
Oncolyic Virotherapy for Prostate Cancer: Lighting a Fire in Winter

Abstract
1. History of Oncolytic Virotherapy
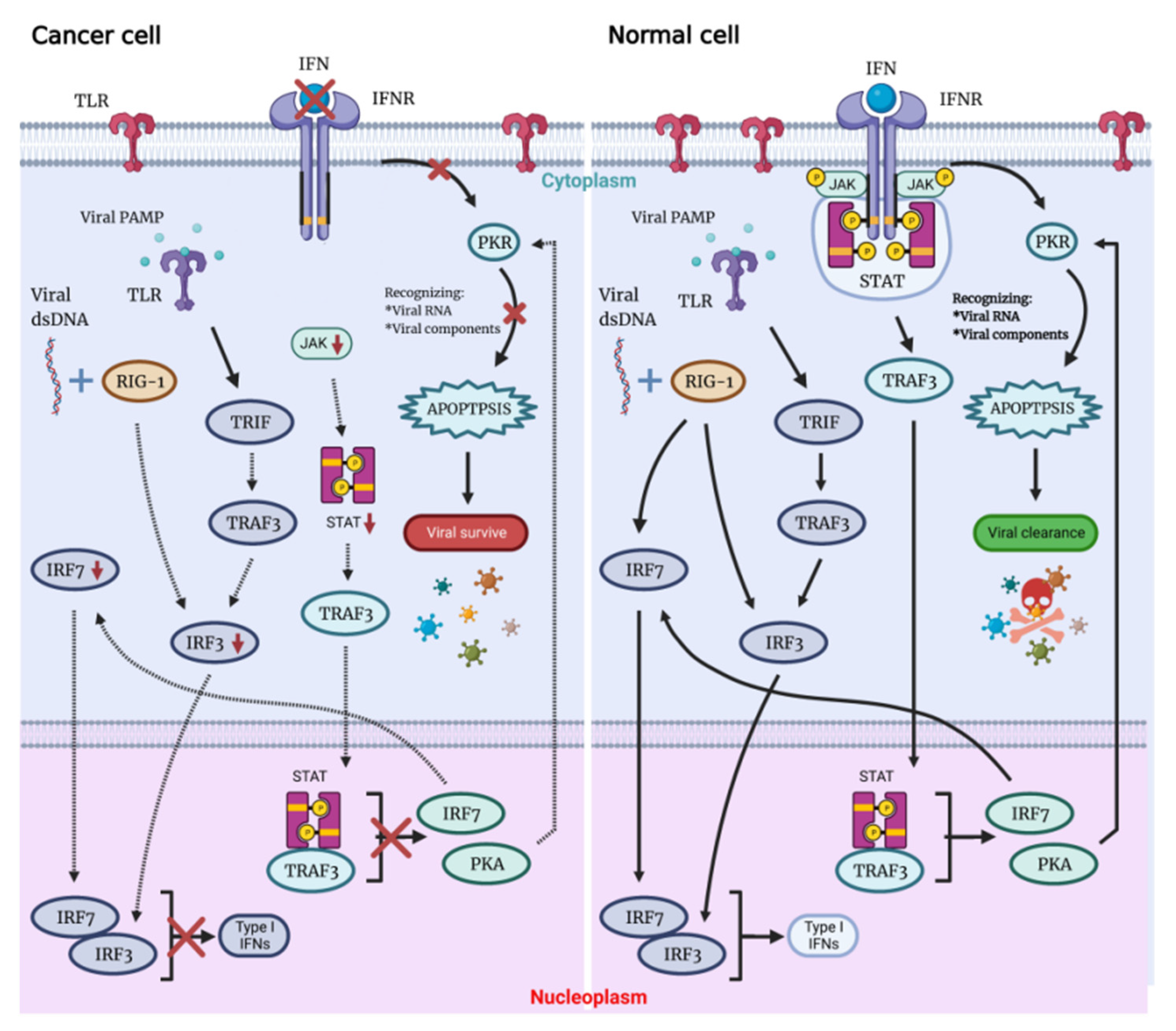
2. General Anti-Tumor Molecular Mechanisms of OVs
2.1. OVs Lyse Tumors Directly but Not Ordinary Cells
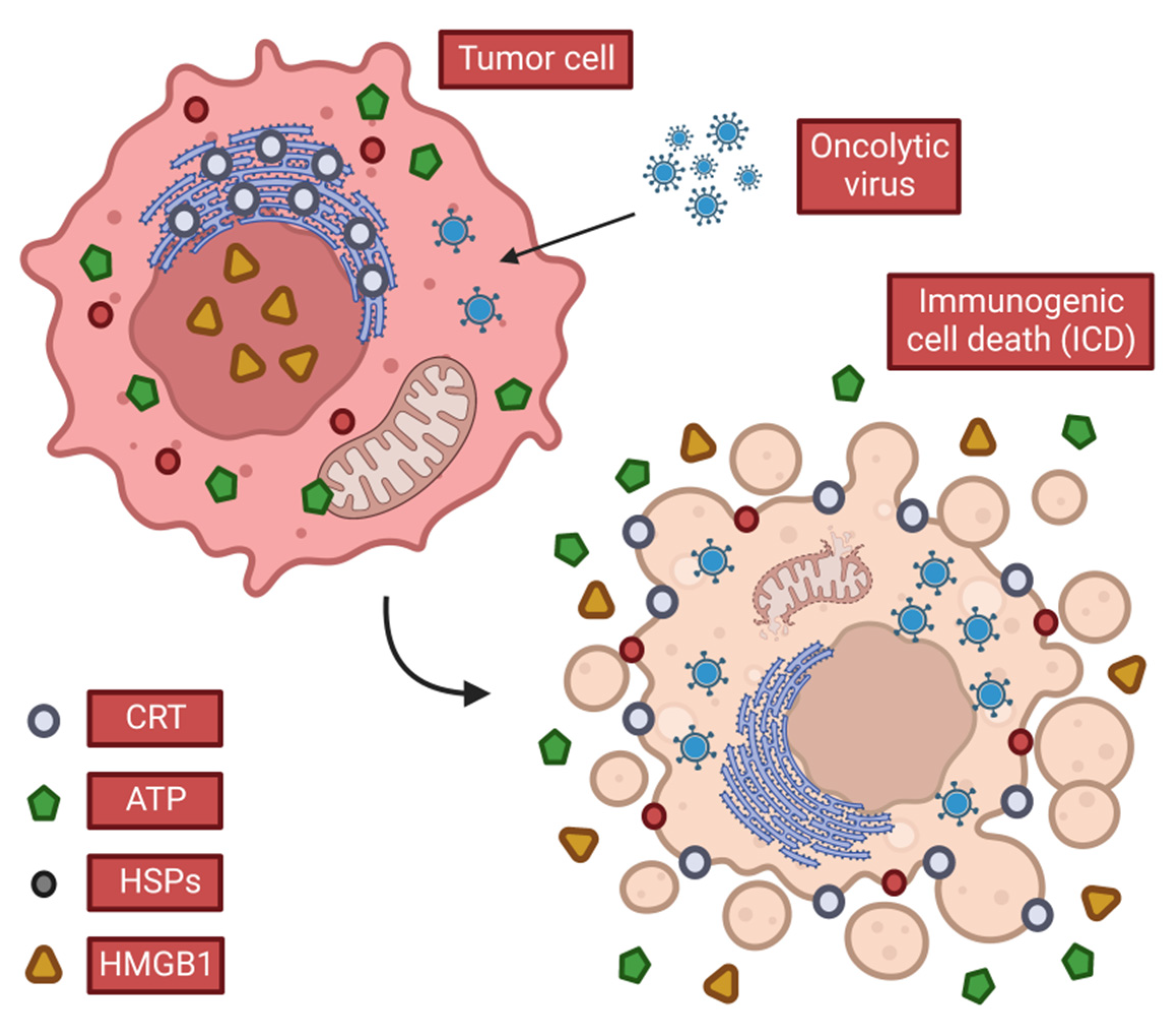
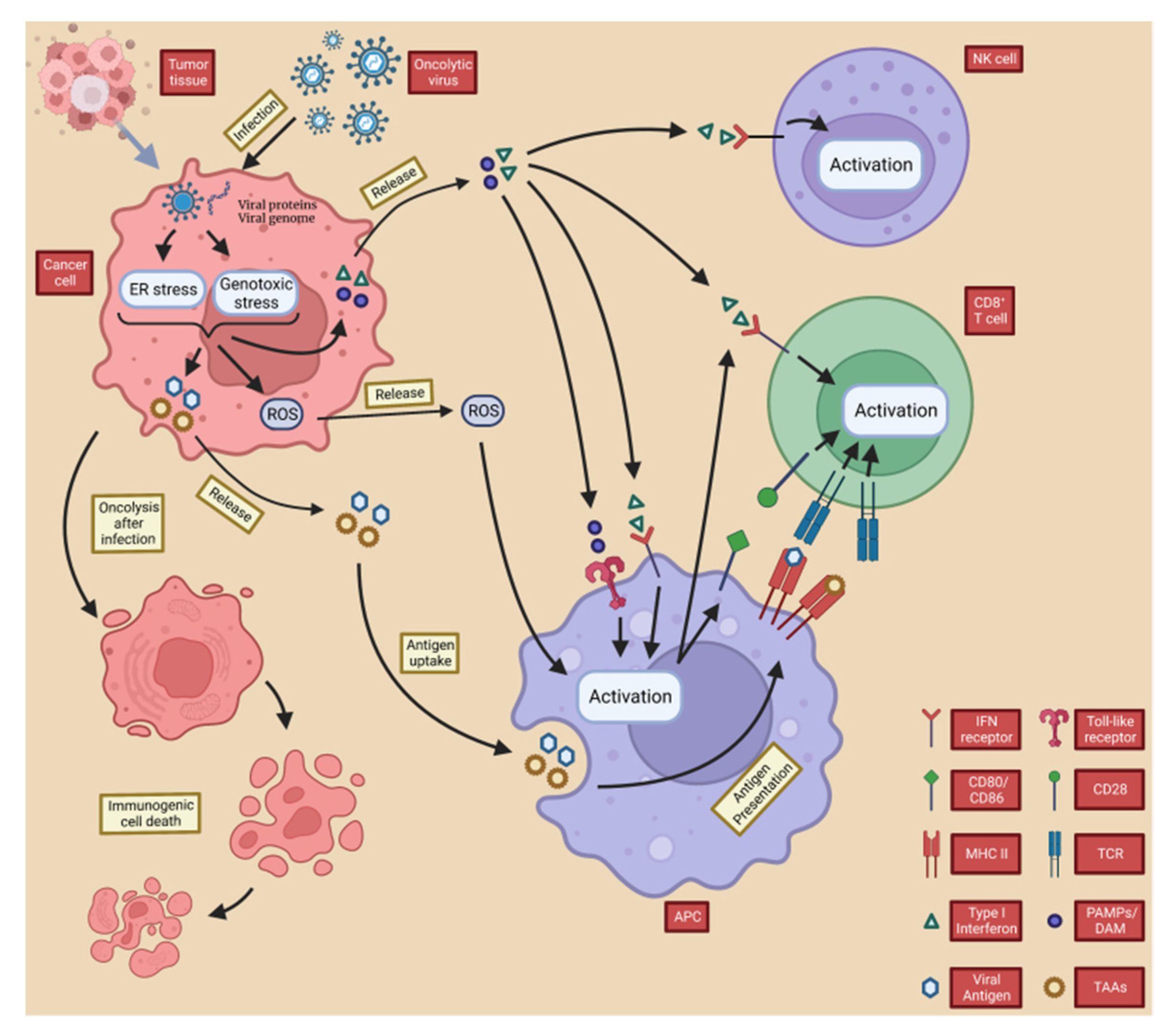
2.2. OVs Induce Anti-Tumor Immunity
2.2.1. OVs Induce Local and Systemic Anti-Tumor Immunity
CALR
ATP
HMGB1
IL-12
2.2.2. OVs Counteract Tumor Immune Evasion
3. Epidemiology and Current Therapy of PCa
3.1. Global Epidemiology of PCa
3.2. Overview of the Current State of PCa Therapy
4. Oncolytic Virotherapy for PCa
4.1. Pre-Clinical Study of OVs for PCa
4.1.1. Preclinical Research to Develop New OV Viruses with Improved Replication and Cell Lysis Capabilities for PCa Treatment
Adenovirus
HSV
Reovirus (Reolysin, Pelareorep)
Newcastle Disease Virus (NDV)
Vaccinia Virus (VV)
Measles Virus (MV)
Other Viruses
4.1.2. Preclinical Research to Enhance the Immunostimulatory Effect of OVs for
Treating PCa

{kind=link}
{kind=link}
{kind=link}
| Name | Change of Construct (Compared to the Original Virus) | Time | Reference |
|---|---|---|---|
| Original virus: Ads | |||
| CV764 (CG7060) | +PSAe, +hK2 | 1999 | [152] |
| CV787 (CG7870) | +PB, +PSAe | 1999 | [153] |
| Ad5-CD/TKrep | +cytidine deaminase (CD), +HSV thymidine kinase (HSV-tk) | 1999 | [165] |
| Ad-OC-E1a | +OC | 2001 | [154] |
| Ad.Δ55.HRE | +hypoxia response element (HRE) | 2004 | [157] |
| Ad-hTERTp-E1A | +hTERTp | 2004 | [160] |
| OAS403 | +hTERTp, +E2F-1 | 2004 | [161] |
| Ad5-yCD/mutTK(SR39)rep-ADP | +yeast cytosine deaminase (yCD)/mutant (SR39), +adenovirus death protein (ADP) | 2006 | [166] |
| CN706/Ad5-PSAe | +PSAe | 2007 | [158] |
| OAS403&OBP-301 | +hTERTp | 2004&2008 | [161,162] |
| Ad.DD3-E1A-IL-24 | +IL-24 | 2010 | [216] |
| Ad.DD3.D55-PTEN | +PTEN | 2012 | [163] |
| Ad.sTβRFc | +sTGFβRIIFc | 2012 | [177] |
| Ad-PL-PPT-E1A | +PPT, +CD40L | 2014 | [229] |
| Ad5/48 | +sTGFβRIIFc | 2014 | [178] |
| Enadenotucirev-BiTE | +BiTE | 2018 | [227] |
| Original virus: HSV | |||
| G207 | −ICP6 | 1995 | [179] |
| G47Δ | −α47 | 2001 | [182] |
| NV1020 | −15-kb region at the UL/S junction | 2002 | [184] |
| NV1034 | +GM-CSF | 2006 | |
| NV1042 | +IL-12 | 2006 | |
| NV1023 | +HSV-1/HSV-2 recombinant | 2007 | [185] |
| bPΔ6-hPAP | +hPAP | 2010 | [186] |
| ARR(2)PB-ICP27 | +ARR(2)PB | 2010 | [187] |
| ARR(2)PB-5’UTR-ICP27 | +ARR(2)PB, +5’UTRs | 2010 | [187] |
| Original virus: Reovirus | |||
| Reovirus | — | 2010 | [193] |
| Reovirus jin-3 | +jin-3 | 2022 | [194] |
| Original virus: NDV | |||
| NDV | — | 2001 | [195] |
| NDV | — | 2013 | [196] |
| Original virus: VV | |||
| GLV-1h68 | +Ruc-GFP, +β-galactosidase, +β-glucuronidase | 2010 | [198] |
| Original virus: MV | |||
| MV-Edm | +J591 scFv | 2009 | [199] |
| MV-Edm | +GFP | 2009 | [200] |
| Original virus: CVA21 | |||
| CVA21 | — | 2008 | [201] |
| CVA21-DAFv | +DAFv | 2008 | [201] |
| Original virus: Echovirus 1 | |||
| EV1 | — | 2008 | [201] |
| Original virus: RSV | |||
| RSV | — | 2009 | [202] |
| Original virus: HVJ-E | |||
| HVJ-E | — | 2009 | [203] |
| Original virus: VSV | |||
| VSV-SV5-F | +SV5-F | 2010 | [204] |
| VSV-GP | +GP | 2018 | [205] |
| Original virus: Poliovirus | |||
| PVSRIPO | — | 2016 | [230] |
4.2. Clinical Trial of OVs on PCa
4.2.1. Clinical Trial of Ads
4.2.2. Clinical Trial of HSV-1
4.2.3. Clinical Trial of Reovirus
4.2.4. Clinical Trial of Vaccinia Virus
4.2.5. Clinical Trial of HVJ-E
| Name | Time | Phase | Tumor Types | Number of Patients | Concentration | Delivery Way | Reference |
|---|---|---|---|---|---|---|---|
| Adenovirus | |||||||
| CV706 (CG7060) | 2001 | I | Local recurrence of PCa after radical radiotherapy | 20 | 1011–1013 | Intratumoral | [232] |
| CV787 (CG7870) | 2006 | I | mCRPC | 23 | 1 × 1010–6 × 1012 | Intravenous | [233] |
| Ad5-CD/TKrep | 2002 | I | Local recurrence of PCa after radical radiotherapy | 16 | 1012 | Intratumoral | [234] |
| Ad6-CD/TKrep | 2003 | I | Newly diagnosed med-high risk PCa | 15 | 1012 | Intratumoral | [235] |
| Ad5-yCD/mutTKSR39rep-ADP | 2007 | I | Newly diagnosed PCa | 9 | 1011–1012 | Intratumoral | [236] |
| HSV-1 | |||||||
| G47Δ | 2013–2016 | I | CRPC | 9 | 3 × 108 pfu | Intratumoral | [238] |
| Reovrius | |||||||
| Pelareorep (REOLYSIN) | 2008 | I | PCa have been treated | 5 | 3 × 108 TCID50 | Intravenous | [239] |
| Pelareorep (REOLYSIN) | 2006 | II | T2 PCa | 6 | - | Intratumoral | [240] |
| Vaccinia viru | |||||||
| rV-PSA | 2000 | I | PCa with elevated PSA after radical surgery or metastasis | 33 | 2.65 × 106–2.65 × 108 | Intratumoral | [241] |
| rV-PSA | 2002 | I | Advanced mPCa | 42 | 2.65 × 105–2.65 × 108 | Subcutaneous | [242] |
| rV-PSA | 2004 | II | Advanced PCa | 64 | 2.34 × 108/1.5 × 109 | Subcutaneous/intramuscular | [243] |
| HVJ-E | |||||||
| HVJ-E (GEN0101) | 2017 | I/II | CRPC | 7 | 3000/10,000 mNAU | Intratumoral/Subcutaneous | [248] |
| HVJ-E (GEN0101) | 2020 | I | mCRPC | 9 | 3000/6000 mNAU | Intratumoral/Subcutaneous | [249] |
5. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kelly, E.; Russell, S.J. History of oncolytic viruses: Genesis to genetic engineering. Mol. Ther. 2007, 15, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Dock, G. The influence of complicating diseases upon leukemia. Am. J. Med. Sci. 1904, 127, 563–592. [Google Scholar] [CrossRef]
- De Pace, N.G. Sulla scomparsa di un enorme cancro vegetante del callo dell’utero senza cura chirurgica. Ginecologia 1912, 9, 82–88. (In Italian) [Google Scholar]
- Bierman, H.R.; Crile, D.M.; Dod, K.S.; Kelly, K.H.; Petrakis, N.I.; White, L.P.; Shimkin, M.B. Remissions in leukemia of childhood following acute infectious disease: Staphylococcus and streptococcus, varicella, and feline panleukopenia. Cancer 1953, 6, 591–605. [Google Scholar] [CrossRef]
- Hoster, H.A.; Zanes, R.P., Jr.; Von Haam, E. Studies in Hodgkin’s syndrome; the association of viral hepatitis and Hodgkin’s disease; A preliminary report. Cancer Res. 1949, 9, 473–480. [Google Scholar]
- Allagui, F.; Achard, C.; Panterne, C.; Combredet, C.; Labarrière, N.; Dréno, B.; Elgaaied, A.B.; Pouliquen, D.; Tangy, F.; Fonteneau, J.F.; et al. Modulation of the Type I Interferon Response Defines the Sensitivity of Human Melanoma Cells to Oncolytic Measles Virus. Curr. Gene Ther. 2017, 16, 419–428. [Google Scholar] [CrossRef]
- Sharma, B.K.; Kakker, N.K.; Bhadouriya, S.; Chhabra, R. Effect of TLR agonist on infections bronchitis virus replication and cytokine expression in embryonated chicken eggs. Mol. Immunol. 2020, 120, 52–60. [Google Scholar] [CrossRef]
- Ichinohe, T. Respective roles of TLR, RIG-I and NLRP3 in influenza virus infection and immunity: Impact on vaccine design. Expert Rev. Vaccines 2010, 9, 1315–1324. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef]
- Huang, B.; Zhao, J.; Unkeless, J.C.; Feng, Z.H.; Xiong, H. TLR signaling by tumor and immune cells: A double-edged sword. Oncogene 2008, 27, 218–224. [Google Scholar] [CrossRef]
- Meurs, E.; Chong, K.; Galabru, J.; Thomas, N.S.; Kerr, I.M.; Williams, B.R.; Hovanessian, A.G. Molecular cloning and characterization of the human double-stranded RNA-activated protein kinase induced by interferon. Cell 1990, 62, 379–390. [Google Scholar] [CrossRef]
- Elde, N.C.; Child, S.J.; Geballe, A.P.; Malik, H.S. Protein kinase R reveals an evolutionary model for defeating viral mimicry. Nature 2009, 457, 485–489. [Google Scholar] [CrossRef] [PubMed]
- Bommareddy, P.K.; Shettigar, M.; Kaufman, H.L. Integrating oncolytic viruses in combination cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 498–513. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.C.; Holl, E.K.; Boczkowski, D.; Dobrikova, E.; Mosaheb, M.; Chandramohan, V.; Bigner, D.D.; Gromeier, M.; Nair, S.K. Cancer immunotherapy with recombinant poliovirus induces IFN-dominant activation of dendritic cells and tumor antigen-specific CTLs. Sci. Transl. Med. 2017, 9, eaan4220. [Google Scholar] [CrossRef]
- Gauvrit, A.; Brandler, S.; Sapede-Peroz, C.; Boisgerault, N.; Tangy, F.; Gregoire, M. Measles virus induces oncolysis of mesothelioma cells and allows dendritic cells to cross-prime tumor-specific CD8 response. Cancer Res. 2008, 68, 4882–4892. [Google Scholar] [CrossRef]
- Zamarin, D.; Holmgaard, R.B.; Subudhi, S.K.; Park, J.S.; Mansour, M.; Palese, P.; Merghoub, T.; Wolchok, J.D.; Allison, J.P. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci. Transl. Med. 2014, 6, 226ra32. [Google Scholar] [CrossRef]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef]
- Tesniere, A.; Apetoh, L.; Ghiringhelli, F.; Joza, N.; Panaretakis, T.; Kepp, O.; Schlemmer, F.; Zitvogel, L.; Kroemer, G. Immunogenic cancer cell death: A key-lock paradigm. Curr. Opin. Immunol. 2008, 20, 504–511. [Google Scholar] [CrossRef]
- Sonnemann, J.; Gressmann, S.; Becker, S.; Wittig, S.; Schmudde, M.; Beck, J.F. The histone deacetylase inhibitor vorinostat induces calreticulin exposure in childhood brain tumour cells in vitro. Cancer Chemother. Pharmacol. 2010, 66, 611–616. [Google Scholar] [CrossRef]
- Panaretakis, T.; Kepp, O.; Brockmeier, U.; Tesniere, A.; Bjorklund, A.C.; Chapman, D.C.; Durchschlag, M.; Joza, N.; Pierron, G.; van Endert, P.; et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J. 2009, 28, 578–590. [Google Scholar] [CrossRef]
- Panaretakis, T.; Joza, N.; Modjtahedi, N.; Tesniere, A.; Vitale, I.; Durchschlag, M.; Fimia, G.M.; Kepp, O.; Piacentini, M.; Froehlich, K.U.; et al. The co-translocation of ERp57 and calreticulin determines the immunogenicity of cell death. Cell Death Differ. 2008, 15, 1499–1509. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Krysko, D.V.; Verfaillie, T.; Kaczmarek, A.; Ferreira, G.B.; Marysael, T.; Rubio, N.; Firczuk, M.; Mathieu, C.; Roebroek, A.J.; et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J. 2012, 31, 1062–1079. [Google Scholar] [CrossRef] [PubMed]
- Gilardini Montani, M.S.; D’Eliseo, D.; Cirone, M.; Di Renzo, L.; Faggioni, A.; Santoni, A.; Velotti, F. Capsaicin-mediated apoptosis of human bladder cancer cells activates dendritic cells via CD91. Nutrition 2015, 31, 578–581. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.P.; Jaiswal, S.; Weissman-Tsukamoto, R.; Alizadeh, A.A.; Gentles, A.J.; Volkmer, J.; Weiskopf, K.; Willingham, S.B.; Raveh, T.; Park, C.Y.; et al. Calreticulin is the dominant pro-phagocytic signal on multiple human cancers and is counterbalanced by CD47. Sci. Transl. Med. 2010, 2, 63ra94. [Google Scholar] [CrossRef]
- Basu, S.; Binder, R.J.; Ramalingam, T.; Srivastava, P.K. CD91 is a common receptor for heat shock proteins gp96, hsp90, hsp70, and calreticulin. Immunity 2001, 14, 303–313. [Google Scholar] [CrossRef]
- Lauber, K.; Ernst, A.; Orth, M.; Herrmann, M.; Belka, C. Dying cell clearance and its impact on the outcome of tumor radiotherapy. Front. Oncol. 2012, 2, 116. [Google Scholar] [CrossRef]
- Krysko, D.V.; Vandenabeele, P. Clearance of dead cells: Mechanisms, immune responses and implication in the development of diseases. Apoptosis 2010, 15, 995–997. [Google Scholar] [CrossRef]
- Krysko, D.V.; Vanden Berghe, T.; Parthoens, E.; D’Herde, K.; Vandenabeele, P. Methods for distinguishing apoptotic from necrotic cells and measuring their clearance. Methods Enzymol. 2008, 442, 307–341. [Google Scholar]
- Krysko, D.V.; Vandenabeele, P. From regulation of dying cell engulfment to development of anti-cancer therapy. Cell Death Differ. 2008, 15, 29–38. [Google Scholar] [CrossRef]
- Krysko, D.V.; D’Herde, K.; Vandenabeele, P. Clearance of apoptotic and necrotic cells and its immunological consequences. Apoptosis 2006, 11, 1709–1726. [Google Scholar] [CrossRef]
- Weiss, E.M.; Frey, B.; Rodel, F.; Herrmann, M.; Schlucker, E.; Voll, R.E.; Fietkau, R.; Gaipl, U.S. Ex vivo- and in vivo-induced dead tumor cells as modulators of antitumor responses. Ann. N. Y. Acad. Sci. 2010, 1209, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Krysko, D.V.; Vandenabeele, P.; Agostinis, P. The emergence of phox-ER stress induced immunogenic apoptosis. Oncoimmunology 2012, 1, 786–788. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Adjemian, S.; Yang, H.; Catani, J.P.; Hannani, D.; Martins, I.; Michaud, M.; Kepp, O.; Sukkurwala, A.Q.; Vacchelli, E.; et al. ATP-dependent recruitment, survival and differentiation of dendritic cell precursors in the tumor bed after anticancer chemotherapy. Oncoimmunology 2013, 2, e24568. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Adjemian, S.; Mattarollo, S.R.; Yamazaki, T.; Aymeric, L.; Yang, H.; Portela Catani, J.P.; Hannani, D.; Duret, H.; Steegh, K.; et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity 2013, 38, 729–741. [Google Scholar] [CrossRef] [PubMed]
- Elliott, M.R.; Chekeni, F.B.; Trampont, P.C.; Lazarowski, E.R.; Kadl, A.; Walk, S.F.; Park, D.; Woodson, R.I.; Ostankovich, M.; Sharma, P.; et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 2009, 461, 282–286. [Google Scholar] [CrossRef]
- McDonald, B.; Pittman, K.; Menezes, G.B.; Hirota, S.A.; Slaba, I.; Waterhouse, C.C.; Beck, P.L.; Muruve, D.A.; Kubes, P. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science 2010, 330, 362–366. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat. Med. 2009, 15, 1170–1178. [Google Scholar] [CrossRef]
- Aymeric, L.; Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Martins, I.; Kroemer, G.; Smyth, M.J.; Zitvogel, L. Tumor cell death and ATP release prime dendritic cells and efficient anticancer immunity. Cancer Res. 2010, 70, 855–858. [Google Scholar] [CrossRef]
- Di Virgilio, F. Liaisons riskeus: P2X(7) and the inflammasome. Trends Pharmacol. Sci. 2007, 28, 465–472. [Google Scholar] [CrossRef]
- Riteau, N.; Gasse, P.; Fauconnier, L.; Gombault, A.; Couegnat, M.; Fick, L.; Kanellopoulos, J.; Quesniaux, V.F.; Marchand-Adam, S.; Crestani, B.; et al. Extracellular ATP is a danger signal activating P2’7 receptor in lung inflammation and fibrosis. Am. J. Respir. Crit. Care Med. 2010, 182, 774–783. [Google Scholar] [CrossRef]
- Cauwels, A.; Rogge, E.; Vandendriessche, B.; Shiva, S.; Brouckaert, P. Extracellular ATP drives systemic inflammation, tissue damage and mortality. Cell Death Dis. 2014, 5, e1102. [Google Scholar] [CrossRef]
- Xiang, Y.; Wang, X.; Yan, C.; Gao, Q.; Li, S.A.; Liu, J.; Zhou, K.; Guo, X.; Lee, W.; Zhang, Y. Adenosine-5′-triphosphate (ATP) protects mice against bacterial infection by activation of the NLRP3 inflammasome. PLoS ONE 2013, 8, e63759. [Google Scholar] [CrossRef]
- Gombault, A.; Baron, L.; Couillin, I. ATP release and purinergic signaling in NLRP3 inflammasome activation. Front. Immunol. 2012, 3, 414. [Google Scholar] [CrossRef] [PubMed]
- Riteau, N.; Baron, L.; Villeret, B.; Guillou, N.; Savigny, F.; Ryffel, B.; Rassendren, F.; Le Bert, M.; Gombault, A.; Couillin, I. ATP release and purinergic signaling: A common pathway for particle-mediated inflammasome activation. Cell Death Dis. 2012, 3, e403. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.S.; Pulskens, W.P.; Sadler, J.J.; Butter, L.M.; Teske, G.J.; Ulland, T.K.; Eisenbarth, S.C.; Florquin, S.; Flavell, R.A.; Leemans, J.C.; et al. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc. Natl. Acad. Sci. USA 2009, 106, 20388–20393. [Google Scholar] [CrossRef]
- England, H.; Summersgill, H.R.; Edye, M.E.; Rothwell, N.J.; Brough, D. Release of interleukin-1alpha or interleukin-1beta depends on mechanism of cell death. J. Biol. Chem. 2014, 289, 15942–15950. [Google Scholar] [CrossRef] [PubMed]
- Steer, S.A.; Scarim, A.L.; Chambers, K.T.; Corbett, J.A. Interleukin-1 stimulates beta-cell necrosis and release of the immunological adjuvant HMGB1. PLoS Med. 2006, 3, e17. [Google Scholar]
- Michaud, M.; Sukkurwala, A.Q.; Martins, I.; Shen, S.; Zitvogel, L.; Kroemer, G. Subversion of the chemotherapy-induced anticancer immune response by the ecto-ATPase CD39. Oncoimmunology 2012, 1, 393–395. [Google Scholar] [CrossRef]
- Aliagas, E.; Vidal, A.; Texido, L.; Ponce, J.; Condom, E.; Martin-Satue, M. High expression of ecto-nucleotidases CD39 and CD73 in human endometrial tumors. Mediat. Inflamm. 2014, 2014, 509027. [Google Scholar] [CrossRef]
- Loi, S.; Pommey, S.; Haibe-Kains, B.; Beavis, P.A.; Darcy, P.K.; Smyth, M.J.; Stagg, J. CD73 promotes anthracycline resistance and poor prognosis in triple negative breast cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 11091–11096. [Google Scholar] [CrossRef]
- Antonioli, L.; Pacher, P.; Vizi, E.S.; Hasko, G. CD39 and CD73 in immunity and inflammation. Trends Mol. Med. 2013, 19, 355–367. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Bruchard, M.; Chalmin, F.; Rebe, C. Production of adenosine by ectonucleotidases: A key factor in tumor immunoescape. J. Biomed. Biotechnol. 2012, 2012, 473712. [Google Scholar] [CrossRef] [PubMed]
- Bastid, J.; Cottalorda-Regairaz, A.; Alberici, G.; Bonnefoy, N.; Eliaou, J.F.; Bensussan, A. ENTPD1CD39 is a promising therapeutic target in oncology. Oncogene 2013, 32, 1743–1751. [Google Scholar] [CrossRef] [PubMed]
- Clayton, A.; Al-Taei, S.; Webber, J.; Mason, M.D.; Tabi, Z. Cancer exosomes express CD39 and CD73, which suppress T cells through adenosine production. J. Immunol. 2011, 187, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Gamboni-Robertson, F.; He, Q.; Svetkauskaite, D.; Kim, J.Y.; Strassheim, D.; Sohn, J.W.; Yamada, S.; Maruyama, I.; Banerjee, A.; et al. High mobility group box 1 protein interacts with multiple Toll-like receptors. Am. J. Physiol. Cell Physiol. 2006, 290, C917–C924. [Google Scholar] [CrossRef]
- Yu, M.; Wang, H.; Ding, A.; Golenbock, D.T.; Latz, E.; Czura, C.J.; Fenton, M.J.; Tracey, K.J.; Yang, H. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock 2006, 26, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Lotze, M.T.; Tracey, K.J. High-mobility group box 1 protein (HMGB1): Nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 2005, 5, 331–342. [Google Scholar] [CrossRef]
- Kokkola, R.; Andersson, A.; Mullins, G.; Ostberg, T.; Treutiger, C.J.; Arnold, B.; Nawroth, P.; Andersson, U.; Harris, R.A.; Harris, H.E. RAGE is the major receptor for the proinflammatory activity of HMGB1 in rodent macrophages. Scand. J. Immunol. 2005, 61, 1–9. [Google Scholar] [CrossRef]
- Dong Xda, E.; Ito, N.; Lotze, M.T.; Demarco, R.A.; Popovic, P.; Shand, S.H.; Watkins, S.; Winikoff, S.; Brown, C.K.; Bartlett, D.L.; et al. High mobility group box I (HMGB1) release from tumor cells after treatment: Implications for development of targeted chemoimmunotherapy. J. Immunother. 2007, 30, 596–606. [Google Scholar] [CrossRef]
- Pathak, S.K.; Skold, A.E.; Mohanram, V.; Persson, C.; Johansson, U.; Spetz, A.L. Activated apoptotic cells induce dendritic cell maturation via engagement of Toll-like receptor 4 (TLR4), dendritic cell-specific intercellular adhesion molecule 3 (ICAM-3)-grabbing nonintegrin (DC-SIGN), and beta2 integrins. J. Biol. Chem. 2012, 287, 13731–13742. [Google Scholar] [CrossRef]
- Bianchi, M.E.; Manfredi, A.A. High-mobility group box 1 (HMGB1) protein at the crossroads between innate and adaptive immunity. Immunol. Rev. 2007, 220, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Curtin, J.F.; Liu, N.; Candolfi, M.; Xiong, W.; Assi, H.; Yagiz, K.; Edwards, M.R.; Michelsen, K.S.; Kroeger, K.M.; Liu, C.; et al. HMGB1 mediates endogenous TLR2 activation and brain tumor regression. PLoS Med. 2009, 6, e10. [Google Scholar] [CrossRef] [PubMed]
- Schiraldi, M.; Raucci, A.; Munoz, L.M.; Livoti, E.; Celona, B.; Venereau, E.; Apuzzo, T.; De Marchis, F.; Pedotti, M.; Bachi, A.; et al. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J. Exp. Med. 2012, 209, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Livesey, K.M.; Cheh, C.W.; Farkas, A.; Loughran, P.; Hoppe, G.; Bianchi, M.E.; Tracey, K.J.; Zeh, H.J., 3rd; et al. Endogenous HMGB1 regulates autophagy. J. Cell Biol. 2010, 190, 881–892. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef]
- Kang, R.; Livesey, K.M.; Zeh, H.J., 3rd; Lotze, M.T.; Tang, D. HMGB1 as an autophagy sensor in oxidative stress. Autophagy 2011, 7, 904–906. [Google Scholar] [CrossRef]
- Lasek, W.; Zagozdzon, R.; Jakobisiak, M. Interleukin 12: Still a promising candidate for tumor immunotherapy? Cancer Immunol. Immunother. 2014, 63, 419–435. [Google Scholar] [CrossRef]
- Colombo, M.; Trinchieri, G. Interleukin-12 in antitumor immunity and immunotherapy. Cytokine Growth Factor Rev. 2002, 13, 155–168. [Google Scholar] [CrossRef]
- Trinchieri, G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat. Rev. Immunol. 2003, 3, 133–146. [Google Scholar] [CrossRef]
- Del Vecchio, M.; Bajetta, E.; Canova, S.; Lotze, M.T.; Wesa, A.; Parmiani, G.; Anichini, A. Interleukin 12: Biological properties and clinical application. Clin. Cancer Res. 2007, 13, 4677–4685. [Google Scholar] [CrossRef] [PubMed]
- Germain, R.N. T-cell development and the CD4–CD8 lineage decision. Nat. Rev. Immunol. 2002, 2, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017, 170, 1109–1119.e10. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Kim, D.W.; DeRaffele, G.; Mitcham, J.; Coffin, R.S.; & Kim-Schulze, S. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann. Surg. Oncol. 2010, 17, 718–730. [Google Scholar] [CrossRef]
- Havunen, R.; Santos, J.M.; Sorsa, S.; Rantapero, T.; Lumen, D.; Siurala, M.; Airaksinen, A.J.; Cervera-Carrascon, V.; Tähtinen, S.; Kanerva, A.; et al. Abscopal Effect in Non-injected Tumors Achieved with Cytokine-Armed Oncolytic Adenovirus. Mol. Ther. Oncolytics 2018, 11, 109–121. [Google Scholar] [CrossRef]
- Ranki, T.; Joensuu, T.; Jäger, E.; Karbach, J.; Wahle, C.; Kairemo, K.; Alanko, T.; Partanen, K.; Turkki, R.; Linder, N.; et al. Local treatment of a pleural mesothelioma tumor with ONCOS-102 induces a systemic antitumor CD8+ T-cell response, prominent infiltration of CD8+ lymphocytes and Th1 type polarization. Oncoimmunology 2014, 3, e958937. [Google Scholar] [CrossRef]
- Leung, E.; Ennis, D.P.; Kennedy, P.R.; Hansell, C.; Dowson, S.; Farquharson, M.; Spiliopoulou, P.; Nautiyal, J.; McNamara, S.; Carlin, L.M.; et al. NK Cells Augment Oncolytic Adenovirus Cytotoxicity in Ovarian Cancer. Mol. Ther. Oncolytics 2020, 16, 289–301. [Google Scholar] [CrossRef]
- Jennings, S.R.; Rice, P.L.; Kloszewski, E.D.; Anderson, R.W.; Thompson, D.L.; Tevethia, S.S. Effect of herpes simplex virus types 1 and 2 on surface expression of class I major histocompatibility complex antigens on infected cells. J. Virol. 1985, 56, 757–766. [Google Scholar] [CrossRef]
- Hill, A.B.; Barnett, B.C.; McMichael, A.J.; McGeoch, D.J. HLA class I molecules are not transported to the cell surface in cells infected with herpes simplex virus types 1 and 2. J. Immunol. 1994, 152, 2736–2741. [Google Scholar]
- Kärre, K.; Ljunggren, H.G.; Piontek, G.; Kiessling, R. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature 1986, 319, 675–678. [Google Scholar] [CrossRef]
- Ljunggren, H.G.; Kärre, K. Host resistance directed selectively against H-2-deficient lymphoma variants. Analysis of the mechanism. J. Exp. Med. 1985, 162, 1745–1759. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F. Molecular mechanisms of IFN-gamma to up-regulate MHC class I antigen processing and presentation. Int. Rev. Immunol. 2009, 28, 239–260. [Google Scholar] [CrossRef] [PubMed]
- Rosa, F.M.; Fellous, M. Regulation of HLA-DR gene by IFN-gamma. Transcriptional and post-transcriptional control. J. Immunol. 1988, 140, 1660–1664. [Google Scholar]
- Zhao, M.Z.; Sun, Y.; Jiang, X.F.; Liu, L.; Liu, L.; Sun, L.X. Promotion on NLRC5 upregulating MHC-I expression by IFN-γ in MHC-I-deficient breast cancer cells. Immunol. Res. 2019, 67, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Doody, G.M.; Stephenson, S.; McManamy, C.; Tooze, R.M. PRDM1/BLIMP-1 modulates IFN-gamma-dependent control of the MHC class I antigen-processing and peptide-loading pathway. J. Immunol. 2007, 179, 7614–7623. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 2015, 35, S185–S198. [Google Scholar] [CrossRef]
- Ruffell, B.; Affara, N.I.; Coussens, L.M. Differential macrophage programming in the tumor microenvironment. Trends Immunol. 2012, 33, 119–126. [Google Scholar] [CrossRef]
- Talmadge, J.E.; Gabrilovich, D.I. History of myeloid-derived suppressor cells. Nat. Rev. Cancer 2013, 13, 739–752. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Pang, Y.; Moses, H.L. TGF-beta and immune cells: An important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010, 31, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Melcher, A.; Parato, K.; Rooney, C.M.; Bell, J.C. Thunder and Lightning: Immunotherapy and Oncolytic Viruses Collide. Mol. Ther. 2011, 19, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Muscolini, M.; Tassone, E.; Hiscott, J. Oncolytic Immunotherapy: Can’t Start a Fire without a Spark. Cytokine Growth Factor Rev. 2020, 56, 94–101. [Google Scholar] [CrossRef]
- Liu, Y.-T.; Sun, Z.-J. Turning cold tumors into hot tumors by improving T-cell infiltration. Theranostics 2021, 11, 5365–5386. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019, 10, 168. [Google Scholar] [CrossRef]
- Di Paolo, N.C.; Miao, E.A.; Iwakura, Y.; Murali-Krishna, K.; Aderem, A.; Flavell, R.A.; Papayannopoulou, T.; Shayakhmetov, D.M. Virus binding to a plasma membrane receptor triggers interleukin-1 α-mediated proinflammatory macrophage response in vivo. Immunity 2009, 31, 110–121. [Google Scholar] [CrossRef]
- Prestwich, R.J.; Errington, F.; Diaz, R.M.; Pandha, H.S.; Harrington, K.J.; Melcher, A.A.; Vile, R.G. The case of oncolytic viruses versus the immune system: Waiting on the judgment of Solomon. Hum. Gene Ther. 2009, 20, 1119–1132. [Google Scholar] [CrossRef]
- Bridle, B.W.; Stephenson, K.B.; Boudreau, J.E.; Koshy, S.; Kazdhan, N.; Pullenayegum, E.; Brunellière, J.; Bramson, J.L.; Lichty, B.D.; Wan, Y. Potentiating cancer immunotherapy using an oncolytic virus. Mol. Ther. 2010, 18, 1430–1439. [Google Scholar] [CrossRef]
- Kanerva, A.; Nokisalmi, P.; Diaconu, I.; Koski, A.; Cerullo, V.; Liikanen, I.; Tähtinen, S.; Oksanen, M.; Heiskanen, R.; Pesonen, S.; et al. Antiviral and antitumor T-cell immunity in patients treated with GM-CSF-coding oncolytic adenovirus. Clin. Cancer Res. 2013, 19, 2734–2744. [Google Scholar] [CrossRef] [PubMed]
- Hargadon, K.M.; Johnson, C.E.; Williams, C.J. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol. 2018, 62, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Tselikas, L.; de Baere, T.; Houot, R. Intratumoral immunotherapy: Using the tumor as the remedy. Ann. Oncol. 2017, 28 (Suppl. S12), xii33–xii43. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-Y.; Hutzen, B.; Wedekind, M.F.; Cripe, T.P. Oncolytic virus and PD-1/PD-L1 blockade combination therapy. Oncolytic Virother. 2018, 7, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Zamarin, D.; Holmgaard, R.B.; Ricca, J.; Plitt, T.; Palese, P.; Sharma, P.; Merghoub, T.; Wolchok, J.D.; Allison, J.P. Intratumoral modulation of the inducible co-stimulator ICOS by recombinant oncolytic virus promotes systemic anti-tumour immunity. Nat. Commun. 2017, 8, 14340. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Gandaglia, G.; Leni, R.; Bray, F.; Fleshner, N.; Freedland, S.J.; Kibel, A.; Stattin, P.; Van Poppel, H.; La Vecchia, C. Epidemiology and Prevention of Prostate Cancer. Eur. Urol. Oncol. 2021, 4, 877–892. [Google Scholar] [CrossRef]
- Zhu, Y.; Mo, M.; Wei, Y.; Wu, J.; Pan, J.; Freedland, S.J.; Zheng, Y.; Ye, D. Epidemiology and genomics of prostate cancer in Asian men. Nat. Rev. Urol. 2021, 18, 282–301. [Google Scholar] [CrossRef]
- Liu, Z.; Jiang, Y.; Fang, Q.; Yuan, H.; Cai, N.; Suo, C.; Ye, W.; Chen, X.; Zhang, T. Future of cancer incidence in Shanghai, China: Predicting the burden upon the ageing population. Cancer Epidemiol. 2019, 60, 8–15. [Google Scholar] [CrossRef]
- Shin, S.; Saito, E.; Sawada, N.; Ishihara, J.; Takachi, R.; Nanri, A.; Shimazu, T.; Yamaji, T.; Iwasaki, M.; Sasazuki, S.; et al. Dietary patterns and prostate cancer risk in Japanese: The Japan Public Health Center-based Prospective Study (JPHC Study). Cancer Causes Control 2018, 29, 589–600. [Google Scholar] [CrossRef]
- GBD 2017 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [Google Scholar] [CrossRef]
- Foreman, K.J.; Marquez, N.; Dolgert, A.; Fukutaki, K.; Fullman, N.; McGaughey, M.; Pletcher, M.A.; Smith, A.E.; Tang, K.; Yuan, C.; et al. Forecasting life expectancy, years of life lost, and all-cause and cause-specific mortality for 250 causes of death: Reference and alternative scenarios for 2016–40 for 195 countries and territories. Lancet 2018, 392, 2052–2090. [Google Scholar] [CrossRef]
- Sandhu, S.; Moore, C.M.; Chiong, E.; Beltran, H.; Bristow, R.G.; Williams, S.G. Prostate cancer. Lancet 2021, 398, 1075–1090. [Google Scholar] [CrossRef]
- Mottet, N.; van den Bergh, R.; Briers, E.; Van den Broeck, T.; Cumberbatch, M.G.; De Santis, M.; Fanti, S.; Fossati, N.; Gandaglia, G.; Gillessen, S.; et al. EAU-EANM-ESTRO-ESUR-SIOG Guidelines on Prostate Cancer—2020 Update. Part 1: Screening, Diagnosis, and Local Treatment with Curative Intent. Eur. Urol. 2021, 79, 243–262. [Google Scholar] [CrossRef] [PubMed]
- Mottet, N.; van den Bergh, R.; Briers, E.; Van den Broeck, T.; Cumberbatch, M.G.; De Santis, M.; Fanti, S.; Fossati, N.; Gandaglia, G.; Gillessen, S.; et al. EAU-EANM-ESTRO-ESUR-SIOG Guidelines on Prostate Cancer. Part II—2020 Update: Treatment of Relapsing and Metastatic Prostate Cancer. Eur. Urol. 2021, 79, 263–282. [Google Scholar] [CrossRef]
- Schaeffer, E.; Srinivas, S.; Antonarakis, E.S.; Armstrong, A.J.; Bekelman, J.E.; Cheng, H.; D’Amico, A.V.; Davis, B.J.; Desai, N.; Dorff, T.; et al. NCCN Guidelines Insights: Prostate Cancer, Version 1.2021. J. Natl. Compr. Cancer Netw. 2021, 19, 134–143. [Google Scholar] [CrossRef]
- Huggins, C.; Hodges, C.V. Studies on Prostatic Cancer. I. The Effect of Castration, of Estrogen and of Androgen Injection on Serum Phosphatases in Metastatic Carcinoma of the Prostate. Cancer Res. 1941, 1, 293–297. [Google Scholar] [CrossRef]
- Henríquez, I.; Roach, M., III; Morgan, T.M.; Bossi, A.; Gómez, J.A.; Abuchaibe, O.; Couñago, F. Current and Emerging Therapies for Metastatic Castration-Resistant Prostate Cancer (mCRPC). Biomedicines 2021, 9, 1247. [Google Scholar] [CrossRef]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmaña, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-repair defects and olaparib in metastatic prostate cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef]
- Clarke, N.; Wiechno, P.; Alekseev, B.; Sala, N.; Jones, R.; Kocak, I.; Chiuri, V.E.; Jassem, J.; Fléchon, A.; Redfern, C.; et al. Olaparib combined with abiraterone in patients with metastatic castration-resistant prostate cancer: A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2018, 19, 975–986. [Google Scholar] [CrossRef]
- Thiery-Vuillemin, A.; de Bono, J.; Hussain, M.; Roubaud, G.; Procopio, G.; Shore, N.; Fizazi, K.; Dos Anjos, G.; Gravis, G.; Joung, J.Y.; et al. Pain and health-related quality of life with olaparib versus physician’s choice of next-generation hormonal drug in patients with metastatic castration-resistant prostate cancer with homologous recombination repair gene alterations (PROfound): An open-label, randomised, phase 3 trial. Lancet Oncol. 2022, 23, 393–405, Erratum in Lancet Oncol. 2022, 23, e161. [Google Scholar] [CrossRef]
- Mateo, J.; Lord, C.J.; Serra, V.; Tutt, A.; Balmaña, J.; Castroviejo-Bermejo, M.; Cruz, C.; Oaknin, A.; Kaye, S.B.; de Bono, J.S. A decade of clinical development of PARP inhibitors in perspective. Ann Oncol. 2019, 30, 1437–1447. [Google Scholar] [CrossRef]
- Messina, C.; Cattrini, C.; Soldato, D.; Vallome, G.; Caffo, O.; Castro, E.; Olmos, D.; Boccardo, F.; Zanardi, E. BRCA Mutations in Prostate Cancer: Prognostic and Predictive Implications. J Oncol. 2020, 2020, 4986365. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Bochum, S.; Berger, S.; Martens, U.M. Olaparib. Recent Results Cancer Res. 2018, 211, 217–233. [Google Scholar] [CrossRef]
- Rucaparib [Package Insert]; Clovis Oncology, Inc.: Boulder, CO, USA, 2020.
- Mateo, J.; Porta, N.; Bianchini, D.; McGovern, U.; Elliott, T.; Jones, R.; Syndikus, I.; Ralph, C.; Jain, S.; Varughese, M.; et al. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): A multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 2020, 21, 162–174. [Google Scholar] [CrossRef]
- de Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef]
- Hussain, M.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Survival with Olaparib in Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 383, 2345–2357. [Google Scholar] [CrossRef]
- Abida, W.; Patnaik, A.; Campbell, D.; Shapiro, J.; Bryce, A.H.; McDermott, R.; Sautois, B.; Vogelzang, N.J.; Bambury, R.M.; Voog, E.; et al. Rucaparib in men with metastatic castration-resistant prostate cancer harboring a BRCA1 or BRCA2 gene alteration. J. Clin. Oncol. 2020, 38, 3763–3772. [Google Scholar] [CrossRef]
- Abida, W.; Campbell, D.; Patnaik, A.; Shapiro, J.D.; Sautois, B.; Vogelzang, N.J.; Voog, E.G.; Bryce, A.H.; McDermott, R.; Ricci, F.; et al. Non-BRCA DNA damage repair gene alterations and response to the PARP inhibitor rucaparib in metastatic castration-resistant prostate cancer: Analysis from the phase II TRITON2 study. Clin. Cancer Res. 2020, 26, 2487–2496. [Google Scholar] [CrossRef] [PubMed]
- Markowski, M.C.; Antonarakis, E.S. BRCA1 versus BRCA2 and PARP inhibitor sensitivity in prostate cancer: More different than alike? J. Clin. Oncol. 2020, 38, 3735–3739. [Google Scholar] [CrossRef] [PubMed]
- FDA Approves Merck’s KEYTRUDA® (Pembrolizumab) for Adult and Pediatric Patients with Unresectable or Metastatic, Microsatellite Instability-High (MSI-H) or Mismatch Repair Deficient Cancer. Available online: https://www.merck.com/news/fda-approves-mercks-keytruda-pembrolizumab-for-adult-and-pediatric-patients-with-unresectable-or-metastatic-microsatellite-instability-high-msi-h-or-mismatch-repair-deficient-cance/ (accessed on 29 December 2020).
- Pembrolizumab [Package Insert]; Merck & Co, Inc.: Whitehouse Station, NJ, USA, 2020.
- Graff, J.N.; Alumkal, J.J.; Drake, C.G.; Thomas, G.V.; Redmond, W.L.; Farhad, M.; Cetnar, J.P.; Ey, F.S.; Bergan, R.C.; Slottke, R.; et al. Early evidence of anti-PD-1 activity in enzalutamide-resistant prostate cancer. Oncotarget 2016, 7, 52810–52817. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Hansen, A.R.; Massard, C.; Ott, P.A.; Haas, N.B.; Lopez, J.S.; Ejadi, S.; Wallmark, J.M.; Keam, B.; Delord, J.P.; Aggarwal, R.; et al. Pembrolizumab for advanced prostate adenocarcinoma: Findings of the KEYNOTE-028 study. Ann. Oncol. 2018, 29, 1807–1813. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.; Cheng, M.L.; Armenia, J.; Middha, S.; Autio, K.A.; Vargas, H.A.; Rathkopf, D.; Morris, M.J.; Danila, D.C.; Slovin, S.F.; et al. Analysis of the prevalence of microsatellite instability in prostate cancer and response to immune checkpoint blockade. JAMA Oncol. 2019, 5, 471–478. [Google Scholar] [CrossRef]
- Tucker, M.D.; Zhu, J.; Marin, D.; Gupta, R.T.; Gupta, S.; Berry, W.R.; Ramalingam, S.; Zhang, T.; Harrison, M.; Wu, Y.; et al. Pembrolizumab in men with heavily treated metastatic castrate-resistant prostate cancer. Cancer Med. 2019, 8, 4644–4655. [Google Scholar] [CrossRef]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of pembrolizumab in patients with noncolorectal high microsatellite instability/mismatch repair-deficient cancer: Results from the phase II KEYNOTE-158 study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef]
- Graham, L.S.; Montgomery, B.; Cheng, H.H.; Yu, E.Y.; Nelson, P.S.; Pritchard, C.; Erickson, S.; Alva, A.; Schweizer, M.T. Mismatch repair deficiency in metastatic prostate cancer: Response to PD-1 blockade and standard therapies. PLoS ONE 2020, 15, e0233260. [Google Scholar] [CrossRef]
- Sena, L.A.; Fountain, J.; Isaacsson Velho, P.; Lim, S.J.; Wang, H.; Nizialek, E.; Rathi, N.; Nussenzveig, R.; Maughan, B.L.; Velez, M.G.; et al. Tumor frameshift mutation proportion predicts response to immunotherapy in mismatch repair-deficient prostate cancer. Oncologist 2021, 26, e270–e278. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Piulats, J.M.; Gross-Goupil, M.; Goh, J.; Ojamaa, K.; Hoimes, C.J.; Vaishampayan, U.; Berger, R.; Sezer, A.; Alanko, T.; et al. Pembrolizumab for treatment-refractory metastatic castration-resistant prostate cancer: Multicohort, open-label phase II KEYNOTE-199 study. J. Clin. Oncol. 2020, 38, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.; Ding, Y.; Xu, N. A Double-Edged Sword Role of Cytokines in Prostate Cancer Immunotherapy. Front. Oncol. 2021, 11, 688489. [Google Scholar] [CrossRef] [PubMed]
- Kwon, E.D.; Drake, C.G.; Scher, H.I.; Fizazi, K.; Bossi, A.; van den Eertwegh, A.J.; Krainer, M.; Houede, N.; Santos, R.; Mahammedi, H.; et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184–043): A multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2014, 15, 700–712. [Google Scholar] [CrossRef]
- Beer, T.M.; Kwon, E.D.; Drake, C.G.; Fizazi, K.; Logothetis, C.; Gravis, G.; Ganju, V.; Polikoff, J.; Saad, F.; Humanski, P.; et al. Randomized, Double-Blind, Phase III Trial of Ipilimumab Versus Placebo in Asymptomatic or Minimally Symptomatic Patients with Metastatic Chemotherapy-Naive Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2017, 35, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, C.C.; Morrissey, C.; Kumar, A.; Zhang, X.; Smith, C.; Coleman, I.; Salipante, S.J.; Milbank, J.; Yu, M.; Grady, W.M.; et al. Complex MSH2 and MSH6 mutations in hypermutated microsatellite unstable advanced prostate cancer. Nat. Commun. 2014, 5, 4988. [Google Scholar] [CrossRef] [PubMed]
- Barata, P.; Agarwal, N.; Nussenzveig, R.; Gerendash, B.; Jaeger, E.; Hatton, W.; Ledet, E.; Lewis, B.; Layton, J.; Babiker, H.; et al. Clinical activity of pembrolizumab in metastatic prostate cancer with microsatellite instability high (MSI-H) detected by circulating tumor DNA. J. Immunother Cancer 2020, 8, e001065. [Google Scholar] [CrossRef]
- Pereira, H.G.; Kelly, B. Dose-response curves of toxic and infective actions of adenovirus in HeLa cell cultures. J. Gen. Microbiol. 1957, 17, 517–524. [Google Scholar] [CrossRef]
- Yu, D.C.; Sakamoto, G.T.; Henderson, D.R. Identification of the transcriptional regulatory sequences of human kallikrein 2 and their use in the construction of calydon virus 764, an attenuated replication competent adenovirus for prostate cancer therapy. Cancer Res. 1999, 59, 1498–1504. [Google Scholar]
- Yu, D.C.; Chen, Y.; Seng, M.; Dilley, J.; Henderson, D.R. The addition of adenovirus type 5 region E3 enables calydon virus 787 to eliminate distant prostate tumor xenografts. Cancer Res. 1999, 59, 4200–4203. [Google Scholar]
- Matsubara, S.; Wada, Y.; Gardner, T.A.; Egawa, M.; Park, M.S.; Hsieh, C.L.; Zhau, H.E.; Kao, C.; Kamidono, S.; Gillenwater, J.Y.; et al. A conditional replication-competent adenoviral vector, Ad-OC-E1a, to cotarget prostate cancer and bone stroma in an experimental model of androgen-independent prostate cancer bone metastasis. Cancer Res. 2001, 61, 6012–6019. [Google Scholar]
- Li, X.; Jung, C.; Liu, Y.H.; Bae, K.H.; Zhang, Y.P.; Zhang, H.J.; Vanderputten, D.; Jeng, M.H.; Gardner, T.A.; Kao, C.; et al. Anti-tumor efficacy of a transcriptional replication-competent adenovirus, Ad-OC-E1a, for osteosarcoma pulmonary metastasis. J. Gene Med. 2006, 8, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.S.; Kraaij, R.; Nilsson, B.; van der Weel, L.; de Ridder, C.M.; Tötterman, T.H.; Essand, M. A Novel TARP-Promoter-Based Adenovirus against Hormone-Dependent and Hormone-Refractory Prostate Cancer. Mol. Ther. 2004, 10, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.K.; Seong, Y.R.; Lee, Y.H.; Kim, M.J.; Hwang, K.S.; Yoo, J.; Choi, S.; Jung, C.R.; Im, D.S. Oncolytic effects of adenovirus mutant capable of replicating in hypoxic and normoxic regions of solid tumor. Mol. Ther. 2004, 10, 938–949. [Google Scholar] [CrossRef] [PubMed]
- Hardcastle, J.; Kurozumi, K.; Chiocca, E.A.; Kaur, B. Oncolytic viruses driven by tumor-specific promoters. Curr. Cancer Drug Targets 2007, 7, 181–189. [Google Scholar] [CrossRef]
- Rodriguez, R.; Schuur, E.R.; Lim, H.Y.; Henderson, G.A.; Simons, J.W.; Henderson, D.R. Prostate attenuated replication competent adenovirus (ARCA) CN706: A selective cytotoxic for prostate-specific antigen-positive prostate cancer cells. Cancer Res. 1997, 57, 2559–2563. [Google Scholar]
- Irving, J.; Wang, Z.; Powell, S.; O’Sullivan, C.; Mok, M.; Murphy, B.; Cardoza, L.; Lebkowski, J.S.; Majumdar, A.S. Conditionally replicative adenovirus driven by the human telomerase promoter provides broad-spectrum antitumor activity without liver toxicity. Cancer Gene Ther. 2004, 11, 174–185. [Google Scholar] [CrossRef][Green Version]
- Ryan, P.C.; Jakubczak, J.L.; Stewart, D.A.; Hawkins, L.K.; Cheng, C.; Clarke, L.M.; Ganesh, S.; Hay, C.; Huang, Y.; Kaloss, M.; et al. Antitumor efficacy and tumor-selective replication with a single intravenous injection of OAS403, an oncolytic adenovirus dependent on two prevalent alterations in human cancer. Cancer Gene Ther. 2004, 11, 555–569. [Google Scholar] [CrossRef]
- Huang, P.; Watanabe, M.; Kaku, H.; Kashiwakura, Y.; Chen, J.; Saika, T.; Nasu, Y.; Fujiwara, T.; Urata, Y.; Kumon, H.; et al. Direct and distant antitumor effects of a telomerase-selective oncolytic adenoviral agent, OBP-301, in a mouse prostate cancer model. Cancer Gene Ther. 2008, 15, 315–322. [Google Scholar] [CrossRef]
- Ding, M.; Cao, X.; Xu, H.N.; Fan, J.K.; Huang, H.L.; Yang, D.Q.; Li, Y.H.; Wang, J.; Li, R.; Liu, X.Y.; et al. Prostate cancer-specific and potent antitumor effect of a DD3-controlled oncolytic virus harboring the PTEN gene. PLoS ONE 2012, 7, e35153. [Google Scholar] [CrossRef]
- Lu, Y. Viral based gene therapy for prostate cancer. Curr. Gene Ther. 2001, 1, 183–200. [Google Scholar] [CrossRef]
- Freytag, S.O.; Rogulski, K.R.; Paielli, D.L.; Gilbert, J.D.; Kim, J.H. A novel three-pronged approach to kill cancer cells selectively: Concomitant viral, double suicide gene, and radiotherapy. Hum. Gene Ther. 1998, 9, 1323–1333. [Google Scholar] [CrossRef] [PubMed]
- Barton, K.N.; Paielli, D.; Zhang, Y.; Koul, S.; Brown, S.L.; Lu, M.; Seely, J.; Kim, J.H.; Freytag, S.O. Second-generation replication-competent oncolytic adenovirus armed with improved suicide genes and ADP gene demonstrates greater efficacy without increased toxicity. Mol. Ther. 2006, 13, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Futakuchi, M.; Ogawa, K.; Asamoto, M.; Nakao, K.; Asai, K.; Shirai, T. Transforming growth factor β derived from bone matrix promotes cell proliferation of prostate cancer and osteoclast activation-associated osteolysis in the bone microenvironment. Cancer Sci. 2008, 99, 316–323. [Google Scholar] [CrossRef]
- Massague, J. TGFβ in cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef]
- Nguyen, D.X.; Bos, P.D.; Massague, J. Metastasis: From dissemination to organ-specific colonization. Nat. Rev. Cancer 2009, 9, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Juarez, P.; Guise, T.A. TGF-β in cancer and bone: Implications for treatment of bone metastases. Bone 2011, 48, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Shariat, S.F.; Shalev, M.; Menesses-Diaz, A.; Kim, I.Y.; Kattan, M.W.; Wheeler, T.M.; Slawin, K.M. Preoperative plasma levels of transforming growth factor β1 (TGF-β1) strongly predict progression in patients undergoing radical prostatectomy. J. Clin. Oncol. 2001, 19, 2856–2864. [Google Scholar] [CrossRef]
- Barrett, J.M.; Rovedo, M.A.; Tajuddin, A.M.; Jilling, T.; Macoska, J.A.; MacDonald, J.; Mangold, K.A.; Kaul, K.L. Prostate cancer cells regulate growth and differentiation of bone marrow endothelial cells through TGFβ and its receptor, TGFβRII. Prostate 2006, 66, 632–650. [Google Scholar] [CrossRef][Green Version]
- Lu, S.; Lee, J.; Revelo, M.; Wang, X.; Lu, S.; Dong, Z. Smad3 is overexpressed in advanced human prostate cancer and necessary for progressive growth of prostate cancer cells in nude mice. Clin. Cancer Res. 2007, 13, 5692–5702. [Google Scholar] [CrossRef]
- Schroten, C.; Dits, N.F.; Steyerberg, E.W.; Kranse, R.; van Leenders, A.G.; Bangma, C.H.; Kraaij, R. The additional value of TGFβ1 and IL-7 to predict the course of prostate cancer progression. Cancer Immunol. Immunother. 2011, 61, 905–915. [Google Scholar] [CrossRef]
- Seth, P.; Wang, Z.G.; Pister, A.; Zafar, M.B.; Kim, S.; Guise, T.; Wakefield, L. Development of oncolytic adenovirus armed with a fusion of soluble transforming growth factor-β receptor II and human immunoglobulin Fc for breast cancer therapy. Hum. Gene Ther. 2006, 17, 1152–1160. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Robbins, J.S.; Pister, A.; Zafar, M.B.; Zhang, Z.W.; Gupta, J.; Lee, K.J.; Newman, K.; Yun, C.O.; Guise, T.; et al. A modified hTERT promoter-directed oncolytic adenovirus replication with concurrent inhibition of TGF-β signaling for breast cancer therapy. Cancer Gene Ther. 2010, 17, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Gupta, J.; Zhang, Z.; Gerseny, H.; Berg, A.; Chen, Y.J.; Zhang, Z.; Du, H.; Brendler, C.B.; Xiao, X.; et al. Systemic Delivery of Oncolytic Adenoviruses Targeting Transforming Growth Factor-β Inhibits Established Bone Metastasis in a Prostate Cancer Mouse Model. Hum. Gene Ther. 2012, 23, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Zhang, Z.; Yang, Y.; Hu, Z.; Wang, C.H.; Morgan, M.; Wu, Y.; Hutten, R.; Xiao, X.; Stock, S.; et al. Ad5/48 hexon oncolytic virus expressing sTGFβRIIFc produces reduced hepatic and systemic toxicities and inhibits prostate cancer bone metastases. Mol. Ther. 2014, 22, 1504–1517. [Google Scholar] [CrossRef] [PubMed]
- Mineta, T.; Rabkin, S.D.; Yazaki, T.; Hunter, W.D.; Martuza, R.L. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat. Med. 1995, 1, 938–943. [Google Scholar] [CrossRef]
- Oyama, M.; Ohigashi, T.; Hoshi, M.; Murai, M.; Uyemura, K.; Yazaki, T. Oncolytic viral therapy for human prostate cancer by conditionally replicating herpes simplex virus 1 vector G207. Jpn. J. Cancer Res. 2000, 91, 1339–1344. [Google Scholar] [CrossRef]
- Walker, J.R.; McGeagh, K.G.; Sundaresan, P.; Jorgensen, T.J.; Rabkin, S.D.; Martuza, R.L. Local and systemic therapy of human prostate adenocarcinoma with the conditionally replicating herpes simplex virus vector G207. Hum. Gene Ther. 1999, 10, 2237–2243. [Google Scholar] [CrossRef]
- Todo, T.; Martuza, R.L.; Rabkin, S.D.; Johnson, P.A. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc. Natl. Acad. Sci. USA 2001, 98, 6396–6401. [Google Scholar] [CrossRef]
- Fukuhara, H.; Martuza, R.L.; Rabkin, S.D.; Ito, Y.; Todo, T. Oncolytic Herpes Simplex Virus Vector G47Δ in Combination with Androgen Ablation for the Treatment of Human Prostate Adenocarcinoma. Clin. Cancer Res. 2005, 11, 7886–7890. [Google Scholar] [CrossRef]
- Cozzi, P.J.; Burke, P.B.; Bhargav, A.; Heston, W.D.; Huryk, B.; Scardino, P.T.; Fong, Y. Oncolytic viral gene therapy for prostate cancer using two attenuated, replication-competent, genetically engineered herpes simplex viruses. Prostate 2002, 53, 95–100. [Google Scholar] [CrossRef]
- Varghese, S.; Rabkin, S.D.; Nielsen, G.P.; MacGarvey, U.; Liu, R.; Martuza, R.L. Systemic therapy of spontaneous prostate cancer in transgenic mice with oncolytic herpes simplex viruses. Cancer Res. 2007, 67, 9371–9379. [Google Scholar] [CrossRef] [PubMed]
- Castelo-Branco, P.; Passer, B.J.; Buhrman, J.S.; Antoszczyk, S.; Marinelli, M.; Zaupa, C.; Rabkin, S.D.; Martuza, R.L. Oncolytic herpes simplex virus armed with xenogeneic homologue of prostatic acid phosphatase enhances antitumor efficacy in prostate cancer. Gene Ther. 2010, 17, 805–810. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y.; Bu, L.X.; DeBenedetti, A.; Williams, B.J.; Rennie, P.S.; Jia, W.W. Transcriptional and translational dual-regulated oncolytic herpes simplex virus type 1 for targeting prostate tumors. Mol. Ther. 2010, 18, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Norman, K.L.; Lee, P.W.K. Reovirus as apotential Anticancer therapeutic. In Replication-Copetent Viruses for Cancer Therapy; Hernaiz Driever, P., Rabkin, S.D., Eds.; Karger: Basel, Switzerland, 2001; Volume 22, pp. 81–99. [Google Scholar]
- Strong, J.E.; Coffey, M.C.; Tang, D.; Sabinin, P.; Lee, P.W. The molecular basis of viral oncolysis: Usurpation of the Ras signaling pathway by reovirus. EMBO J. 1998, 17, 3351–3362. [Google Scholar] [CrossRef]
- Norman, K.L.; Hirasawa, K.; Yang, A.-D.; Shields, M.A.; Lee, P.W.K. Reovirus oncolysis: The Ras/RalGEF/p38 pathway dictates host cell permissiveness to reovirus infection. Proc. Natl. Acad. Sci. USA 2004, 101, 11099–11104. [Google Scholar] [CrossRef]
- Coffey, M.C.; Strong, J.E.; Forsyth, P.A.; Lee, P.W. Reovirus therapy of tumors with activated Ras pathway. Science 1998, 282, 1332–1334. [Google Scholar] [CrossRef]
- Stoeckel, J.; Hay, J.G. Drug evaluation: Reolysin—Wild-type reovirus as a cancer therapeutic. Curr. Opin. Mol. Ther. 2006, 8, 249–260. [Google Scholar]
- Thirukkumaran, C.M.; Nodwell, M.J.; Hirasawa, K.; Shi, Z.Q.; Diaz, R.; Luider, J.; Johnston, R.N.; Forsyth, P.A.; Magliocco, A.M.; Lee, P.; et al. Oncolytic viral therapy for prostate cancer: Efficacy of reovirus as a biological therapeutic. Cancer Res. 2010, 70, 2435–2444. [Google Scholar] [CrossRef]
- van de Merbel, A.F.; van der Horst, G.; van der Mark, M.H.; Bots, S.; van den Wollenberg, D.; de Ridder, C.; Stuurman, D.; Aalders, T.; Erkens-Schulz, S.; van Montfoort, N.; et al. Reovirus mutant jin-3 exhibits lytic and immune-stimulatory effects in preclinical human prostate cancer models. Cancer Gene Ther. 2022, 29, 793–802. [Google Scholar] [CrossRef]
- Phuangsab, A.; Lorence, R.M.; Reichard, K.W.; Peeples, M.E.; Walter, R.J. Newcastle disease virus therapy of human tumor xenografts: Antitumor effects of local or systemic administration. Cancer Lett. 2001, 172, 27–36. [Google Scholar] [CrossRef]
- Shobana, R.; Samal, S.K.; Elankumaran, S. Prostate-specific antigen-retargeted recombinant newcastle disease virus for prostate cancer virotherapy. J. Virol. 2013, 87, 3792–3800. [Google Scholar] [CrossRef] [PubMed]
- Hodge, J.W.; Schlom, J.; Donohue, S.J.; Tomaszewski, J.E.; Wheeler, C.W.; Levine, B.S.; Gritz, L.; Panicali, D.; Kantor, J.A. A recombinant vaccinia virus expressing human prostate-specific antigen (PSA): Safety and immunogenicity in a non-human primate. Int. J. Cancer 1995, 63, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Gentschev, I.; Donat, U.; Hofmann, E.; Weibel, S.; Adelfinger, M.; Raab, V.; Heisig, M.; Chen, N.; Yu, Y.A.; Stritzker, J.; et al. Regression of human prostate tumors and metastases in nude mice following treatment with the recombinant oncolytic vaccinia virus GLV-1h68. J. Biomed. Biotechnol. 2010, 2010, 489759. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Hasegawa, K.; Russell, S.J.; Sadelain, M.; Peng, K.W. Prostate-specific membrane antigen retargeted measles virotherapy for the treatment of prostate cancer. Prostate 2009, 69, 1128–1141. [Google Scholar] [CrossRef]
- Msaouel, P.; Iankov, I.D.; Allen, C.; Morris, J.C.; von Messling, V.; Cattaneo, R.; Koutsilieris, M.; Russell, S.J.; Galanis, E. Engineered measles virus as a novel oncolytic therapy against prostate cancer. Prostate 2009, 69, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Berry, L.J.; Au, G.G.; Barry, R.D.; Shafren, D.R. Potent oncolytic activity of human enteroviruses against human prostate cancer. Prostate 2008, 68, 577–587. [Google Scholar] [CrossRef] [PubMed]
- Echchgadda, I.; Kota, S.; DeLa Cruz, I.; Sabbah, A.; Chang, T.; Harnack, R.; Mgbemena, V.; Chatterjee, B.; Bose, S. Anticancer oncolytic activity of respiratory syncytial virus. Cancer Gene Ther. 2009, 16, 923–935. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Miyamoto, Y.; Inoue, T.; Kaneda, Y. Efficient eradication of hormone-resistant human prostate cancers by inactivated Sendai virus particle. Int. J. Cancer 2009, 124, 2478–2487. [Google Scholar] [CrossRef]
- Chang, G.; Xu, S.; Watanabe, M.; Jayakar, H.R.; Whitt, M.A.; Gingrich, J.R. Enhanced oncolytic activity of vesicular stomatitis virus encoding SV5-F protein against prostate cancer. J. Urol. 2010, 183, 1611–1618. [Google Scholar] [CrossRef]
- Urbiola, C.; Santer, F.R.; Petersson, M.; van der Pluijm, G.; Horninger, W.; Erlmann, P.; Wollmann, G.; Kimpel, J.; Culig, Z.; von Laer, D. Oncolytic activity of the rhabdovirus VSV-GP against prostate cancer. Int. J. Cancer 2018, 143, 1786–1796. [Google Scholar] [CrossRef]
- Martikainen, M.; Ruotsalainen, J.; Tuomela, J.; Härkönen, P.; Essand, M.; Heikkilä, J.; Hinkkanen, A. Oncolytic alphavirus SFV-VA7 efficiently eradicates subcutaneous and orthotopic human prostate tumours in mice. Br. J. Cancer 2017, 117, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Gujar, S.A.; Pan, D.A.; Marcato, P.; Garant, K.A.; Lee, P.W. Oncolytic Virus-initiated Protective Immunity Against Prostate Cancer. Mol. Ther. 2011, 19, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, R.; Mhashilkar, A.M.; Tanaka, F.; Saito, Y.; Branch, C.D.; Sieger, K.; Mumm, J.B.; Stewart, A.L.; Boquoi, A.; Dumoutier, L.; et al. Melanoma differentiation-associated gene 7/interleukin (IL)-24 is a novel ligand that regulates angiogenesis via the IL-22 receptor. Cancer Res. 2003, 63, 5105–5113. [Google Scholar] [PubMed]
- Su, Z.; Emdad, L.; Sauane, M.; Lebedeva, I.V.; Sarkar, D.; Gupta, P.; James, C.D.; Randolph, A.; Valerie, K.; Walter, M.R.; et al. Unique aspects of mda-7/IL-24 antitumor bystander activity: Establishing a role for secretion of MDA-7/IL-24 protein by normal cells. Oncogene 2005, 24, 7552–7566. [Google Scholar] [CrossRef]
- Su, Z.Z.; Lebedeva, I.V.; Sarkar, D.; Gopalkrishnan, R.V.; Sauane, M.; Sigmon, C.; Yacoub, A.; Valerie, K.; Dent, P.; Fisher, P.B. Melanoma differentiation associated gene-7, mda-7/IL-24, selectively induces growth suppression, apoptosis and radiosensitization in malignant gliomas in a p53-independent manner. Oncogene 2003, 22, 1164–1180. [Google Scholar] [CrossRef]
- Jiang, H.; Su, Z.Z.; Lin, J.J.; Goldstein, N.I.; Young, C.S.; Fisher, P.B. The melanoma differentiation associated gene mda-7 suppresses cancer cell growth. Proc. Natl. Acad. Sci. USA 1996, 93, 9160–9165. [Google Scholar] [CrossRef]
- Huang, E.Y.; Madireddi, M.T.; Gopalkrishnan, R.V.; Leszczyniecka, M.; Su, Z.; Lebedeva, I.V.; Kang, D.; Jiang, H.; Lin, J.J.; Alexandre, D.; et al. Genomic structure, chromosomal localization and expression profile of a novel melanoma differentiation associated (mda-7) gene with cancer specific growth suppressing and apoptosis inducing properties. Oncogene 2001, 20, 7051–7063. [Google Scholar] [CrossRef]
- Lebedeva, I.V.; Sauane, M.; Gopalkrishnan, R.V.; Sarkar, D.; Su, Z.Z.; Gupta, P.; Nemunaitis, J.; Cunningham, C.; Yacoub, A.; Dent, P.; et al. Mda-7/IL-24: Exploiting cancer’s Achilles’ heel. Mol. Ther. 2005, 11, 4–18. [Google Scholar] [CrossRef]
- Gupta, P.; Su, Z.Z.; Lebedeva, I.V.; Sarkar, D.; Sauane, M.; Emdad, L.; Bachelor, M.A.; Grant, S.; Curiel, D.T.; Dent, P.; et al. Mda-7/IL-24: Multifunctional cancer-specific apoptosis-inducing cytokine. Pharmacol. Ther. 2006, 111, 596–628. [Google Scholar] [CrossRef]
- Fan, J.K.; Wei, N.; Ding, M.; Gu, J.F.; Liu, X.R.; Li, B.H.; Qi, R.; Huang, W.D.; Li, Y.H.; Xiong, X.Q.; et al. Targeting Gene-ViroTherapy for prostate cancer by DD3-driven oncolytic virus-harboring interleukin-24 gene. Int. J. Cancer 2010, 127, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.; Sachet, M.; Zinngrebe, J.; Aschacher, T.; Krainer, M.; Hegedus, B.; Walczak, H.; Bergmann, M. IL-24 sensitizes tumor cells to TLR3-mediated apoptosis. Cell Death Differ. 2013, 20, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Varghese, S.; Rabkin, S.D.; Liu, R.; Nielsen, P.G.; Ipe, T.; Martuza, R.L. Enhanced therapeutic efficacy of IL-12, but not GM-CSF, expressing oncolytic herpes simplex virus for transgenic mouse derived prostate cancers. Cancer Gene Ther. 2006, 13, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Freytag, S.O.; Barton, K.N.; Zhang, Y. Efficacy of oncolytic adenovirus expressing suicide genes and interleukin-12 in preclinical model of prostate cancer. Gene Ther. 2013, 20, 1131–1139. [Google Scholar] [CrossRef] [PubMed]
- Passer, B.J.; Cheema, T.; Wu, S.; Wu, C.L.; Rabkin, S.D.; Martuza, R.L. Combination of vinblastine and oncolytic herpes simplex virus vector expressing IL-12 therapy increases antitumor and antiangiogenic effects in prostate cancer models. Cancer Gene Ther. 2013, 20, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Varghese, S.; Rabkin, S.D.; Nielsen, P.G.; Wang, W.; Martuza, R.L. Systemic oncolytic herpes virus therapy of poorly immunogenic prostate cancer metastatic to lung. Clin. Cancer Res. 2006, 12, 2919–2927. [Google Scholar] [CrossRef] [PubMed]
- Varghese, S.; Rabkin, S.D. Oncolytic herpes simplex virus vectors for cancer virotherapy. Cancer Gene Ther. 2002, 9, 967–978. [Google Scholar] [CrossRef]
- Efferson, C.L.; Tsuda, N.; Kawano, K.; Nistal-Villán, E.; Sellappan, S.; Yu, D.; Murray, J.L.; García-Sastre, A.; Ioannides, C.G. Prostate tumor cells infected with a recombinant influenza virus expressing a truncated NS1 protein activate cytolytic CD8+ cells to recognize noninfected tumor cells. J. Virol. 2006, 80, 383–394. [Google Scholar] [CrossRef]
- Zafar, S.; Basnet, S.; Launonen, I.M.; Quixabeira, D.; Santos, J.; Hemminki, O.; Malmstedt, M.; Cervera-Carrascon, V.; Aronen, P.; Kalliokoski, R.; et al. Oncolytic Adenovirus Type 3 Coding for CD40L Facilitates Dendritic Cell Therapy of Prostate Cancer in Humanized Mice and Patient Samples. Hum. Gene Ther. 2021, 32, 192–202. [Google Scholar] [CrossRef]
- Su, Z.; Dannull, J.; Yang, B.K.; Dahm, P.; Coleman, D.; Yancey, D.; Sichi, S.; Niedzwiecki, D.; Boczkowski, D.; Gilboa, E.; et al. Telomerase mRNA-transfected dendritic cells stimulate antigen-specific CD8+ and CD4+ T cell responses in patients with metastatic prostate cancer. J. Immunol. 2005, 174, 3798–3807. [Google Scholar] [CrossRef]
- Diaconu, I.; Cerullo, V.; Hirvinen, M.L.; Escutenaire, S.; Ugolini, M.; Pesonen, S.K.; Bramante, S.; Parviainen, S.; Kanerva, A.; Loskog, A.S.; et al. Immune response is an important aspect of the antitumor effect produced by a CD40L-encoding oncolytic adenovirus. Cancer Res. 2012, 72, 2327–2338. [Google Scholar] [CrossRef] [PubMed]
- Freedman, J.D.; Duffy, M.R.; Lei-Rossmann, J.; Muntzer, A.; Scott, E.M.; Hagel, J.; Campo, L.; Bryant, R.J.; Verrill, C.; Lambert, A.; et al. An Oncolytic Virus Expressing a T-cell Engager Simultaneously Targets Cancer and Immunosuppressive Stromal Cells. Cancer Res. 2018, 78, 6852–6865. [Google Scholar] [CrossRef]
- Fearon, D.T. The carcinoma-associated fibroblast expressing fibroblast activation protein and escape from immune surveillance. Cancer Immunol. Res. 2014, 2, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.F.; Xue, S.Y.; Lu, Z.Z.; Xiao, F.J.; Yin, Y.; Zhang, Q.W.; Wu, C.T.; Wang, H.; Wang, L.S. Antitumor effects of oncolytic adenovirus armed with PSA-IZ-CD40L fusion gene against prostate cancer. Gene Ther. 2014, 21, 723–731. [Google Scholar] [CrossRef] [PubMed]
- Holl, E.K.; Brown, M.C.; Boczkowski, D.; McNamara, M.A.; George, D.J.; Bigner, D.D.; Gromeier, M.; Nair, S.K. Recombinant oncolytic poliovirus, PVSRIPO, has potent cytotoxic and innate inflammatory effects, mediating therapy in human breast and prostate cancer xenograft models. Oncotarget 2016, 7, 79828–79841. [Google Scholar] [CrossRef]
- Wang, X.; Shao, X.; Gu, L.; Jiang, K.; Wang, S.; Chen, J.; Fang, J.; Guo, X.; Yuan, M.; Shi, J.; et al. Targeting STAT3 enhances NDV-induced immunogenic cell death in prostate cancer cells. J. Cell. Mol. Med. 2020, 24, 4286–4297. [Google Scholar] [CrossRef]
- DeWeese, T.L.; van der Poel, H.; Li, S.; Mikhak, B.; Drew, R.; Goemann, M.; Hamper, U.; DeJong, R.; Detorie, N.; Rodriguez, R.; et al. A phase I trial of CV706, a replication-competent, PSA selective oncolytic adenovirus, for the treatment of locally recurrent prostate cancer following radiation therapy. Cancer Res. 2001, 61, 7464–7472. [Google Scholar]
- Small, E.J.; Carducci, M.A.; Burke, J.M.; Rodriguez, R.; Fong, L.; van Ummersen, L.; Yu, D.C.; Aimi, J.; Ando, D.; Working, P.; et al. A phase I trial of intravenous CG7870, a replication-selective, prostate-specific antigen-targeted oncolytic adenovirus, for the treatment of hormone-refractory, metastatic prostate cancer. Mol. Ther. 2006, 14, 107–117. [Google Scholar] [CrossRef]
- Freytag, S.O.; Khil, M.; Stricker, H.; Peabody, J.; Menon, M.; DePeralta-Venturina, M.; Nafziger, D.; Pegg, J.; Paielli, D.; Brown, S.; et al. Phase I study of replication-competent adenovirus-mediated double suicide gene therapy for the treatment of locally recurrent prostate cancer. Cancer Res. 2002, 62, 4968–4976. [Google Scholar]
- Freytag, S.O.; Stricker, H.; Pegg, J.; Paielli, D.; Pradhan, D.G.; Peabody, J.; DePeralta-Venturina, M.; Xia, X.; Brown, S.; Lu, M.; et al. Phase I study of replication-competent adenovirus-mediated double-suicide gene therapy in combination with conventional-dose three-dimensional conformal radiation therapy for the treatment of newly diagnosed, intermediate- to high-risk prostate cancer. Cancer Res. 2003, 63, 7497–7506. [Google Scholar]
- Freytag, S.O.; Movsas, B.; Aref, I.; Stricker, H.; Peabody, J.; Pegg, J.; Zhang, Y.; Barton, K.N.; Brown, S.L.; Lu, M.; et al. Phase I Trial of Replication-competent Adenovirus-mediated Suicide Gene Therapy Combined with IMRT for Prostate Cancer. Mol. Ther. 2007, 15, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Freytag, S.O.; Stricker, H.; Peabody, J.; Pegg, J.; Paielli, D.; Movsas, B.; Barton, K.N.; Brown, S.L.; Lu, M.; Kim, J.H. Five-year follow-up of trial of replication-competent adenovirus-mediated suicide gene therapy for treatment of prostate cancer. Mol. Ther. 2007, 15, 636–642. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, S.; Fukuhara, H.; Homma, Y.; Todo, T. Current status of clinical trials assessing oncolytic virus therapy for urological cancers. Int. J. Urol. 2017, 24, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Vidal, L.; Pandha, H.S.; Yap, T.A.; White, C.L.; Twigger, K.; Vile, R.G.; Melcher, A.; Coffey, M.; Harrington, K.J.; DeBono, J.S. A phase I study of intravenous oncolytic reovirus type 3 Dearing in patients with advanced cancer. Clin. Cancer Res. 2008, 14, 7127–7137. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, R.; Tran, H.; Selvaggi, G.; Hagerman, A.; Thompson, B.; Coffey, M. The oncolytic virus, pelareorep, as a novel anticancer agent: A review. Invest. New Drugs. 2015; 33, 761–774. [Google Scholar]
- Eder, J.P.; Kantoff, P.W.; Roper, K.; Xu, G.X.; Bubley, G.J.; Boyden, J.; Gritz, L.; Mazzara, G.; Oh, W.K.; Arlen, P.; et al. A phase I trial of a recombinant vaccinia virus expressing prostate-specific antigen in advanced prostate cancer. Clin. Cancer Res. 2000, 6, 1632–1638. [Google Scholar] [PubMed]
- Gulley, J.; Chen, A.P.; Dahut, W.; Arlen, P.M.; Bastian, A.; Steinberg, S.M.; Tsang, K.; Panicali, D.; Poole, D.; Schlom, J.; et al. Phase I study of a vaccine using recombinant vaccinia virus expressing PSA (rV-PSA) in patients with metastatic androgen-independent prostate cancer. Prostate 2002, 53, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Wang, W.; Manola, J.; DiPaola, R.S.; Ko, Y.J.; Sweeney, C.; Whiteside, T.L.; Schlom, J.; Wilding, G.; Weiner, L.M. Phase II randomized study of vaccine treatment of advanced prostate cancer (E7897): A trial of the Eastern Cooperative Oncology Group. J. Clin. Oncol. 2004, 22, 2122–2132. [Google Scholar] [CrossRef] [PubMed]
- DiPaola, R.S.; Plante, M.; Kaufman, H.; Petrylak, D.P.; Israeli, R.; Lattime, E.; Manson, K.; Schuetz, T. A phase I trial of pox PSA vaccines (PROSTVAC-VF) with B7-1, ICAM-1, and LFA-3 co-stimulatory molecules (TRICOM) in patients with prostate cancer. J. Transl. Med. 2006, 4, 1. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Schuetz, T.J.; Blumenstein, B.A.; Glode, L.M.; Bilhartz, D.L.; Wyand, M.; Manson, K.; Panicali, D.L.; Laus, R.; Schlom, J.; et al. Overall survival analysis of a phase II randomized controlled trial of a Poxviral-based PSA-targeted immunotherapy in metastatic castration-resistant prostate cancer. J. Clin. Oncol. 2010, 28, 1099–1105. [Google Scholar] [CrossRef]
- Parsons, J.K.; Pinto, P.A.; Pavlovich, C.P.; Uchio, E.; Kim, H.L.; Nguyen, M.N.; Gulley, J.L.; Jamieson, C.; Hsu, P.; Wojtowicz, M.; et al. A Randomized, Double-blind, Phase II Trial of PSA-TRICOM (PROSTVAC) in Patients with Localized Prostate Cancer: The Immunotherapy to Prevent Progression on Active Surveillance Study. Eur. Urol. Focus 2018, 4, 636–638. [Google Scholar] [CrossRef]
- Gulley, J.L.; Borre, M.; Vogelzang, N.J.; Ng, S.; Agarwal, N.; Parker, C.C.; Pook, D.W.; Rathenborg, P.; Flaig, T.W.; Carles, J.; et al. Phase III Trial of PROSTVAC in Asymptomatic or Minimally Symptomatic Metastatic Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2019, 37, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Nakai, Y.; Kawashima, A.; Ujike, T.; Nagahara, A.; Nakajima, T.; Inoue, T.; Lee, C.M.; Uemura, M.; Miyagawa, Y.; et al. Phase I/II clinical trial to assess safety and efficacy of intratumoral and subcutaneous injection of HVJ-E in castration-resistant prostate cancer patients. Cancer Gene Ther. 2017, 24, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Kato, T.; Hatano, K.; Kawashima, A.; Ujike, T.; Uemura, M.; Imamura, R.; Okihara, K.; Ukimura, O.; Miki, T.; et al. Intratumoral and s.c. injection of inactivated hemagglutinating virus of Japan envelope (GEN0101) in metastatic castration-resistant prostate cancer. Cancer Sci. 2020, 111, 1692–1698. [Google Scholar] [CrossRef] [PubMed]
- TTopalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Isaacsson Velho, P.; Antonarakis, E.S. PD-1/PD-L1 pathway inhibitors in advanced prostate cancer. Expert Rev. Clin. Pharmacol. 2018, 11, 475–486. [Google Scholar] [CrossRef]
- Xu, Y.; Song, G.; Xie, S.; Jiang, W.; Chen, X.; Chu, M.; Hu, X.; Wang, Z.W. The roles of PD-1/PD-L1 in the prognosis and immunotherapy of prostate cancer. Mol. Ther. 2021, 29, 1958–1969. [Google Scholar] [CrossRef]
- Majidpoor, J.; Mortezaee, K. The efficacy of PD-1/PD-L1 blockade in cold cancers and future perspectives. Clin. Immunol. 2021, 226, 108707. [Google Scholar] [CrossRef]
- Sharma, M.; Yang, Z.; Miyamoto, H. Immunohistochemistry of immune checkpoint markers PD-1 and PD-L1 in prostate cancer. Medicine 2019, 98, e17257. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef]
- Higano, C.S.; Small, E.J.; Schellhammer, P.; Yasothan, U.; Gubernick, S.; Kirkpatrick, P.; Kantoff, P.W. Sipuleucel-T. Nat. Rev. Drug Discov. 2010, 9, 513–514. [Google Scholar] [CrossRef]
- Yi, R.; Chen, B.; Duan, P.; Zheng, C.; Shen, H.; Liu, Q.; Yuan, C.; Ou, W.; Zhou, Z. Sipuleucel-T and Androgen Receptor-Directed Therapy for Castration-Resistant Prostate Cancer: A Meta-Analysis. J. Immunol. Res. 2016, 2016, 4543861. [Google Scholar] [CrossRef] [PubMed]
- Handy, C.E.; Antonarakis, E.S. Sipuleucel-T for the treatment of prostate cancer: Novel insights and future directions. Future Oncol. 2018, 14, 907–917. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, S.I.M.; Ju, X.; Horvath, L.G.; Clark, G.J. Moving on From Sipuleucel-T: New Dendritic Cell Vaccine Strategies for Prostate Cancer. Front. Immunol. 2021, 12, 641307. [Google Scholar] [CrossRef] [PubMed]



| Name | Baltimore Classification | Family | Capsid Shape |
|---|---|---|---|
| DNA virus | |||
| Adenovirus | Group I: dsDNA | Adenoviridae | Icosahedral |
| Herpes simplex virus | Group I: dsDNA | Herpesviridae | Icosahedral |
| Vaccinia virus | Group I: dsDNA | Poxviridae | Complex |
| RNA virus | |||
| Reovirus | Group III: dsRNA | Reoviridae | Icosahedral |
| Measles virus | Group V: ss(−) RNA | Paramyxoviridae | Icosahedral |
| Newcastle disease virus | Group V: ss(−) RNA | Paramyxoviridae | Helical |
| Vesicular stomatitis virus | Group V ss(−) RNA | Rhabdoviridae | Helical |
| Coxsackievirus | Group IV: ssRNA | Picornaviridae | Icosahedral |
| Poliovirus | Group IV: ss(+) RNA | Picornaviridae | Icosahedral |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, G.; Liu, Y.; Liu, S.; Lin, Y.; Hu, C. Oncolyic Virotherapy for Prostate Cancer: Lighting a Fire in Winter. Int. J. Mol. Sci. 2022, 23, 12647. https://doi.org/10.3390/ijms232012647
Wang G, Liu Y, Liu S, Lin Y, Hu C. Oncolyic Virotherapy for Prostate Cancer: Lighting a Fire in Winter. International Journal of Molecular Sciences. 2022; 23(20):12647. https://doi.org/10.3390/ijms232012647
Chicago/Turabian StyleWang, Gongwei, Ying Liu, Shuoru Liu, Yuan Lin, and Cheng Hu. 2022. "Oncolyic Virotherapy for Prostate Cancer: Lighting a Fire in Winter" International Journal of Molecular Sciences 23, no. 20: 12647. https://doi.org/10.3390/ijms232012647
APA StyleWang, G., Liu, Y., Liu, S., Lin, Y., & Hu, C. (2022). Oncolyic Virotherapy for Prostate Cancer: Lighting a Fire in Winter. International Journal of Molecular Sciences, 23(20), 12647. https://doi.org/10.3390/ijms232012647