
Epigenetics and Pregnancy: Conditional Snapshot or Rolling Event



Abstract
1. Epigenetic Reorganization during Pregnancy and Embryogenesis Processes
1.1. Epigenetics
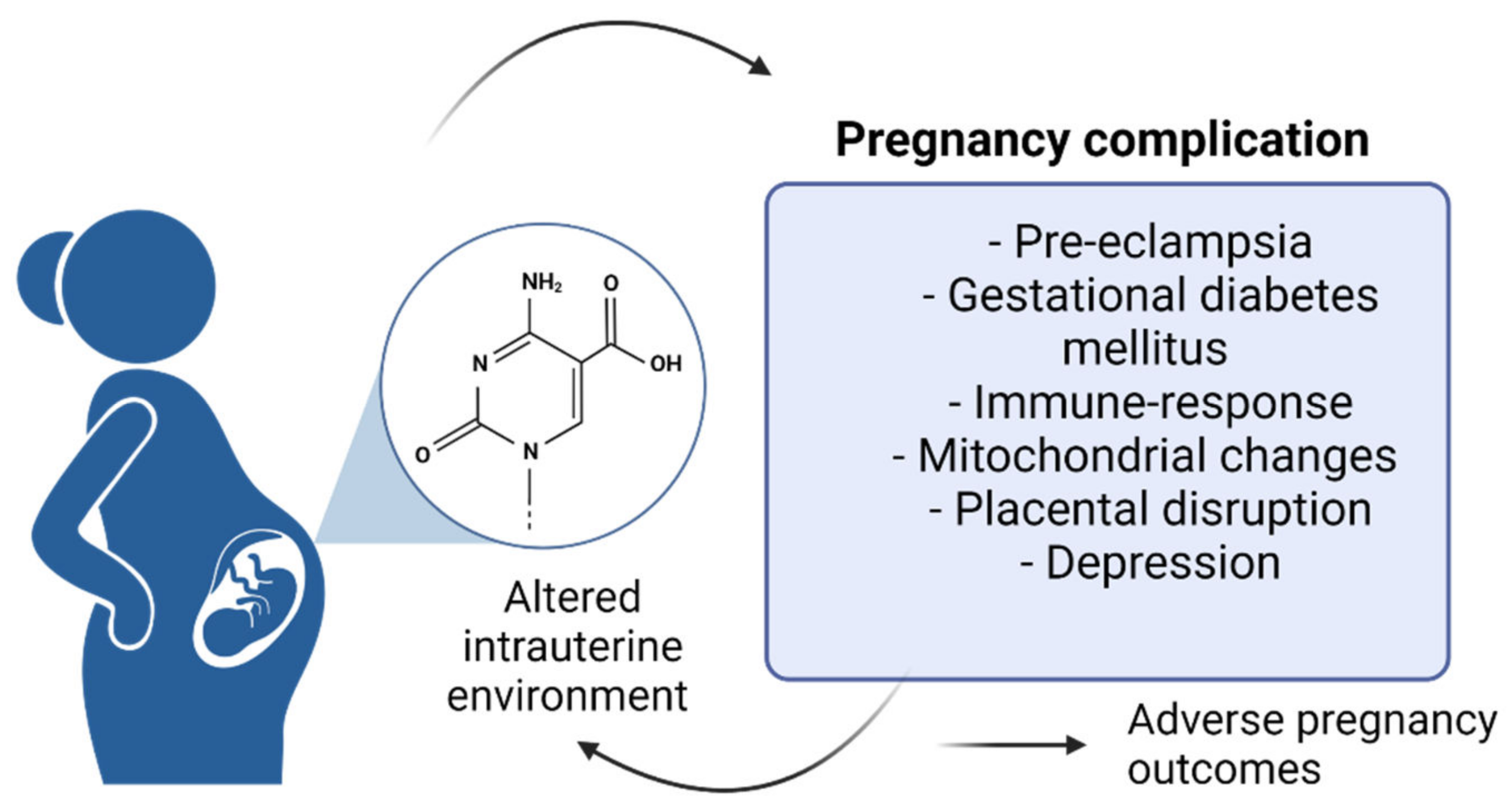
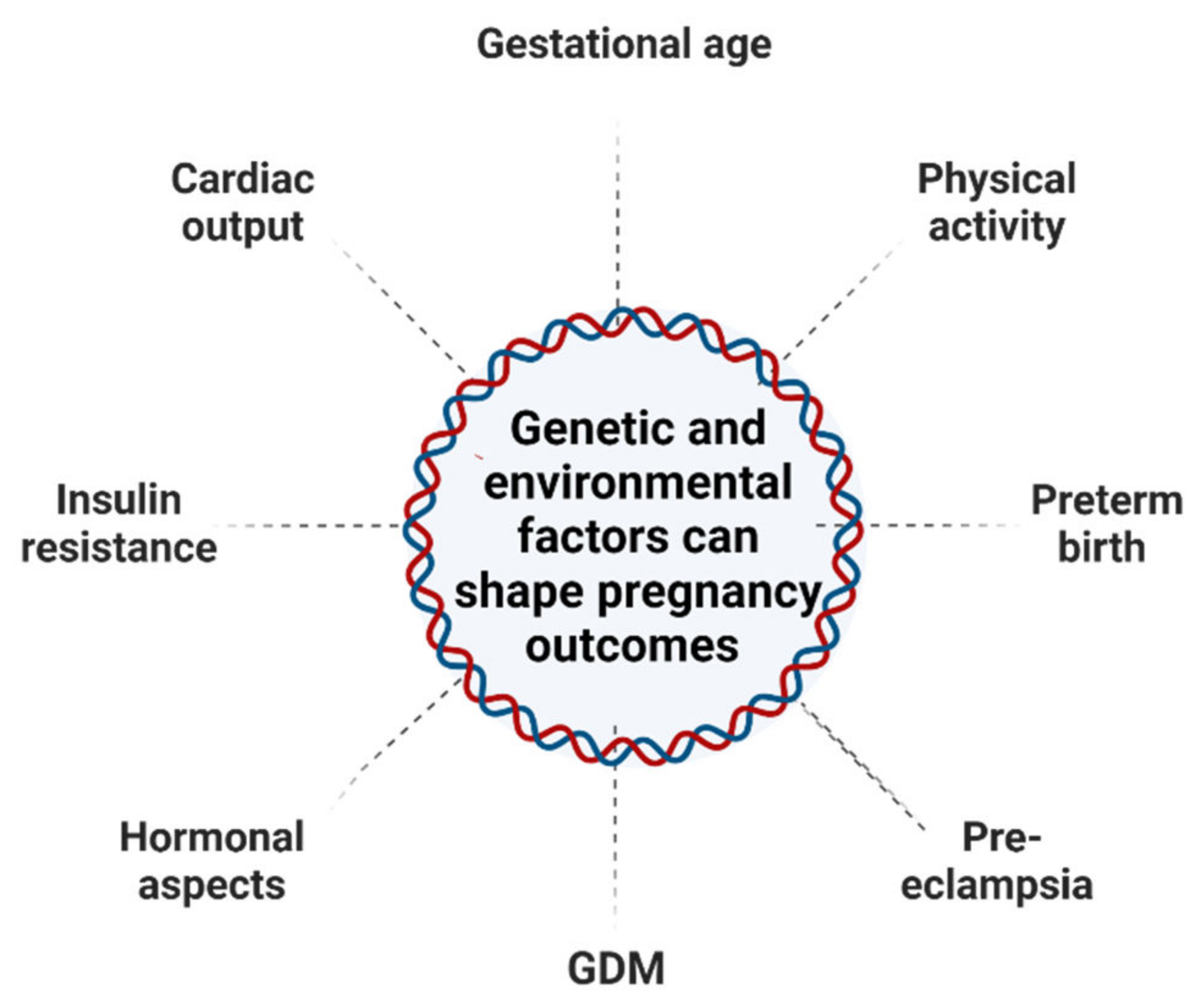
1.2. Pregnancy and Epigenetic Changes
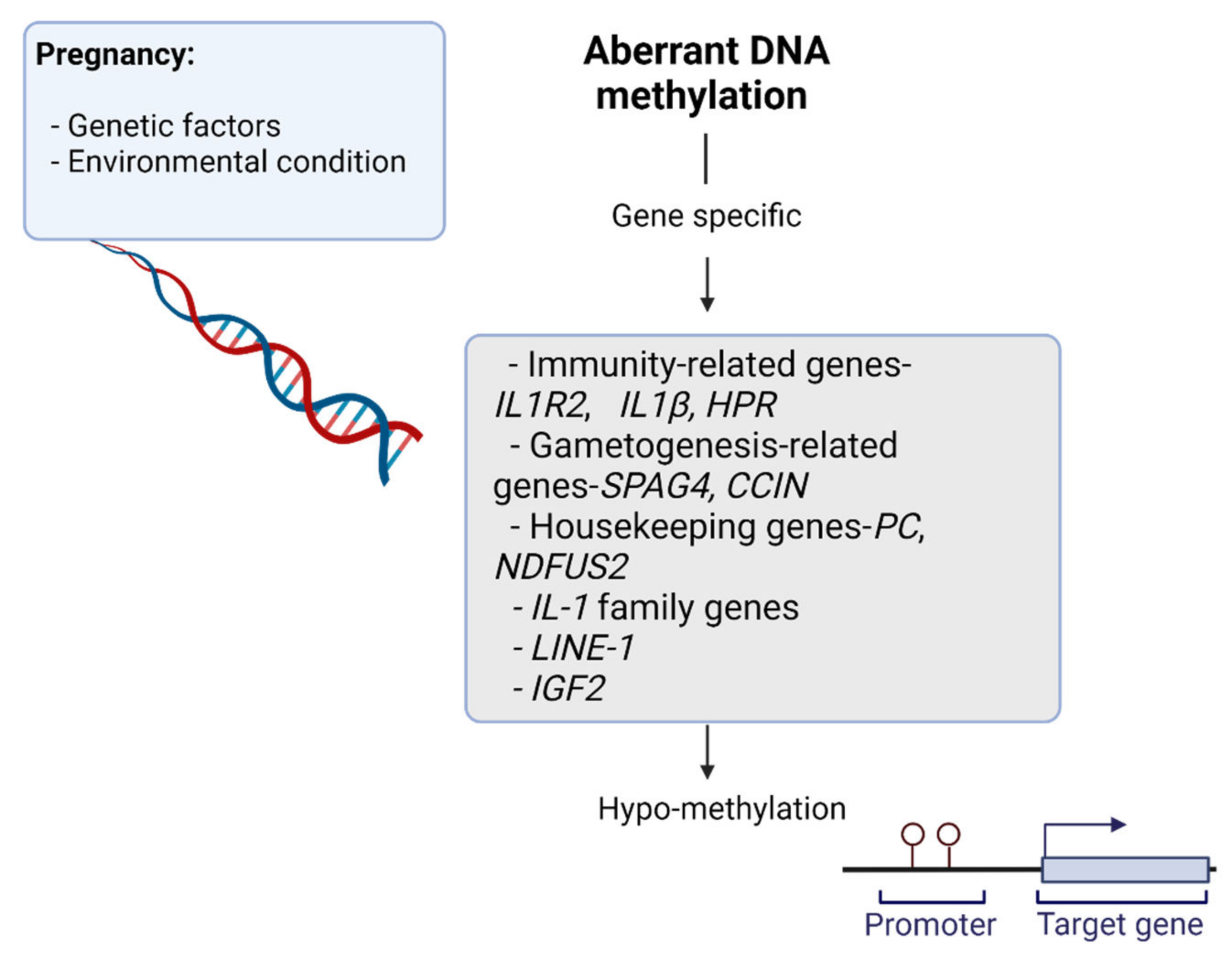
1.2.1. DNA Methylation
1.2.2. Pregnancy and the Immune System
1.2.3. Pregnancy and Stress
1.2.4. Pregnancy and Histone Modification
1.2.5. Pregnancy and Non-Coding RNA
1.3. Embryo Development
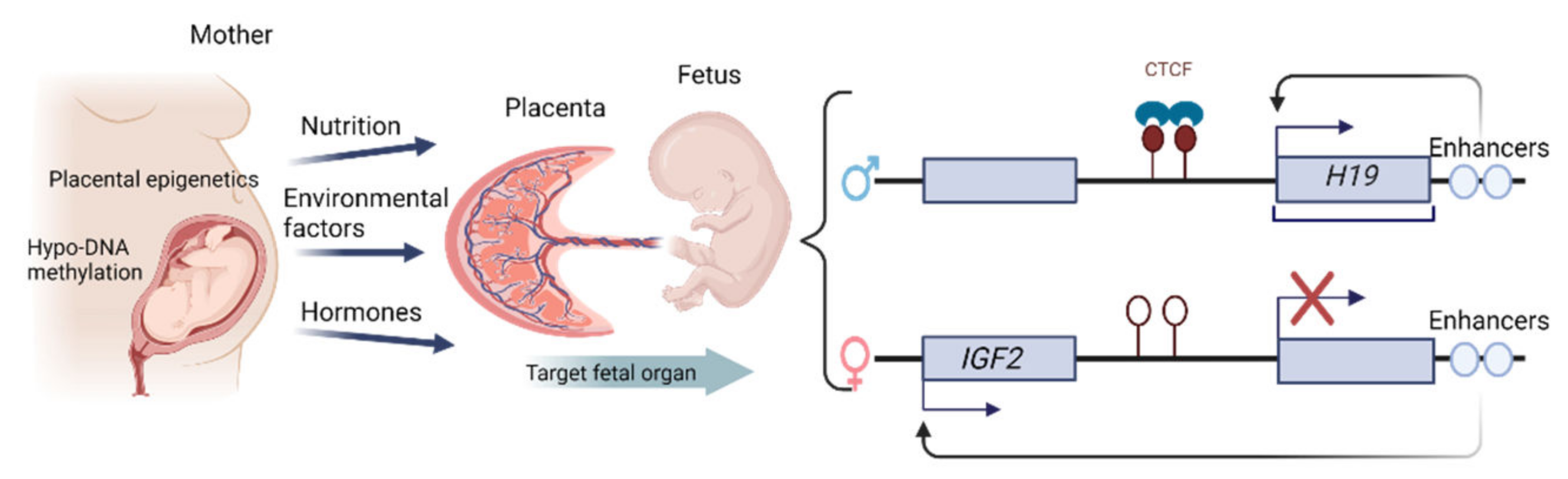
1.4. Epigenetic Changes in the Placenta
2. Epigenetic Changes Characterizing Maternal Pregnancy-Associated Disorders
2.1. The Epigenetics of Pre-Eclampsia
2.2. DNA Methylation in Placental Pathology and Senescence
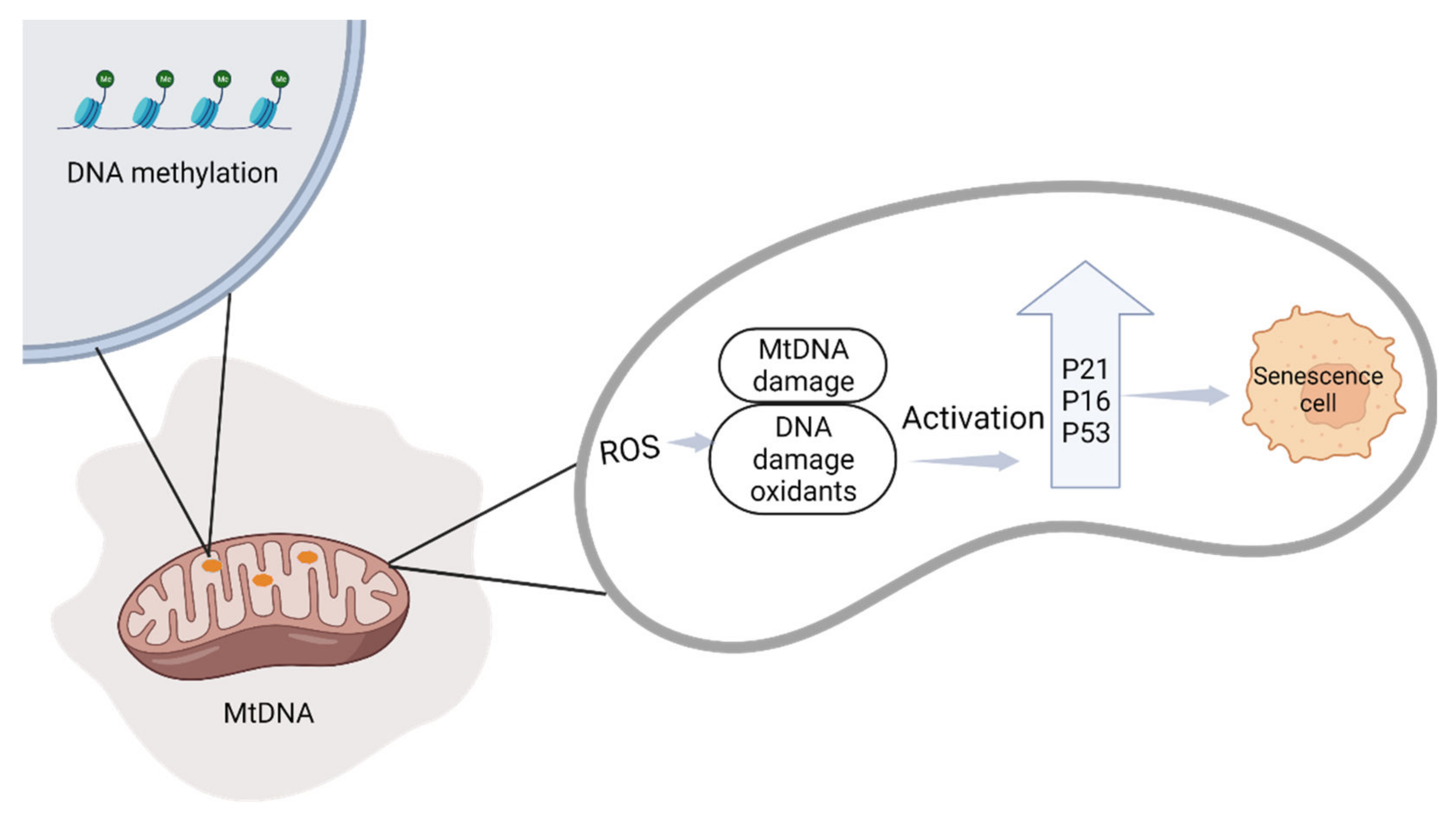
2.2.1. Mitochondrial Changes during Pregnancy
2.2.2. The Relationship between Cellular Senescence and Adverse Pregnancy Outcomes
2.3. Epigenetic Modifications Associated with Obesity and Gestational Diabetes
2.4. Epigenetic Modifications and Prenatal Maternal Depression
2.5. Nutrition as an Epigenetic Stimulus during Pregnancy
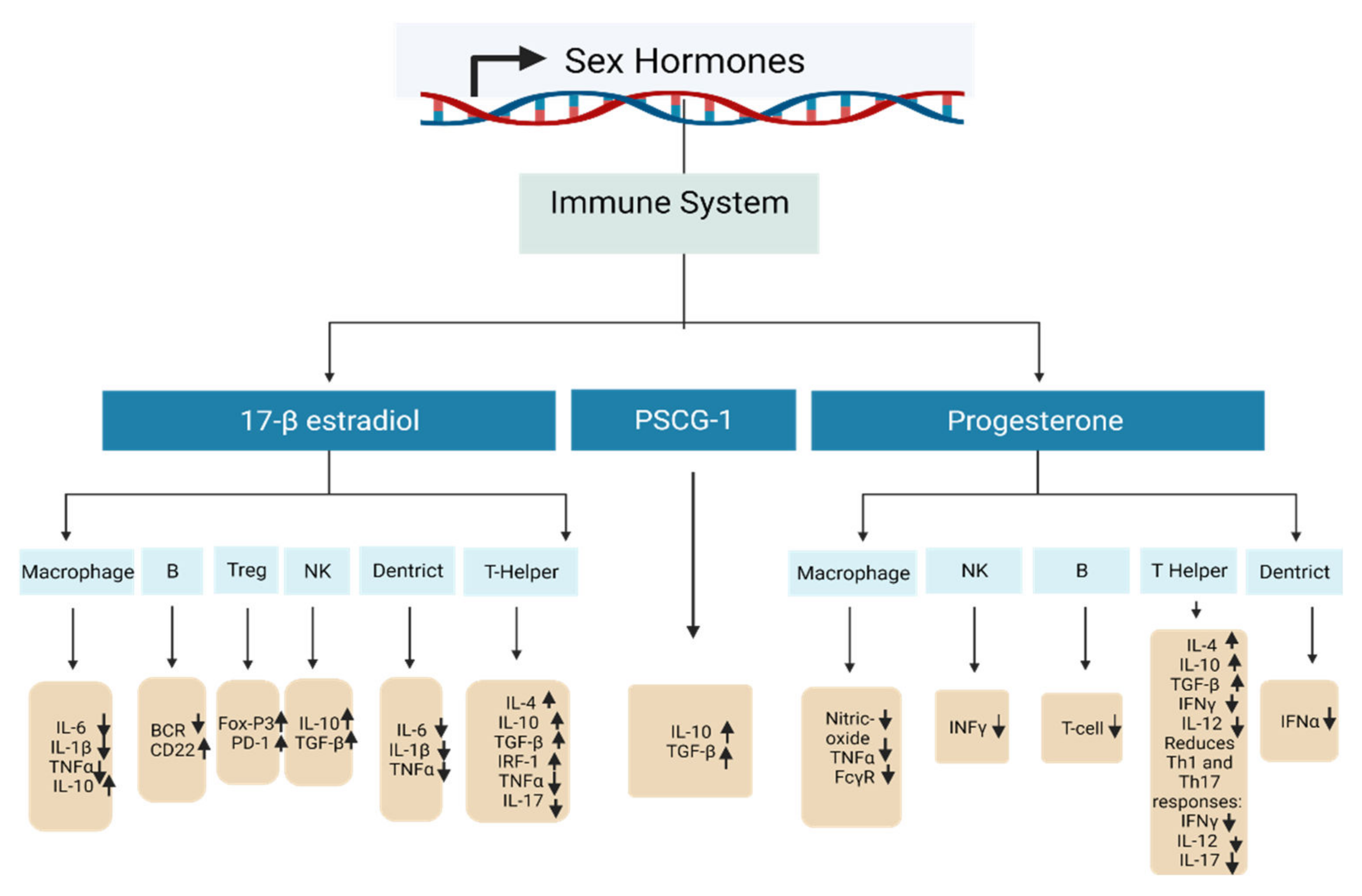
2.6. Significance of Maternal Immune Responses during Pregnancy
2.6.1. Pregnancy-Related Maternal Immunological Adaptation
2.6.2. Autophagy
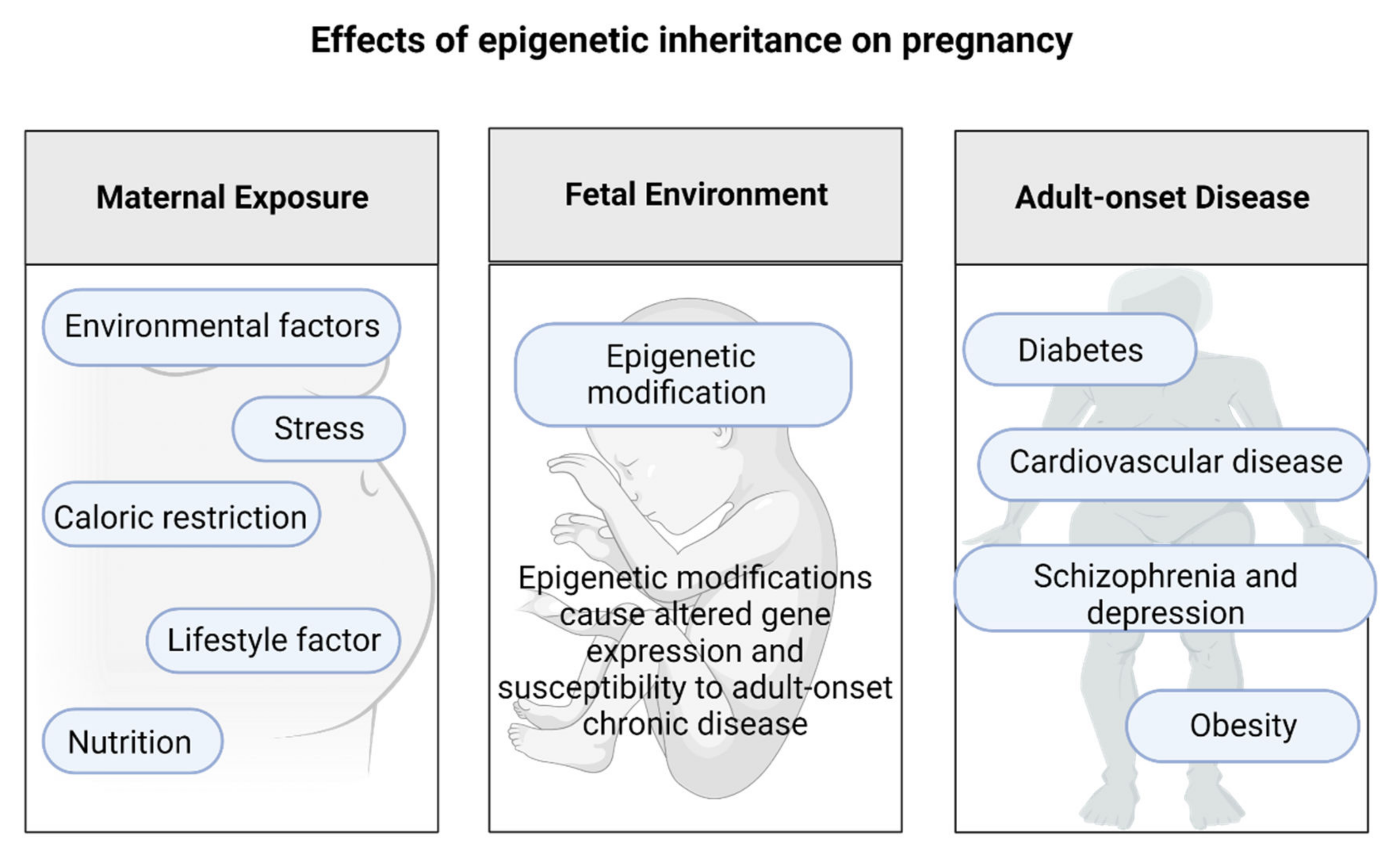
3. Epigenetic Changes during Pregnancy That Affect Offspring Health Later in Life
The Effects of Epigenetic Inheritance on Pregnancy
4. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Richards, E.J. Inherited epigenetic variation—Revisiting soft inheritance. Nat. Rev. Genet. 2006, 7, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. Perceptions of epigenetics. Nature 2007, 447, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Best, J.D.; Carey, N. The Epigenetics of Normal Pregnancy. Obstet. Med. 2013, 6. [Google Scholar] [CrossRef] [PubMed]
- Millán-Zambrano, G.; Burton, A.; Bannister, A.J.; Schneider, R. Histone post-translational modifications—Cause and consequence of genome function. Nat. Rev. Genet. 2022, 23, 563–580. [Google Scholar] [CrossRef]
- Yao, Q.; Chen, Y.; Zhou, X. The roles of microRNAs in epigenetic regulation. Curr. Opin. Chem. Biol. 2019, 51, 11–17. [Google Scholar] [CrossRef]
- Aldhous, M.C.; Hor, K.; Reynolds, R.M. Epigenetics and Diet in Pregnancy. In Handbook of Nutrition and Pregnancy; Humana Press: Cham, Switzerland, 2018; pp. 163–181. [Google Scholar] [CrossRef]
- Roseboom, T.J.; Painter, R.C.; Van Abeelen, A.F.M.; Veenendaal, M.V.E.; De Rooij, S.R. Hungry in the womb: What are the consequences? Lessons from the Dutch famine. Maturitas 2011, 70. [Google Scholar] [CrossRef]
- Serpeloni, F.; Radtke, K.; de Assis, S.G.; Henning, F.; Nätt, D.; Elbert, T. Grandmaternal stress during pregnancy and DNA methylation of the third generation: An epigenome-wide association study. Transl. Psychiatry 2017, 7, e1202. [Google Scholar] [CrossRef]
- Woods, S.M.; Melville, J.L.; Guo, Y.; Fan, M.Y.; Gavin, A. Psychosocial stress during pregnancy. Am. J. Obstet. Gynecol. 2010, 202. [Google Scholar] [CrossRef]
- Badon, S.E.; Littman, A.J.; Chan, K.C.G.; Tadesse, M.G.; Stapleton, P.L.; Bammler, T.K.; Sorensen, T.K.; Williams, M.A.; Enquobahrie, D.A. Physical activity and epigenetic biomarkers in maternal blood during pregnancy. Epigenomics 2018, 10, 11. [Google Scholar] [CrossRef]
- Wu, P.; Farrell, W.E.; Haworth, K.E.; Emes, R.D.; Kitchen, M.O.; Glossop, J.R.; Hanna, F.W.; Fryer, A.A. Maternal genome-wide DNA methylation profiling in gestational diabetes shows distinctive disease-associated changes relative to matched healthy pregnancies. Epigenetics 2018, 13, 122–128. [Google Scholar] [CrossRef]
- Michalczyk, A.A.; Janus, E.D.; Judge, A.; Ebeling, P.R.; Best, J.D.; Ackland, M.J.; Asproloupos, D.; Dunbar, J.A.; Ackland, M.L. Transient epigenomic changes during pregnancy and early postpartum in women with and without type 2 diabetes. Epigenomics 2018, 10, 4. [Google Scholar] [CrossRef]
- Zuccarello, D.; Sorrentino, U.; Brasson, V.; Marin, L.; Piccolo, C.; Capalbo, A.; Andrisani, A.; Cassina, M. Epigenetics of pregnancy: Looking beyond the DNA code. J. Assist. Reprod. Genet. 2022, 39, 801–816. [Google Scholar] [CrossRef]
- Legoff, L.; D’Cruz, S.C.; Tevosian, S.; Primig, M.; Smagulova, F. Transgenerational inheritance of environmentally induced epigenetic alterations during mammalian development. Cells 2019, 8, 1559. [Google Scholar] [CrossRef]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef]
- Tammen, S.A.; Friso, S.; Choi, S.W. Epigenetics: The link between nature and nurture. Mol. Aspects Med. 2013, 34, 753–764. [Google Scholar] [CrossRef]
- Pauwels, S.; Duca, R.C.; Devlieger, R.; Freson, K.; Straetmans, D.; Van Herck, E.; Huybrechts, I.; Koppen, G.; Godderis, L. Maternal methyl-group donor intake and global DNA (Hydroxy)methylation before and during pregnancy. Nutrients 2016, 8, 474. [Google Scholar] [CrossRef]
- Gruzieva, O.; Merid, S.K.; Chen, S.; Mukherjee, N.; Hedman, A.M.; Almqvist, C.; Andolf, E.; Jiang, Y.; Kere, J.; Scheynius, A.; et al. DNA Methylation Trajectories During Pregnancy. Epigenetics Insights 2019, 12. [Google Scholar] [CrossRef]
- Anderson, C.M.; Ralph, J.L.; Wright, M.L.; Linggi, B.; Ohm, J.E. DNA Methylation as a Biomarker for Preeclampsia. Biol. Res. Nurs. 2014, 16. [Google Scholar] [CrossRef]
- Enquobahrie, D.A.; Moore, A.; Muhie, S.; Tadesse, M.G.; Lin, S.; Williams, M.A. Early pregnancy maternal blood DNA methylation in repeat pregnancies and change in gestational diabetes mellitus status—A pilot study. Reprod. Sci. 2015, 22, 904–910. [Google Scholar] [CrossRef]
- Burris, H.H.; Rifas-Shiman, S.L.; Baccarelli, A.; Tarantini, L.; Boeke, C.E.; Kleinman, K.; Litonjua, A.A.; Rich-Edwards, J.W.; Gillman, M.W. Associations of LINE-1 DNA Methylation with Preterm Birth in a Prospective Cohort Study. J. Dev. Orig. Health Dis. 2012, 3, 173–181. [Google Scholar] [CrossRef]
- Lumey, L.H.; Stein, A.D.; Kahn, H.S.; Van der Pal-de Bruin, K.M.; Blauw, G.J.; Zybert, P.A.; Susser, E.S. Cohort profile: The Dutch Hunger Winter families study. Int. J. Epidemiol. 2007, 36, 1196–1204. [Google Scholar] [CrossRef]
- Heijmans, B.T.; Tobi, E.W.; Stein, A.D.; Putter, H.; Blauw, G.J.; Susser, E.S.; Slagboom, P.E.; Lumey, L.H. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. USA 2008, 105, 17046–17049. [Google Scholar] [CrossRef]
- Smith, F.M.; Garfield, A.S.; Ward, A. Regulation of growth and metabolism by imprinted genes. Cytogenet. Genome Res. 2006, 113, 279–291. [Google Scholar] [CrossRef]
- Shen, W.-B.; Ni, J.; Yao, R.; Goetzinger, K.R.; Harman, C.; Reece, E.A.; Wang, B.; Yang, P. Maternal obesity increases DNA methylation and decreases RNA methylation in the human placenta. Reprod. Toxicol. 2022, 107, 90–96. [Google Scholar] [CrossRef]
- El Hajj, N.; Schneider, E.; Lehnen, H.; Haaf, T. Epigenetics and life-long consequences of an adverse nutritional and diabetic intrauterine environment. Reproduction 2014, 148, R111–R120. [Google Scholar] [CrossRef]
- Wongpaiboonwattana, W.; Tosukhowong, P.; Dissayabutra, T.; Mutirangura, A.; Boonla, C. Oxidative stress induces hypomethylation of LINE-1 and hypermethylation of the RUNX3 promoter in a bladder cancer cell line. Asian Pacific J. Cancer Prev. 2013, 14, 3773–3778. [Google Scholar] [CrossRef]
- Huh, S.J.; Clement, K.; Jee, D.; Merlini, A.; Choudhury, S.; Maruyama, R.; Yoo, R.; Chytil, A.; Boyle, P.; Ran, F.A.; et al. Age- and pregnancy-associated dna methylation changes in mammary epithelial cells. Stem Cell Rep. 2015, 4. [Google Scholar] [CrossRef]
- Erlebacher, A. Immunology of the maternal-fetal interface. Annu. Rev. Immunol. 2013, 31, 387–411. [Google Scholar] [CrossRef]
- Svensson-Arvelund, J.; Ernerudh, J.; Buse, E.; Cline, J.M.; Haeger, J.D.; Dixon, D.; Markert, U.R.; Pfarrer, C.; de Vos, P.; Faas, M.M. The Placenta in Toxicology. Part II:Systemic and Local Immune Adaptations in Pregnancy. Toxicol. Pathol. 2014, 42. [Google Scholar] [CrossRef]
- Babenko, O.; Kovalchuk, I.; Metz, G.A.S. Stress-induced perinatal and transgenerational epigenetic programming of brain development and mental health. Neurosci. Biobehav. Rev. 2015, 48, 70–91. [Google Scholar] [CrossRef]
- Oberlander, T.F.; Weinberg, J.; Papsdorf, M.; Grunau, R.; Misri, S.; Devlin, A.M. Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics 2008, 3, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Malentacchi, F.; Fambrini, M.; Harrath, A.H.; Huang, H.; Petraglia, F. Epigenetics of Estrogen and Progesterone Receptors in Endometriosis. Reprod. Sci. 2020, 27, 1967–1974. [Google Scholar] [CrossRef] [PubMed]
- Glasmacher, B.; Evertz, F.; Bernemann, I.; Sun, H.; Pogozhikh, D.; Spindler, R.; Hofmann, N. New Cryopreservation Strategies: A View from Biothermal and Biomedical Process Technology. In Proceedings of the 7th International Conference of Boar Semen Preservation, Bonn, Germany, 14–17 August 2011; Volume 63. [Google Scholar]
- Knight, A.K.; Conneely, K.N.; Kilaru, V.; Cobb, D.; Payne, J.L.; Meilman, S.; Corwin, E.J.; Kaminsky, Z.A.; Dunlop, A.L.; Smith, A.K. SLC9B1 methylation predicts fetal intolerance of labor. Epigenetics 2018, 13, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. 2013, 14, 204. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, K.; Lillycrop, K.A.; Silver, M.J. Fetal programming and epigenetics. Curr. Opin. Endocr. Metab. Res. 2020, 13, 1–6. [Google Scholar] [CrossRef]
- Bestor, T.H. The DNA methyltransferases of mammals. Hum. Mol. Genet. 2000, 9. [Google Scholar] [CrossRef]
- Pozharny, Y.; Lambertini, L.; Clunie, G.; Ferrara, L.; Lee, M.J. Epigenetics in women’s health care. Mt. Sinai J. Med. 2010, 77, 2395–2402. [Google Scholar] [CrossRef]
- Smew, A.I.; Hedman, A.M.; Chiesa, F.; Ullemar, V.; Andolf, E.; Pershagen, G.; Almqvist, C. Limited association between markers of stress during pregnancy and fetal growth in ‘Born into Life’, a new prospective birth cohort. Acta Paediatr. Int. J. Paediatr. 2018, 107, 1003–1010. [Google Scholar] [CrossRef]
- Mastorakos, G.; Ilias, I. Maternal and fetal hypothalamic-pituitary-adrenal axes during pregnancy and postpartum. Ann. N. Y. Acad. Sci. 2003, 997, 136–149. [Google Scholar] [CrossRef]
- Obel, C.; Hedegaard, M.; Henriksen, T.B.; Secher, N.J.; Olsen, J.; Levine, S. Stress and salivary cortisol during pregnancy. Psychoneuroendocrinology 2005, 30, 647–656. [Google Scholar] [CrossRef]
- Bolten, M.I.; Wurmser, H.; Buske-Kirschbaum, A.; Papoušek, M.; Pirke, K.M.; Hellhammer, D. Cortisol levels in pregnancy as a psychobiological predictor for birth weight. Arch. Women’s Ment. Health 2011, 14, 33–41. [Google Scholar] [CrossRef]
- Lim, S.; Smith, K.R.; Lim, S.T.S.; Tian, R.; Lu, J.; Tan, M. Regulation of mitochondrial functions by protein phosphorylation and dephosphorylation. Cell Biosci. 2016, 6, 1–15. [Google Scholar] [CrossRef]
- Qiu, C.; Hevner, K.; Enquobahrie, D.A.; William, M.A. A case-control study of maternal blood mitochondrial DNA copy number and preeclampsia risk. Int. J. Mol. Epidemiol. Genet. 2012, 3, 237–244. [Google Scholar]
- Genest, D.S.; Falcao, S.; Gutkowska, J.; Lavoie, J.L. Impact of exercise training on preeclampsia: Potential preventive mechanisms. Hypertension 2012, 60, 1104–1109. [Google Scholar] [CrossRef]
- Gao, W.L.; Li, D.; Xiao, Z.X.; Liao, Q.P.; Yang, H.X.; Li, Y.X.; Ji, L.; Wang, Y.L. Detection of global DNA methylation and paternally imprinted H19 gene methylation in preeclamptic placentas. Hypertens. Res. 2011, 34, 655–661. [Google Scholar] [CrossRef]
- Kulkarni, A.; Chavan-Gautam, P.; Mehendale, S.; Yadav, H.; Joshi, S. Global DNA methylation patterns in placenta and its association with maternal hypertension in Pre-eclampsia. DNA Cell Biol. 2011, 30, 79–84. [Google Scholar] [CrossRef]
- Kamrani, A.; Alipourfard, I.; Ahmadi-Khiavi, H.; Yousefi, M.; Rostamzadeh, D.; Izadi, M.; Ahmadi, M. The role of epigenetic changes in preeclampsia. BioFactors 2019, 45, 712–724. [Google Scholar] [CrossRef]
- Julian, C.G.; Pedersen, B.S.; Salmon, C.S.; Yang, I.V.; Gonzales, M.; Vargas, E.; Moore, L.G.; Schwartz, D.A. Unique DNA Methylation Patterns in Offspring of Hypertensive Pregnancy. Clin. Transl. Sci. 2015, 8, 740–745. [Google Scholar] [CrossRef]
- Huang, A.; Wu, H.; Iriyama, T.; Zhang, Y.; Sun, K.; Song, A.; Liu, H.; Peng, Z.; Tang, L.; Lee, M.; et al. Elevated Adenosine Induces Placental DNA Hypomethylation Independent of A2B Receptor Signaling in Preeclampsia. Hypertension 2017, 70, 209–218. [Google Scholar] [CrossRef]
- Langbein, M.; Strick, R.; Strissel, P.L.; Vogt, N.; Parsch, H.; Beckmann, M.W.; Schild, R.L. Impaired cytotrophoblast cell-cell fusion is associated with reduced syncytin and increased apoptosis in patients with placental dysfunction. Mol. Reprod. Dev. 2008, 75, 175–183. [Google Scholar] [CrossRef]
- Ruebner, M.; Strissel, P.L.; Langbein, M.; Fahlbusch, F.; Wachter, D.L.; Faschingbauer, F.; Beckmann, M.W.; Strick, R. Impaired cell fusion and differentiation in placentae from patients with intrauterine growth restriction correlate with reduced levels of HERV envelope genes. J. Mol. Med. 2010, 88, 1143–1156. [Google Scholar] [CrossRef]
- Anderson, O.S.; Sant, K.E.; Dolinoy, D.C. Nutrition and epigenetics: An interplay of dietary methyl donors, one-carbon metabolism and DNA methylation. J. Nutr. Biochem. 2012, 23, 853–859. [Google Scholar] [CrossRef]
- White, W.M.; Brost, B.C.; Sun, Z.; Rose, C.; Craici, I.; Wagner, S.J.; Turner, S.; Garovic, V.D. Normal early pregnancy: A transient state of epigenetic change favoring hypomethylation. Epigenetics 2012, 7, 729–734. [Google Scholar] [CrossRef][Green Version]
- Weng, X.; Liu, F.; Zhang, H.; Kan, M.; Wang, T.; Dong, M.; Liu, Y. Genome-wide DNA methylation profiling in infants born to gestational diabetes mellitus. Diabetes Res. Clin. Pract. 2018, 142, 10–18. [Google Scholar] [CrossRef]
- White, W.M.; Sun, Z.; Borowski, K.S.; Brost, B.C.; Davies, N.P.; Rose, C.H.; Garovic, V.D. Preeclampsia/Eclampsia candidate genes show altered methylation in maternal leukocytes of preeclamptic women at the time of delivery. Hypertens. Pregnancy 2016, 35, 394–404. [Google Scholar] [CrossRef]
- Majchrzak-Celińska, A.; Kosicka, K.; Paczkowska, J.; Główka, F.K.; Brȩborowicz, G.H.; Krzyścin, M.; Siemiątkowska, A.; Szaumkessel, M.; Baer-Dubowska, W. HSD11B2, RUNX3, and LINE-1 Methylation in Placental DNA of Hypertensive Disorders of Pregnancy Patients. Reprod. Sci. 2017, 24, 1520–1531. [Google Scholar] [CrossRef]
- Seth, S.; Lewis, A.J.; Saffery, R.; Lappas, M.; Galbally, M. Maternal prenatal mental health and placental 11β-HSD2 gene expression: Initial findings from the mercy pregnancy and emotionalwellbeing study. Int. J. Mol. Sci. 2015, 16, 27482–27496. [Google Scholar] [CrossRef]
- Ma, M.; Zhou, Q.J.; Xiong, Y.; Li, B.; Li, X.T. Preeclampsia is associated with hypermethylation of IGF-1 promoter mediated by DNMT1. Am. J. Transl. Res. 2018, 10, 16–39. [Google Scholar]
- Alahari, S.; Garcia, J.; Post, M.; Caniggia, I. The von Hippel Lindau tumour suppressor gene is a novel target of E2F4-mediated transcriptional repression in preeclampsia. Biochim. Biophys. Acta—Mol. Basis Dis. 2018, 1864, 3298–3308. [Google Scholar] [CrossRef]
- Chiu, R.W.K.; Chim, S.S.C.; Wong, I.H.N.; Wong, C.S.C.; Lee, W.S.; To, K.F.; Tong, J.H.M.; Yuen, R.K.C.; Shum, A.S.W.; Chan, J.K.C.; et al. Hypermethylation of RASSF1A in human and rhesus placentas. Am. J. Pathol. 2007, 170, 941–950. [Google Scholar] [CrossRef]
- Manna, S.; McCarthy, C.; McCarthy, F.P. Placental ageing in adverse pregnancy outcomes: Telomere shortening, cell senescence, and mitochondrial dysfunction. Oxid. Med. Cell. Longev. 2019, 2019, 3095383. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.J. Mitochondria in early development: Linking the microenvironment, metabolism and the epigenome. Reproduction 2019, 157, R159–R179. [Google Scholar] [CrossRef] [PubMed]
- Colleoni, F.; Lattuada, D.; Garretto, A.; Massari, M.; Mand, C.; Somigliana, E.; Cetin, I. Maternal blood mitochondrial DNA content during normal and intrauterine growth restricted (IUGR) pregnancy. Am. J. Obstet. Gynecol. 2010, 203, 365.e1–365.e6. [Google Scholar] [CrossRef] [PubMed]
- Priliani, L.; Prado, E.L.; Restuadi, R.; Waturangi, D.E.; Shankar, A.H.; Malik, S.G. Maternal multiple micronutrient supplementation stabilizes mitochondrial DNA copy number in pregnant women in Lombok, Indonesia. J. Nutr. 2019, 149, 1309–1316. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.J.; Hesson, L.B.; Youngson, N.A. Non-CpG methylation biases bisulphite PCR towards low or unmethylated mitochondrial DNA: Recommendations for the field. Environ. Epigenetics 2020, 6, dvaa001. [Google Scholar] [CrossRef] [PubMed]
- Lopes, A.F.C. Mitochondrial metabolism and DNA methylation: A review of the interaction between two genomes. Clin. Epigenetics 2020, 12, 182. [Google Scholar] [CrossRef] [PubMed]
- Gorman, G.S.; Schaefer, A.M.; Ng, Y.; Gomez, N.; Blakely, E.L.; Alston, C.L.; Feeney, C.; Horvath, R.; Yu-Wai-Man, P.; Chinnery, P.F.; et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann. Neurol. 2015, 77, 753–759. [Google Scholar] [CrossRef]
- Sharma, N.; Pasala, M.S.; Prakash, A. Mitochondrial DNA: Epigenetics and environment. Environ. Mol. Mutagen. 2019, 60, 668–682. [Google Scholar] [CrossRef]
- Zhao, Y.; Sun, L.; Wang, R.R.; Hu, J.F.; Cui, J. The effects of mitochondria-associated long noncoding RNAs in cancer mitochondria: New players in an old arena. Crit. Rev. Oncol. Hematol. 2018, 131, 76–82. [Google Scholar] [CrossRef]
- Bienertova-Vasku, J.; Sana, J.; Slaby, O. The role of microRNAs in mitochondria in cancer. Cancer Lett. 2013, 336, 1–7. [Google Scholar] [CrossRef]
- Pirini, F.; Guida, E.; Lawson, F.; Mancinelli, A.; Guerrero-Preston, R. Nuclear and mitochondrial DNA alterations in newborns with prenatal exposure to cigarette smoke. Int. J. Environ. Res. Public Health 2015, 12, 1135–1155. [Google Scholar] [CrossRef]
- Armstrong, D.A.; Green, B.B.; Blair, B.A.; Guerin, D.J.; Litzky, J.F.; Chavan, N.R.; Pearson, K.J.; Marsit, C.J. Maternal smoking during pregnancy is associated with mitochondrial DNA methylation. Environ. Epigenetics 2016, 2, dvw020. [Google Scholar] [CrossRef]
- Vos, S.; Nawrot, T.S.; Martens, D.S.; Byun, H.M.; Janssen, B.G. Mitochondrial DNA methylation in placental tissue: A proof of concept study by means of prenatal environmental stressors. Epigenetics 2020, 16, 121–131. [Google Scholar] [CrossRef]
- Shock, L.S.; Thakkar, P.V.; Peterson, E.J.; Moran, R.G.; Taylor, S.M. DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc. Natl. Acad. Sci. USA 2011, 108, 3630–3635. [Google Scholar] [CrossRef]
- Patil, V.; Cuenin, C.; Chung, F.; Aguilera, J.R.R.; Fernandez-Jimenez, N.; Romero-Garmendia, I.; Bilbao, J.R.; Cahais, V.; Rothwell, J.; Herceg, Z. Human mitochondrial DNA is extensively methylated in a non-CpG context. Nucleic Acids Res. 2019, 47, 10072–10085. [Google Scholar] [CrossRef]
- Patil, V.; Ward, R.L.; Hesson, L.B. The evidence for functional non-CpG methylation in mammalian cells. Epigenetics 2014, 9, 823–828. [Google Scholar] [CrossRef]
- Bicci, I.; Calabrese, C.; Golder, Z.J.; Gomez-Duran, A.; Chinnery, P.F. Single-molecule mitochondrial DNA sequencing shows no evidence of CpG methylation in human cells and tissues. Nucleic Acids Res. 2021, 49, 12757–12768. [Google Scholar] [CrossRef]
- Bartho, L.A.; Fisher, J.J.; Cuffe, J.S.M.; Perkins, A.V. Mitochondrial transformations in the aging human placenta. Am. J. Physiol. 2020, 319, E981–E994. [Google Scholar] [CrossRef]
- Schroeder, D.I.; Blair, J.D.; Lott, P.; Yu, H.O.K.; Hong, D.; Crary, F.; Ashwood, P.; Walker, C.; Korf, I.; Robinson, W.P.; et al. The human placenta methylome. Proc. Natl. Acad. Sci. USA 2013, 110, 6037–6042. [Google Scholar] [CrossRef]
- Novakovic, B.; Gordon, L.; Wong, N.C.; Moffett, A.; Manuelpillai, U.; Craig, J.M.; Sharkey, A.; Saffery, R. Wide-ranging DNA methylation differences of primary trophoblast cell populations and derived cell lines: Implications and opportunities for understanding trophoblast function. Mol. Hum. Reprod. 2011, 17, 344–353. [Google Scholar] [CrossRef]
- Smith, Z.D.; Shi, J.; Gu, H.; Donaghey, J.; Clement, K.; Cacchiarelli, D.; Gnirke, A.; Michor, F.; Meissner, A. Epigenetic restriction of extraembryonic lineages mirrors the somatic transition to cancer. Nature 2017, 549, 543–547. [Google Scholar] [CrossRef]
- Hemberger, M.; Hanna, C.W.; Dean, W. Mechanisms of early placental development in mouse and humans. Nat. Rev. Genet. 2020, 21, 27–43. [Google Scholar] [CrossRef]
- Wiley, C.D.; Campisi, J. From Ancient Pathways to Aging Cells—Connecting Metabolism and Cellular Senescence. Cell Metab. 2016, 23, 1013–1021. [Google Scholar] [CrossRef]
- Suvakov, S.; Ghamrawi, R.; Cubro, H.; Tu, H.; White, W.M.; Tobah, Y.S.B.; Milic, N.M.; Grande, J.P.; Cunningham, J.M.; Chebib, F.T.; et al. Epigenetic and senescence markers indicate an accelerated ageing-like state in women with preeclamptic pregnancies. EBioMedicine 2021, 70. [Google Scholar] [CrossRef]
- Shrestha, D.; Workalemahu, T.; Tekola-Ayele, F. Maternal dyslipidemia during early pregnancy and epigenetic ageing of the placenta. Epigenetics 2019, 14, 1030–1039. [Google Scholar] [CrossRef]
- Tekola-Ayele, F.; Workalemahu, T.; Gorfu, G.; Shrestha, D.; Tycko, B.; Wapner, R.; Zhang, C.; Louis, G.M.B. Sex differences in the associations of placental epigenetic aging with fetal growth. Aging 2019, 11, 5412–5432. [Google Scholar] [CrossRef]
- Mayne, B.T.; Leemaqz, S.Y.; Smith, A.K.; Breen, J.; Roberts, C.T.; Bianco-Miotto, T. Accelerated placental aging in early onset preeclampsia pregnancies identified by DNA methylation. Epigenomics 2017, 9, 279–289. [Google Scholar] [CrossRef]
- Londero, A.P.; Orsaria, M.; Marzinotto, S.; Grassi, T.; Fruscalzo, A.; Calcagno, A.; Bertozzi, S.; Nardini, N.; Stella, E.; Lellé, R.J.; et al. Placental aging and oxidation damage in a tissue micro-array model: An immunohistochemistry study. Histochem. Cell Biol. 2016, 146, 191–204. [Google Scholar] [CrossRef]
- Giller, A.; Andrawus, M.; Gutman, D.; Atzmon, G. Pregnancy as a model for aging. Ageing Res. Rev. 2020, 62, 101093. [Google Scholar] [CrossRef]
- Poganik, J.; Zhang, B.; Baht, G.; Kerepesi, C.; Yim, S.H.; Lu, A.; Haghani, A.; Gong, T.; Hedman, A.; Andolf, E.; et al. Biological age is increased by stress and restored upon recovery. bioRxiv 2022. [Google Scholar] [CrossRef]
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Sultana, Z.; Maiti, K.; Aitken, J.; Morris, J.; Dedman, L.; Smith, R. Oxidative stress, placental ageing-related pathologies and adverse pregnancy outcomes. Am. J. Reprod. Immunol. 2017, 77, e12653. [Google Scholar] [CrossRef] [PubMed]
- Franzago, M.; Fraticelli, F.; Stuppia, L.; Vitacolonna, E. Nutrigenetics, epigenetics and gestational diabetes: Consequences in mother and child. Epigenetics 2019, 14, 215–235. [Google Scholar] [CrossRef] [PubMed]
- Das, J.; Maitra, A. Maternal DNA Methylation During Pregnancy: A Review. Reprod. Sci. 2021, 28, 2758–2769. [Google Scholar] [CrossRef]
- De Barros, M.C.; Lopes, M.A.B.; Francisco, R.P.V.; Sapienza, A.D.; Zugaib, M. Resistance exercise and glycemic control in women with gestational diabetes mellitus. Am. J. Obstet. Gynecol. 2010, 203, 556.e1–556.e6. [Google Scholar] [CrossRef]
- Gupta, Y.; Kalra, B. Screening and diagnosis of gestational diabetes mellitus. J. Pak. Med. Assoc. 2016, 66. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, C. Prevalence of Gestational Diabetes and Risk of Progression to Type 2 Diabetes: A Global Perspective. Curr. Diab. Rep. 2016, 16, 7. [Google Scholar] [CrossRef]
- Agarwal, P.; Morriseau, T.S.; Kereliuk, S.M.; Doucette, C.A.; Wicklow, B.A.; Dolinsky, V.W. Maternal obesity, diabetes during pregnancy and epigenetic mechanisms that influence the developmental origins of cardiometabolic disease in the offspring. Crit. Rev. Clin. Lab. Sci. 2018, 55, 71–101. [Google Scholar] [CrossRef]
- Zhang, C.; Ning, Y. Effect of dietary and lifestyle factors on the risk of gestational diabetes: Review of epidemiologic evidence. Am. J. Clin. Nutr. 2011, 94, 1975S–1979S. [Google Scholar] [CrossRef]
- Sletner, L.; Moen, A.E.F.; Yajnik, C.S.; Lekanova, N.; Sommer, C.; Birkeland, K.I.; Jenum, A.K.; Böttcher, Y. Maternal Glucose and LDL-Cholesterol Levels Are Related to Placental Leptin Gene Methylation, and, Together With Nutritional Factors, Largely Explain a Higher Methylation Level Among Ethnic South Asians. Front. Endocrinol. 2021, 12. [Google Scholar] [CrossRef]
- Kang, J.; Lee, C.N.; Li, H.Y.; Hsu, K.H.; Lin, S.Y. Genome-wide DNA methylation variation in maternal and cord blood of gestational diabetes population. Diabetes Res. Clin. Pract. 2017, 132, 127–136. [Google Scholar] [CrossRef]
- Ruchat, S.M.; Houde, A.A.; Voisin, G.; St-Pierre, J.; Perron, P.; Baillargeon, J.P.; Gaudet, D.; Hivert, M.F.; Brisson, D.; Bouchard, L. Gestational diabetes mellitus epigenetically affects genes predominantly involved in metabolic diseases. Epigenetics 2013, 8, 935–943. [Google Scholar] [CrossRef]
- Nemoda, Z.; Szyf, M. Epigenetic Alterations and Prenatal Maternal Depression. Birth Defects Res. 2017, 109, 888–897. [Google Scholar] [CrossRef]
- Wilson, S.L.; Liu, Y.; Robinson, W.P. Placental telomere length decline with gestational age differs by sex and TERT, DNMT1, and DNMT3A DNA methylation. Placenta 2016, 48, 26–33. [Google Scholar] [CrossRef]
- Non, A.L.; Binder, A.M.; Kubzansky, L.D.; Michels, K.B. Genome-wide DNA methylation in neonates exposed to maternal depression, anxiety, or SSRI medication during pregnancy. Epigenetics 2014, 9, 964–972. [Google Scholar] [CrossRef]
- Bagot, R.C.; Labonté, B.; Peña, C.J.; Nestler, E.J. Epigenetic signaling in psychiatric disorders: Stress and depression. Dialogues Clin. Neurosci. 2014, 16, 281–295. [Google Scholar] [CrossRef]
- Liu, M.; Liu, F. Transcriptional and post-translational regulation of adiponectin. Biochem. J. 2010, 425, 41–52. [Google Scholar] [CrossRef]
- Guintivano, J.; Arad, M.; Gould, T.D.; Payne, J.L.; Kaminsky, Z.A. Antenatal prediction of postpartum depression with blood DNA methylation biomarkers. Mol. Psychiatry 2014, 19, 560–567. [Google Scholar] [CrossRef]
- Choi, S.W.; Friso, S. Epigenetics: A new bridge between nutrition and health. Adv. Nutr. 2010, 1, 8–16. [Google Scholar] [CrossRef]
- Davis, C.D.; Ross, S.A. Dietary components impact histone modifications and cancer risk. Nutr. Rev. 2007, 65, 88–94. [Google Scholar] [CrossRef]
- Mitsuya, K.; Parker, A.N.; Liu, L.; Ruan, J.; Vissers, M.C.M.; Myatt, L. Alterations in the placental methylome with maternal obesity and evidence for metabolic regulation. PLoS ONE 2017, 12, e0186115. [Google Scholar] [CrossRef]
- Reichetzeder, C. Overweight and obesity in pregnancy: Their impact on epigenetics. Eur. J. Clin. Nutr. 2021, 75, 1710–1722. [Google Scholar] [CrossRef]
- Jansson, N.; Rosario, F.J.; Gaccioli, F.; Lager, S.; Jones, H.N.; Roos, S.; Jansson, T.; Powell, T.L. Activation of placental mTOR signaling and amino acid transporters in obese women giving birth to large babies. J. Clin. Endocrinol. Metab. 2013, 98, 105–113. [Google Scholar] [CrossRef]
- Jacobs, D.I.; Hansen, J.; Fu, A.; Stevens, R.G.; Tjonneland, A.; Vogel, U.B.; Zheng, T.; Zhu, Y. Methylation alterations at imprinted genes detected among long-term shiftworkers. Environ. Mol. Mutagen. 2013, 54, 141–146. [Google Scholar] [CrossRef]
- Aon, M.A.; Cortassa, S.; Juhaszova, M.; Sollott, S.J. Mitochondrial health, the epigenome and healthspan. Clin. Sci. 2016, 130, 1285–1305. [Google Scholar] [CrossRef]
- Smolders, J.; Menheere, P.; Kessels, A.; Damoiseaux, J.; Hupperts, R. Association of vitamin D metabolite levels with relapse rate and disability in multiple sclerosis. Mult. Scler. 2008, 14, 1220–1224. [Google Scholar] [CrossRef]
- Faulk, C.; Dolinoy, D.C. Timing is everything: The when and how of environmentally induced changes in the epigenome of animals. Epigenetics 2011, 6, 791–797. [Google Scholar] [CrossRef]
- Zenclussen, A.C. Adaptive Immune Responses During Pregnancy. Am. J. Reprod. Immunol. 2013, 69, 291–303. [Google Scholar] [CrossRef]
- Mor, G.; Cardenas, I. The Immune System in Pregnancy: A Unique Complexity. Am. J. Reprod. Immunol. 2010, 63, 425–433. [Google Scholar] [CrossRef]
- Pacini, G.; Paolino, S.; Andreoli, L.; Tincani, A.; Gerosa, M.; Caporali, R.; Iagnocco, A.; Ospelt, C.; Smith, V.; Cutolo, M. Epigenetics, pregnancy and autoimmune rheumatic diseases. Autoimmun. Rev. 2020, 19, 102685. [Google Scholar] [CrossRef]
- Straub, R.H. The complex role of estrogens in inflammation. Endocr. Rev. 2007, 28, 521–574. [Google Scholar] [CrossRef] [PubMed]
- Merrheim, J.; Villegas, J.; Van Wassenhove, J.; Khansa, R.; Berrih-Aknin, S.; le Panse, R.; Dragin, N. Estrogen, estrogen-like molecules and autoimmune diseases. Autoimmun. Rev. 2020, 19, 102468. [Google Scholar] [CrossRef] [PubMed]
- Schaub, B.; Liu, J.; Höppler, S.; Schleich, I.; Huehn, J.; Olek, S.; Wieczorek, G.; Illi, S.; von Mutius, E. Maternal farm exposure modulates neonatal immune mechanisms through regulatory T cells. J. Allergy Clin. Immunol. 2009, 123, 774–782.e5. [Google Scholar] [CrossRef] [PubMed]
- Polanczyk, M.J.; Hopke, C.; Huan, J.; Vandenbark, A.A.; Offner, H. Enhanced FoxP3 expression and Treg cell function in pregnant and estrogen-treated mice. J. Neuroimmunol. 2005, 170, 85–92. [Google Scholar] [CrossRef]
- Kovats, S. Estrogen receptors regulate innate immune cells and signaling pathways. Cell. Immunol. 2015, 294, 63–69. [Google Scholar] [CrossRef]
- Moulton, V.R. Sex hormones in acquired immunity and autoimmune disease. Front. Immunol. 2018, 9, 2279. [Google Scholar] [CrossRef]
- Hughes, G.C.; Choubey, D. Modulation of autoimmune rheumatic diseases by oestrogen and progesterone. Nat. Rev. Rheumatol. 2014, 10, 740–751. [Google Scholar] [CrossRef]
- Tan, I.J.; Peeva, E.; Zandman-Goddard, G. Hormonal modulation of the immune system—A spotlight on the role of progestogens. Autoimmun. Rev. 2015, 14, 536–542. [Google Scholar] [CrossRef]
- Bupp, M.R.G.; Jorgensen, T.N. Androgen-induced immunosuppression. Front. Immunol. 2018, 9, 794. [Google Scholar] [CrossRef]
- Motrán, C.C.; Díaz, F.L.; Gruppi, A.; Slavin, D.; Chatton, B.; Bocco, J.L. Human pregnancy-specific glycoprotein 1a (PSG1a) induces alternative activation in human and mouse monocytes and suppresses the accessory cell-dependent T cell proliferation. J. Leukoc. Biol. 2002, 72, 512–521. [Google Scholar] [CrossRef]
- Zhou, G.Q.; Hammarström, S. Pregnancy-specific glycoprotein (PSG) in baboon (Papio hamadryas): Family size, domain structure, and prediction of a functional region in primate PSGS. Biol. Reprod. 2001, 64, 90–99. [Google Scholar] [CrossRef][Green Version]
- Singh, N.P.; Miranda, K.; Singh, U.P.; Nagarkatti, P.; Nagarkatti, M. Diethylstilbestrol (DES) induces autophagy in thymocytes by regulating Beclin-1 expression through epigenetic modulation. Toxicology 2018, 410, 49–58. [Google Scholar] [CrossRef]
- Block, T.; El-Osta, A. Epigenetic programming, early life nutrition and the risk of metabolic disease. Atherosclerosis 2017, 266, 31–40. [Google Scholar] [CrossRef]
- Signorelli, P.; Avagliano, L.; Virgili, E.; Gagliostro, V.; Doi, P.; Braidotti, P.; Bulfamante, G.P.; Ghidoni, R.; Marconi, A.M. Autophagy in term normal human placentas. Placenta 2011, 32, 482–485. [Google Scholar] [CrossRef]
- Kanninen, T.T.; De Andrade Ramos, B.R.; Jaffe, S.; Bongiovanni, A.M.; Linhares, I.M.; Di Renzo, G.C.; Witkin, S.S. Inhibition of autophagy by sera from pregnant women. Reprod. Sci. 2013, 20, 1327–1331. [Google Scholar] [CrossRef]
- Finer, S.; Mathews, C.; Lowe, R.; Smart, M.; Hillman, S.; Foo, L.; Sinha, A.; Williams, D.; Rakyan, V.K.; Hitman, G.A. Maternal gestational diabetes is associated with genome-wide DNA methylation variation in placenta and cord blood of exposed offspring. Hum. Mol. Genet. 2014, 24, 3021–3029. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B. Autophagy in mammalian development and differentiation. Nat. Cell Biol. 2010, 12, 823–830. [Google Scholar] [CrossRef]
- Oh, S.Y.; Choi, S.J.; Kim, H.K.; Cho, E.; Kim, J.H.; Roh, C.R. Autophagy-related proteins, LC3 and beclin-1, in placentas from pregnancies complicated by preeclampsia. Reprod. Sci. 2008, 15, 912–920. [Google Scholar] [CrossRef]
- Tikhodeyev, O.N. The mechanisms of epigenetic inheritance: How diverse are they? Biol. Rev. 2018, 93, 1987–2005. [Google Scholar] [CrossRef]
- King, S.E.; Skinner, M.K. Epigenetic Transgenerational Inheritance of Obesity Susceptibility. Trends Endocrinol. Metab. 2020, 31, 478–494. [Google Scholar] [CrossRef]
- Bleker, L.S.; De Rooij, S.R.; Painter, R.C.; Ravelli, A.C.J.; Roseboom, T.J. Cohort profile: The Dutch famine birth cohort (DFBC)—A prospective birth cohort study in the Netherlands. BMJ Open 2021, 11, e042078. [Google Scholar] [CrossRef]
- Schulz, L.C. The Dutch hunger winter and the developmental origins of health and disease. Proc. Natl. Acad. Sci. USA 2010, 107, 16757–16758. [Google Scholar] [CrossRef]
- Preece, M.A.; Green, A. Pregnancy and inherited metabolic disorders: Maternal and fetal complications. Ann. Clin. Biochem. 2002, 39, 444–455. [Google Scholar] [CrossRef]
- Houseman, E.A.; Accomando, W.P.; Koestler, D.C.; Christensen, B.C.; Marsit, C.J.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform. 2012, 13, 86. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Genes | DNA Methylation Changes | Functional Roles | Pregnancy Effects | References |
|---|---|---|---|---|
| 11β-HSD2 | Hypo-methylation | Plays a critical role in hypertension, plays an important role in the regulation of blood pressure by preventing the activation of the mineralocorticoid receptor in tissues such as the placenta. | Pregnancy and maternal distress affect placental HSD11B4; during the third trimester, the impact of depression and anxiety symptoms is greater than in the first trimester. | [58,59] |
| RUNX3 | Hypo-methylation | Regularly deleted or transcriptionally silenced in cancer; was selected for analysis because tumor progression and gestation reveal several general features, for example, immune tolerance and invasion. | Significant increases in RUNX3 mRNA expression levels were reported among female smokers relative to nonsmoking women. RUNX3 has been established to be fundamental for promoting Th1 phenotypes through IL-4 repression. | [58] |
| LINE-1-elements | Hypo-methylation | Prevents activation of the placental mineralocorticoid receptor. Associated with cardiovascular disease and with risk factors for both cardiovascular disease and preterm birth. | Significant decreases in LINE-1 element methylation levels were observed in placentas during the third trimester relative to the first trimester. Hypomethylation of LINE-1 elements has been associated to pathological processes, as well as tumorigenesis, abnormal placental function, birth defects, aging, and chronic diseases. | [58] |
| IGF-1 | Hyper-methylation | Involved in placental formation and fetal growth; associated with increased DNMT1 expression. | Maternal IGF-1 levels are negatively correlated with pregnancies complicated by pre-eclampsia. Higher maternal IGF-1 concentrations have been reported. | [60] |
| VHL | Hyper-methylation | Codes for a tumor suppressor protein that is critical for normal placental development. | Gestation in patients with VHL disease stimulates cerebellar hemangioblastoma progression and affects the high VHL disorder-related pregnancy complication rate. | [61] |
| RASSF1A | Hyper-methylation | Activated by promoter hyper-methylation in many tumor types. | Increased levels in the placenta in complicated pregnancies. | [62] |
| Genes | DNA Methylation Changes | Mechanism of Genes | References |
|---|---|---|---|
| LINE-1-elements, RUNX3, HSD11B2 | Hypo-methylation | Inhibits the activation of mineralocorticoid receptor in placenta. | [58] |
| IGF-1 | No difference | Formation of placenta and growth of fetus. | [60] |
| DDAH1 | Hyper-methylation | Contribute nitric oxide generation. | [57] |
| VHL | Hyper-methylation | Proper placental development. | [61] |
| TERT | Hypo-methylated | Reverse transcriptase activity. | [105] |
| ADORA2B | Hyper-methylation | Hyper-methylation of this gene associated with hypoxia and PE and sensitive to atmospheric pollutants. | [105] |
| CALCA | Hyper-methylation | Ca++ regulation in placenta. | |
| AGT | Hypo-methylation | Produces angiotensinogen. | [57] |
| MMP9 | Hyper-methylation | Trophoblast cell migration | [91] |
| DNMT1, DNMT3A | Hypo-methylated | DNA methyltransferases. | [106] |
| SPESP1 | Hyper-methylated | Need for successful fertilization. | [80] |
| Sex Hormone | DNA Methylation Changes | Target Gene | Biological Effect | References |
|---|---|---|---|---|
| 17-β-estradiol—high concentration | A crucial element in the passive and active DNA demethylation activities both on the DNA and histones. | T helper cell | Amplifies Th2 responses: raises IL-4, IL-10, TGF-β, promotes IRF-1, inhibits TNFα and IL-17. | [122,123,124] |
| Treg cell | Promotes differentiation and activity: stimulates FoxP3 and PD-1. | [125,126] | ||
| Nk cell | Declines activity: raises IL-10, TGF-β. | [123,127] | ||
| B cell | Amplifies survival of autoreactive B cells: reduces BCR, increases CD22. | [122] | ||
| Macrophages | Decreases activity: decreases IL-6, IL-1β and TNFα, raises IL-10. | [123,128] | ||
| Dendritic cells | Reduces activity: reduces IL-6, IL-1β and TNFα. | [122,123] | ||
| Progesterone | DNA methylation status is still not yet confirmed. | T helper cell | Promotes Th2 response: increases IL-4, IL-10, TGF-β and decreases IFNγ and IL-12. Reduces Th1 and Th17 responses: decreases IFNγ, IL-12, and IL-17. | [122,129,130] |
| NK cells | Reduces activity: reduces INFγ. | [122,130] | ||
| B cell | Decreases class-switch recombination and T cells. | [122,129,130] | ||
| Macrophages | Lowers activity: reduces nitric oxide production, TNFα, and FcγR. | [130] | ||
| Dendritic cells | Decreases activity: decreases TLR-mediated IFNα production. | [131] | ||
| PSG1a | Highly expressed in myoblasts and strongly downregulated after differentiation. | Enhances IL-10 and TGF-β production. | [132,133] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andrawus, M.; Sharvit, L.; Atzmon, G. Epigenetics and Pregnancy: Conditional Snapshot or Rolling Event. Int. J. Mol. Sci. 2022, 23, 12698. https://doi.org/10.3390/ijms232012698
Andrawus M, Sharvit L, Atzmon G. Epigenetics and Pregnancy: Conditional Snapshot or Rolling Event. International Journal of Molecular Sciences. 2022; 23(20):12698. https://doi.org/10.3390/ijms232012698
Chicago/Turabian StyleAndrawus, Mariana, Lital Sharvit, and Gil Atzmon. 2022. "Epigenetics and Pregnancy: Conditional Snapshot or Rolling Event" International Journal of Molecular Sciences 23, no. 20: 12698. https://doi.org/10.3390/ijms232012698
APA StyleAndrawus, M., Sharvit, L., & Atzmon, G. (2022). Epigenetics and Pregnancy: Conditional Snapshot or Rolling Event. International Journal of Molecular Sciences, 23(20), 12698. https://doi.org/10.3390/ijms232012698