, N.K.; Papazian, N.; Rombouts, E.J.; Weijer, K.; Grogan, J.L.; Fibbe, W.E.; Cornelissen, J.J.; Spits, H. Human fetal lymphoid tissue-inducer cells are interleukin 17-producing precursors to RORC+ CD127+ natural killer-like cells. Nat. Immunol. 2009, 10, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Cella, M.; Fuchs, A.; Vermi, W.; Facchetti, F.; Otero, K.; Lennerz, J.K.; Doherty, J.M.; Mills, J.C.; Colonna, M. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature 2009, 457, 722–725. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.L.; Kasper, D.L. Interactions between the intestinal microbiota and innate lymphoid cells. Gut Microbes 2014, 5, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Wang, L.; Liu, F. Immunological Impact of Intestinal T Cells on Metabolic Diseases. Front. Immunol. 2021, 12, 639902. [Google Scholar] [CrossRef]
- Sun, T.; Nguyen, A.; Gommerman, J.L. Dendritic Cell Subsets in Intestinal Immunity and Inflammation. J. Immunol. 2020, 204, 1075–1083. [Google Scholar] [CrossRef]
- Stagg, A.J. Intestinal Dendritic Cells in Health and Gut Inflammation. Front. Immunol. 2018, 9, 2883. [Google Scholar] [CrossRef]
- Kadowaki, N. Dendritic cells: A conductor of T cell differentiation. Allergol. Int. 2007, 56, 193–199. [Google Scholar] [CrossRef]
- Coombes, J.L.; Siddiqui, K.R.; Arancibia-Carcamo, C.V.; Hall, J.; Sun, C.M.; Belkaid, Y.; Powrie, F. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J. Exp. Med. 2007, 204, 1757–1764. [Google Scholar] [CrossRef]
- Tezuka, H.; Ohteki, T. Regulation of IgA Production by Intestinal Dendritic Cells and Related Cells. Front. Immunol. 2019, 10, 1891. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Yu, X.; Feng, J. Iron homeostasis disorder in piglet intestine. Metallomics 2020, 12, 1494–1507. [Google Scholar] [CrossRef]
- Pereira, P.M.; Vicente, A.F. Meat nutritional composition and nutritive role in the human diet. Meat Sci. 2013, 93, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Simpson, R.J.; McKie, A.T. Regulation of intestinal iron absorption: The mucosa takes control? Cell Metab. 2009, 10, 84–87. [Google Scholar] [CrossRef] [PubMed]
- McKie, A.T.; Barrow, D.; Latunde-Dada, G.O.; Rolfs, A.; Sager, G.; Mudaly, E.; Mudaly, M.; Richardson, C.; Barlow, D.; Bomford, A.; et al. An iron-regulated ferric reductase associated with the absorption of dietary iron. Science 2001, 291, 1755–1759. [Google Scholar] [CrossRef]
- Kuhn, L.C. Iron regulatory proteins and their role in controlling iron metabolism. Metallomics 2015, 7, 232–243. [Google Scholar] [CrossRef]
- Chen, H.; Attieh, Z.K.; Su, T.; Syed, B.A.; Gao, H.; Alaeddine, R.M.; Fox, T.C.; Usta, J.; Naylor, C.E.; Evans, R.W.; et al. Hephaestin is a ferroxidase that maintains partial activity in sex-linked anemia mice. Blood 2004, 103, 3933–3939. [Google Scholar] [CrossRef]
- Chen, A.C.; Donovan, A.; Ned-Sykes, R.; Andrews, N.C. Noncanonical role of transferrin receptor 1 is essential for intestinal homeostasis. Proc. Natl. Acad. Sci. USA 2015, 112, 11714–11719. [Google Scholar] [CrossRef]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [PubMed]
- Theil, E.C.; Chen, H.; Miranda, C.; Janser, H.; Elsenhans, B.; Nunez, M.T.; Pizarro, F.; Schumann, K. Absorption of iron from ferritin is independent of heme iron and ferrous salts in women and rat intestinal segments. J. Nutr. 2012, 142, 478–483. [Google Scholar] [CrossRef]
- Andrews, S.C.; Robinson, A.K.; Rodriguez-Quinones, F. Bacterial iron homeostasis. FEMS Microbiol. Rev. 2003, 27, 215–237. [Google Scholar] [CrossRef]
- Yilmaz, B.; Li, H. Gut Microbiota and Iron: The Crucial Actors in Health and Disease. Pharmaceuticals 2018, 11, 98. [Google Scholar] [CrossRef]
- Skaar, E.P. The battle for iron between bacterial pathogens and their vertebrate hosts. PLoS Pathog. 2010, 6, e1000949. [Google Scholar] [CrossRef] [PubMed]
- Neilands, J.B. Siderophores: Structure and function of microbial iron transport compounds. J. Biol. Chem. 1995, 270, 26723–26726. [Google Scholar] [CrossRef]
- Wandersman, C.; Stojiljkovic, I. Bacterial heme sources: The role of heme, hemoprotein receptors and hemophores. Curr. Opin. Microbiol. 2000, 3, 215–220. [Google Scholar] [CrossRef]
- Stojiljkovic, I.; Hantke, K. Transport of haemin across the cytoplasmic membrane through a haemin-specific periplasmic binding-protein-dependent transport system in Yersinia enterocolitica. Mol. Microbiol. 1994, 13, 719–732. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Francis, J.; Doster, R.S.; Haley, K.P.; Craft, K.M.; Moore, R.E.; Chambers, S.A.; Aronoff, D.M.; Osteen, K.; Damo, S.M.; et al. Lactoferrin: A Critical Mediator of Both Host Immune Response and Antimicrobial Activity in Response to Streptococcal Infections. ACS Infect. Dis. 2020, 6, 1615–1623. [Google Scholar] [CrossRef] [PubMed]
- Troxell, B.; Hassan, H.M. Transcriptional regulation by Ferric Uptake Regulator (Fur) in pathogenic bacteria. Front. Cell Infect. Microbiol. 2013, 3, 59. [Google Scholar] [CrossRef] [PubMed]
- Fleming, R.E.; Ponka, P. Iron overload in human disease. N. Engl. J. Med. 2012, 366, 348–359. [Google Scholar] [CrossRef]
- Fang, S.; Zhuo, Z.; Yu, X.; Wang, H.; Feng, J. Oral administration of liquid iron preparation containing excess iron induces intestine and liver injury, impairs intestinal barrier function and alters the gut microbiota in rats. J. Trace Elem. Med. Biol. 2018, 47, 12–20. [Google Scholar] [CrossRef]
- Fuqua, B.K.; Vulpe, C.D.; Anderson, G.J. Intestinal iron absorption. J. Trace Elem. Med. Biol. 2012, 26, 115–119. [Google Scholar] [CrossRef]
- Li, Y.; Hansen, S.L.; Borst, L.B.; Spears, J.W.; Moeser, A.J. Dietary Iron Deficiency and Oversupplementation Increase Intestinal Permeability, Ion Transport, and Inflammation in Pigs. J. Nutr. 2016, 146, 1499–1505. [Google Scholar] [CrossRef]
- Tenenbein, M.; Littman, C.; Stimpson, R.E. Gastrointestinal pathology in adult iron overdose. J. Toxicol. Clin. Toxicol. 1990, 28, 311–320. [Google Scholar] [CrossRef]
- Gupta, A. Ferric citrate hydrate as a phosphate binder and risk of aluminum toxicity. Pharmaceuticals 2014, 7, 990–998. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.M.; Eng, R.H.; Campos, J.M.; Chmel, H. D-lactic acid measurements in the diagnosis of bacterial infections. J. Clin. Microbiol. 1989, 27, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Li, R.; Liu, Y.; Liu, M.; Liang, H. Protective Effect of Aplysin Supplementation on Intestinal Permeability and Microbiota in Rats Treated with Ethanol and Iron. Nutrients 2018, 10, 681. [Google Scholar] [CrossRef] [PubMed]
- Flint, H.J.; Duncan, S.H.; Scott, K.P.; Louis, P. Interactions and competition within the microbial community of the human colon: Links between diet and health. Environ. Microbiol. 2007, 9, 1101–1111. [Google Scholar] [CrossRef]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef]
- Lee, S.H.; Shinde, P.; Choi, J.; Park, M.; Ohh, S.; Kwon, I.K.; Pak, S.I.; Chae, B.J. Effects of dietary iron levels on growth performance, hematological status, liver mineral concentration, fecal microflora, and diarrhea incidence in weanling pigs. Biol. Trace Elem. Res. 2008, 126 (Suppl. 1), S57–S68. [Google Scholar] [CrossRef]
- Tompkins, G.R.; O’Dell, N.L.; Bryson, I.T.; Pennington, C.B. The effects of dietary ferric iron and iron deprivation on the bacterial composition of the mouse intestine. Curr. Microbiol. 2001, 43, 38–42. [Google Scholar] [CrossRef]
- Jaeggi, T.; Kortman, G.A.; Moretti, D.; Chassard, C.; Holding, P.; Dostal, A.; Boekhorst, J.; Timmerman, H.M.; Swinkels, D.W.; Tjalsma, H.; et al. Iron fortification adversely affects the gut microbiome, increases pathogen abundance and induces intestinal inflammation in Kenyan infants. Gut 2015, 64, 731–742. [Google Scholar] [CrossRef]
- Zimmermann, M.B.; Chassard, C.; Rohner, F.; N’Goran, E.K.; Nindjin, C.; Dostal, A.; Utzinger, J.; Ghattas, H.; Lacroix, C.; Hurrell, R.F. The effects of iron fortification on the gut microbiota in African children: A randomized controlled trial in Cote d’Ivoire. Am. J. Clin. Nutr. 2010, 92, 1406–1415. [Google Scholar] [CrossRef]
- Qi, X.; Zhang, Y.; Guo, H.; Hai, Y.; Luo, Y.; Yue, T. Mechanism and intervention measures of iron side effects on the intestine. Crit. Rev. Food Sci. Nutr. 2020, 60, 2113–2125. [Google Scholar] [CrossRef] [PubMed]
- Kortman, G.A.; Boleij, A.; Swinkels, D.W.; Tjalsma, H. Iron availability increases the pathogenic potential of Salmonella typhimurium and other enteric pathogens at the intestinal epithelial interface. PLoS ONE 2012, 7, e29968. [Google Scholar] [CrossRef] [PubMed]
- Lia, M.; Yin, Q.; Dang, X.; Chang, J.; Zuo, R.; Zheng, Q. Effect of different iron loads on serum and tissue biochemical parameters and liver hepcidin mRNA abundance of neonatal piglets. Arch. Anim. Nutr. 2011, 65, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Stuart, K.A.; Anderson, G.J.; Frazer, D.M.; Powell, L.W.; McCullen, M.; Fletcher, L.M.; Crawford, D.H. Duodenal expression of iron transport molecules in untreated haemochromatosis subjects. Gut 2003, 52, 953–959. [Google Scholar] [CrossRef]
- Mete, A.; Jalving, R.; van Oost, B.A.; van Dijk, J.E.; Marx, J.J. Intestinal over-expression of iron transporters induces iron overload in birds in captivity. Blood Cells Mol. Dis. 2005, 34, 151–156. [Google Scholar] [CrossRef]
- Canonne-Hergaux, F.; Levy, J.E.; Fleming, M.D.; Montross, L.K.; Andrews, N.C.; Gros, P. Expression of the DMT1 (NRAMP2/DCT1) iron transporter in mice with genetic iron overload disorders. Blood 2001, 97, 1138–1140. [Google Scholar] [CrossRef]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef]
- Nakamura, T.; Naguro, I.; Ichijo, H. Iron homeostasis and iron-regulated ROS in cell death, senescence and human diseases. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 1398–1409. [Google Scholar] [CrossRef]
- Mantzaris, M.D.; Bellou, S.; Skiada, V.; Kitsati, N.; Fotsis, T.; Galaris, D. Intracellular labile iron determines H2O2-induced apoptotic signaling via sustained activation of ASK1/JNK-p38 axis. Free Radic. Biol. Med. 2016, 97, 454–465. [Google Scholar] [CrossRef]
- Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S.M.; Ahmad, M.; Alnemri, E.S.; Wang, X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997, 91, 479–489. [Google Scholar] [CrossRef]
- Conrad, M.; Angeli, J.P.; Vandenabeele, P.; Stockwell, B.R. Regulated necrosis: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2016, 15, 348–366. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017, 547, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Angeli, J.P.F.; Shah, R.; Pratt, D.A.; Conrad, M. Ferroptosis Inhibition: Mechanisms and Opportunities. Trends Pharmacol. Sci. 2017, 38, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Hirschhorn, T.; Stockwell, B.R. The development of the concept of ferroptosis. Free Radic. Biol. Med. 2019, 133, 130–143. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef]
- Ng, S.W.; Norwitz, S.G.; Norwitz, E.R. The Impact of Iron Overload and Ferroptosis on Reproductive Disorders in Humans: Implications for Preeclampsia. Int. J. Mol. Sci. 2019, 20, 3283. [Google Scholar] [CrossRef]
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Porto Freitas, F.; Seibt, T.; et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 2018, 172, 409–422.e21. [Google Scholar] [CrossRef]
- Ye, Z.; Liu, W.; Zhuo, Q.; Hu, Q.; Liu, M.; Sun, Q.; Zhang, Z.; Fan, G.; Xu, W.; Ji, S.; et al. Ferroptosis: Final destination for cancer? Cell Prolif. 2020, 53, e12761. [Google Scholar] [CrossRef]
- Gao, M.; Jiang, X. To eat or not to eat-the metabolic flavor of ferroptosis. Curr. Opin. Cell Biol. 2018, 51, 58–64. [Google Scholar] [CrossRef]
- Feng, H.; Stockwell, B.R. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol. 2018, 16, e2006203. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Qu, M.; Zhang, H.; Chen, Z.; Sun, X.; Zhu, S.; Nan, K.; Chen, W.; Miao, C. The Role of Ferroptosis in Acute Respiratory Distress Syndrome. Front. Med. 2021, 8, 651552. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Tao, J.; Yang, Y.; Tan, S.; Liu, H.; Jiang, J.; Zheng, F.; Wu, B. Ferroptosis involves in intestinal epithelial cell death in ulcerative colitis. Cell Death Dis. 2020, 11, 86. [Google Scholar] [CrossRef]
- Wang, S.; Liu, W.; Wang, J.; Bai, X. Curculigoside inhibits ferroptosis in ulcerative colitis through the induction of GPX4. Life Sci. 2020, 259, 118356. [Google Scholar] [CrossRef]
- Mayr, L.; Grabherr, F.; Schwarzler, J.; Reitmeier, I.; Sommer, F.; Gehmacher, T.; Niederreiter, L.; He, G.W.; Ruder, B.; Kunz, K.T.R.; et al. Dietary lipids fuel GPX4-restricted enteritis resembling Crohn’s disease. Nat. Commun. 2020, 11, 1775. [Google Scholar] [CrossRef]
- Xu, C.; Liu, Z.; Xiao, J. Ferroptosis: A Double-Edged Sword in Gastrointestinal Disease. Int. J. Mol. Sci. 2021, 22, 12403. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, P.; Chen, W.; Chen, G. Ferroptosis mediated DSS-induced ulcerative colitis associated with Nrf2/HO-1 signaling pathway. Immunol. Lett. 2020, 225, 9–15. [Google Scholar] [CrossRef]
- Ito, M.; Tanaka, T.; Nangaku, M. Nuclear factor erythroid 2-related factor 2 as a treatment target of kidney diseases. Curr. Opin. Nephrol. Hypertens. 2020, 29, 128–135. [Google Scholar] [CrossRef]
- Chang, L.C.; Chiang, S.K.; Chen, S.E.; Yu, Y.L.; Chou, R.H.; Chang, W.C. Heme oxygenase-1 mediates BAY 11-7085 induced ferroptosis. Cancer Lett. 2018, 416, 124–137. [Google Scholar] [CrossRef]
- Adedoyin, O.; Boddu, R.; Traylor, A.; Lever, J.M.; Bolisetty, S.; George, J.F.; Agarwal, A. Heme oxygenase-1 mitigates ferroptosis in renal proximal tubule cells. Am. J. Physiol. Renal Physiol. 2018, 314, F702–F714. [Google Scholar] [CrossRef]
- Arnold, M.; Sierra, M.S.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global patterns and trends in colorectal cancer incidence and mortality. Gut 2017, 66, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zhu, S.; Song, X.; Sun, X.; Fan, Y.; Liu, J.; Zhong, M.; Yuan, H.; Zhang, L.; Billiar, T.R.; et al. The Tumor Suppressor p53 Limits Ferroptosis by Blocking DPP4 Activity. Cell Rep. 2017, 20, 1692–1704. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Zhang, R.; Liu, S.; Duan, T.; Zhai, L.; Zhang, M.; Han, X.; Xiang, Y.; Huang, X.; Lin, H.; et al. RSL3 Drives Ferroptosis Through GPX4 Inactivation and ROS Production in Colorectal Cancer. Front. Pharmacol. 2018, 9, 1371. [Google Scholar] [CrossRef]
- Guo, J.; Xu, B.; Han, Q.; Zhou, H.; Xia, Y.; Gong, C.; Dai, X.; Li, Z.; Wu, G. Ferroptosis: A Novel Anti-tumor Action for Cisplatin. Cancer Res. Treat. 2018, 50, 445–460. [Google Scholar] [CrossRef]
- Shen, L.D.; Qi, W.H.; Bai, J.J.; Zuo, C.Y.; Bai, D.L.; Gao, W.D.; Zong, X.L.; Hao, T.T.; Ma, Y.; Cao, G.C. Resibufogenin inhibited colorectal cancer cell growth and tumorigenesis through triggering ferroptosis and ROS production mediated by GPX4 inactivation. Anat. Rec. 2021, 304, 313–322. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, W.; Liu, F.; Wang, Q.; Song, M.; Yu, Q.; Tang, K.; Teng, T.; Wu, D.; Wang, X.; et al. IMCA Induces Ferroptosis Mediated by SLC7A11 through the AMPK/mTOR Pathway in Colorectal Cancer. Oxidative Med. Cell. Longev. 2020, 2020, 1675613. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, X.; Wei, C.; Zheng, D.; Lu, X.; Yang, Y.; Luo, A.; Zhang, K.; Duan, X.; Wang, Y. Targeting SLC7A11 specifically suppresses the progression of colorectal cancer stem cells via inducing ferroptosis. Eur. J. Pharm. Sci. 2020, 152, 105450. [Google Scholar] [CrossRef]
- Acquaviva, R.; Sorrenti, V.; Santangelo, R.; Cardile, V.; Tomasello, B.; Malfa, G.; Vanella, L.; Amodeo, A.; Genovese, C.; Mastrojeni, S.; et al. Effects of an extract of Celtis aetnensis (Tornab.) Strobl twigs on human colon cancer cell cultures. Oncol. Rep. 2016, 36, 2298–2304. [Google Scholar] [CrossRef]
- Busserolles, J.; Megias, J.; Terencio, M.C.; Alcaraz, M.J. Heme oxygenase-1 inhibits apoptosis in Caco-2 cells via activation of Akt pathway. Int. J. Biochem. Cell Biol. 2006, 38, 1510–1517. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Alam, J.; Choi, A.M. Heme oxygenase-1/carbon monoxide: From basic science to therapeutic applications. Physiol. Rev. 2006, 86, 583–650. [Google Scholar] [CrossRef]
- Loboda, A.; Jozkowicz, A.; Dulak, J. HO-1/CO system in tumor growth, angiogenesis and metabolism—Targeting HO-1 as an anti-tumor therapy. Vascul Pharmacol. 2015, 74, 11–22. [Google Scholar] [CrossRef]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef] [PubMed]
- Nitti, M.; Piras, S.; Marinari, U.M.; Moretta, L.; Pronzato, M.A.; Furfaro, A.L. HO-1 Induction in Cancer Progression: A Matter of Cell Adaptation. Antioxidants 2017, 6, 29. [Google Scholar] [CrossRef] [PubMed]
- Hassannia, B.; Wiernicki, B.; Ingold, I.; Qu, F.; Van Herck, S.; Tyurina, Y.Y.; Bayir, H.; Abhari, B.A.; Angeli, J.P.F.; Choi, S.M.; et al. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J. Clin. Investig. 2018, 128, 3341–3355. [Google Scholar] [CrossRef] [PubMed]
- Malfa, G.A.; Tomasello, B.; Acquaviva, R.; Genovese, C.; La Mantia, A.; Cammarata, F.P.; Ragusa, M.; Renis, M.; Di Giacomo, C. Betula etnensis Raf. (Betulaceae) Extract Induced HO-1 Expression and Ferroptosis Cell Death in Human Colon Cancer Cells. Int. J. Mol. Sci. 2019, 20, 2723. [Google Scholar] [CrossRef]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef]
- Park, S.; Oh, J.; Kim, M.; Jin, E.J. Bromelain effectively suppresses Kras-mutant colorectal cancer by stimulating ferroptosis. Anim. Cells Syst. 2018, 22, 334–340. [Google Scholar] [CrossRef]
- Xu, L.; Li, T.; Chen, Q.; Liu, Z.; Chen, Y.; Hu, K.; Zhang, X. The alpha2AR/Caveolin-1/p38MAPK/NF-kappaB axis explains dexmedetomidine protection against lung injury following intestinal ischaemia-reperfusion. J. Cell Mol. Med. 2021, 25, 6361–6372. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhou, Y.; Gu, X.; Zhang, X.; Jia, Z. NLRX1/FUNDC1/NIPSNAP1-2 axis regulates mitophagy and alleviates intestinal ischaemia/reperfusion injury. Cell Prolif. 2021, 54, e12986. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Cui, R.; Wang, C.; Feng, Y.; Li, Z.; Tong, Y.; Qu, K.; Liu, C.; Zhang, J. Metformin protects against intestinal ischemia-reperfusion injury and cell pyroptosis via TXNIP-NLRP3-GSDMD pathway. Redox Biol. 2020, 32, 101534. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Sun, R.; Wang, G.; Chen, Z.; Li, Y.; Zhao, Y.; Liu, D.; Zhao, H.; Zhang, F.; Yao, J.; et al. SIRT3-mediated deacetylation of PRDX3 alleviates mitochondrial oxidative damage and apoptosis induced by intestinal ischemia/reperfusion injury. Redox Biol. 2020, 28, 101343. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Xu, B.; Sun, Y.; Lian, M.; Li, Y.; Lin, Y.; Chen, D.; Diao, Y.; Almoiliqy, M.; Wang, L. Paeoniflorin protects against intestinal ischemia/reperfusion by activating LKB1/AMPK and promoting autophagy. Pharmacol. Res. 2019, 146, 104308. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Mao, Z.; Xu, L.; Yin, L.; Tao, X.; Tang, Z.; Qi, Y.; Sun, P.; Peng, J. Protective effect of dioscin against intestinal ischemia/reperfusion injury via adjusting miR-351-5p-mediated oxidative stress. Pharmacol. Res. 2018, 137, 56–63. [Google Scholar] [CrossRef]
- Stefanutti, G.; Pierro, A.; Parkinson, E.J.; Smith, V.V.; Eaton, S. Moderate hypothermia as a rescue therapy against intestinal ischemia and reperfusion injury in the rat. Crit. Care Med. 2008, 36, 1564–1572. [Google Scholar] [CrossRef]
- Li, Y.; Cao, Y.; Xiao, J.; Shang, J.; Tan, Q.; Ping, F.; Huang, W.; Wu, F.; Zhang, H.; Zhang, X. Inhibitor of apoptosis-stimulating protein of p53 inhibits ferroptosis and alleviates intestinal ischemia/reperfusion-induced acute lung injury. Cell Death Differ. 2020, 27, 2635–2650. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef]
- Borella, E.; Oosterholt, S.; Magni, P.; Della Pasqua, O. Characterisation of individual ferritin response in patients receiving chelation therapy. Br. J. Clin. Pharmacol. 2022, 88, 3683–3694. [Google Scholar] [CrossRef]
- Di Maggio, R.; Maggio, A. The new era of chelation treatments: Effectiveness and safety of 10 different regimens for controlling iron overloading in thalassaemia major. Br. J. Haematol. 2017, 178, 676–688. [Google Scholar] [CrossRef]
- Williams, A.; Meyer, D. Desferrioxamine as immunomodulatory agent during microorganism infection. Curr. Pharm. Des. 2009, 15, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues de Morais, T.; Gambero, A. Iron chelators in obesity therapy—Old drugs from a new perspective? Eur. J. Pharmacol. 2019, 861, 172614. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Han, N.R.; Kim, H.M.; Jeong, H.J. The Iron Chelator and Anticancer Agent Dp44mT Relieves Allergic Inflammation in Mice With Allergic Rhinitis. Inflammation 2018, 41, 1744–1754. [Google Scholar] [CrossRef]
- Choi, E.Y.; Lee, S.; Oh, H.M.; Kim, Y.D.; Choi, E.J.; Kim, S.H.; Kim, S.W.; Choi, S.C.; Jun, C.D. Involvement of protein kinase Cdelta in iron chelator-induced IL-8 production in human intestinal epithelial cells. Life Sci. 2007, 80, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Choi, S.C.; Choi, E.Y.; Lee, M.H.; Seo, G.S.; Kim, E.C.; Yang, B.J.; Lee, M.S.; Shin, Y.I.; Park, K.I.; et al. Iron chelator induces MIP-alpha/CCL20 in human intestinal epithelial cells: Implication for triggering mucosal adaptive immunity. Exp. Mol. Med. 2005, 37, 297–310. [Google Scholar] [CrossRef]
- Millar, A.D.; Rampton, D.S.; Blake, D.R. Effects of iron and iron chelation in vitro on mucosal oxidant activity in ulcerative colitis. Aliment. Pharmacol. Ther. 2000, 14, 1163–1168. [Google Scholar] [CrossRef] [PubMed]
- Kannan, N.; Guruvayoorappan, C. Protective effect of Bauhinia tomentosa on acetic acid induced ulcerative colitis by regulating antioxidant and inflammatory mediators. Int. Immunopharmacol. 2013, 16, 57–66. [Google Scholar] [CrossRef]
- Trivedi, P.P.; Jena, G.B. Melatonin reduces ulcerative colitis-associated local and systemic damage in mice: Investigation on possible mechanisms. Dig. Dis. Sci. 2013, 58, 3460–3474. [Google Scholar] [CrossRef]
- Marnett, L.J. The COXIB experience: A look in the rearview mirror. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 265–290. [Google Scholar] [CrossRef]
- Tian, T.; Wang, Z.; Zhang, J. Pathomechanisms of Oxidative Stress in Inflammatory Bowel Disease and Potential Antioxidant Therapies. Oxidative Med. Cell. Longev. 2017, 2017, 4535194. [Google Scholar] [CrossRef]
- Terry, P.D.; Villinger, F.; Bubenik, G.A.; Sitaraman, S.V. Melatonin and ulcerative colitis: Evidence, biological mechanisms, and future research. Inflamm. Bowel Dis. 2009, 15, 134–140. [Google Scholar] [CrossRef]
- Arab, H.H.; Salama, S.A.; Eid, A.H.; Omar, H.A.; Arafa el, S.A.; Maghrabi, I.A. Camel’s milk ameliorates TNBS-induced colitis in rats via downregulation of inflammatory cytokines and oxidative stress. Food Chem. Toxicol. 2014, 69, 294–302. [Google Scholar] [CrossRef]
- Sun, Y.; Chen, P.; Zhai, B.; Zhang, M.; Xiang, Y.; Fang, J.; Xu, S.; Gao, Y.; Chen, X.; Sui, X.; et al. The emerging role of ferroptosis in inflammation. Biomed. Pharmacother. 2020, 127, 110108. [Google Scholar] [CrossRef]
- Linkermann, A.; Stockwell, B.R.; Krautwald, S.; Anders, H.J. Regulated cell death and inflammation: An auto-amplification loop causes organ failure. Nat. Rev. Immunol. 2014, 14, 759–767. [Google Scholar] [CrossRef]
- Masaldan, S.; Belaidi, A.A.; Ayton, S.; Bush, A.I. Cellular Senescence and Iron Dyshomeostasis in Alzheimer’s Disease. Pharmaceuticals 2019, 12, 93. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huo, C.; Li, G.; Hu, Y.; Sun, H. The Impacts of Iron Overload and Ferroptosis on Intestinal Mucosal Homeostasis and Inflammation. Int. J. Mol. Sci. 2022, 23, 14195. https://doi.org/10.3390/ijms232214195
Huo C, Li G, Hu Y, Sun H. The Impacts of Iron Overload and Ferroptosis on Intestinal Mucosal Homeostasis and Inflammation. International Journal of Molecular Sciences. 2022; 23(22):14195. https://doi.org/10.3390/ijms232214195
Chicago/Turabian StyleHuo, Caiyun, Guiping Li, Yanxin Hu, and Huiling Sun. 2022. "The Impacts of Iron Overload and Ferroptosis on Intestinal Mucosal Homeostasis and Inflammation" International Journal of Molecular Sciences 23, no. 22: 14195. https://doi.org/10.3390/ijms232214195
APA StyleHuo, C., Li, G., Hu, Y., & Sun, H. (2022). The Impacts of Iron Overload and Ferroptosis on Intestinal Mucosal Homeostasis and Inflammation. International Journal of Molecular Sciences, 23(22), 14195. https://doi.org/10.3390/ijms232214195