


Single-Cell DNA Methylation Analysis of Chicken Lampbrush Chromosomes

 , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
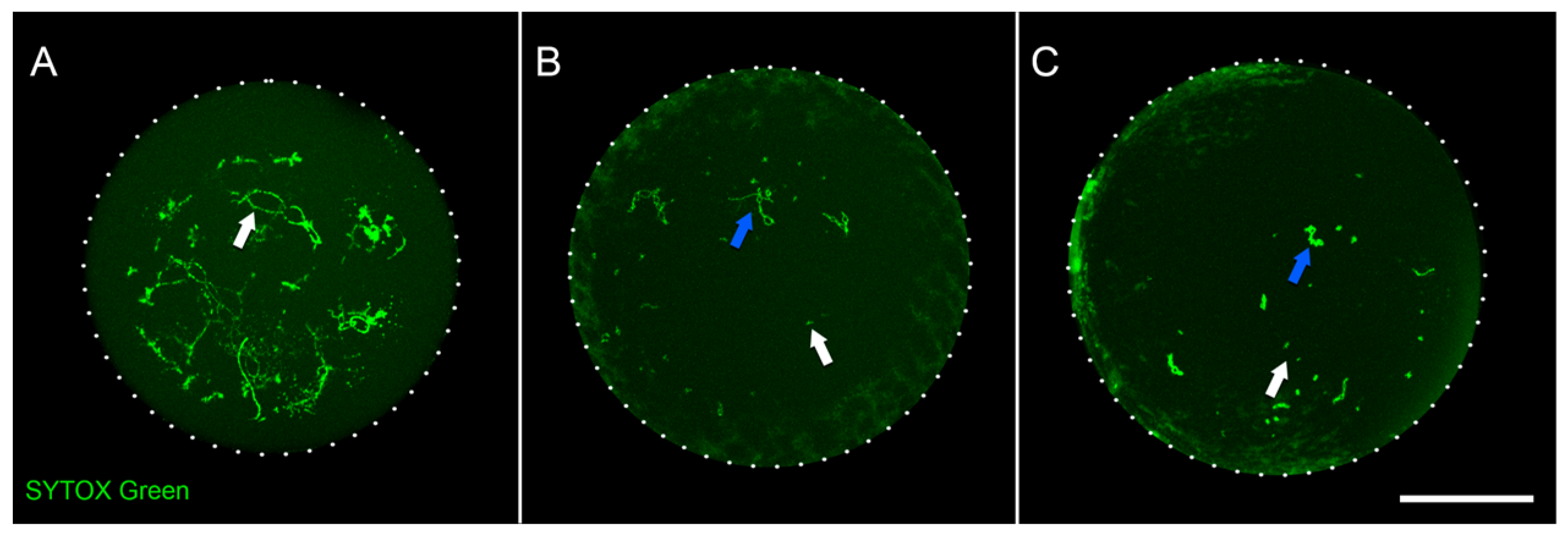
2.1. Chicken Oocytes Isolation and Bisulfite Sequencing
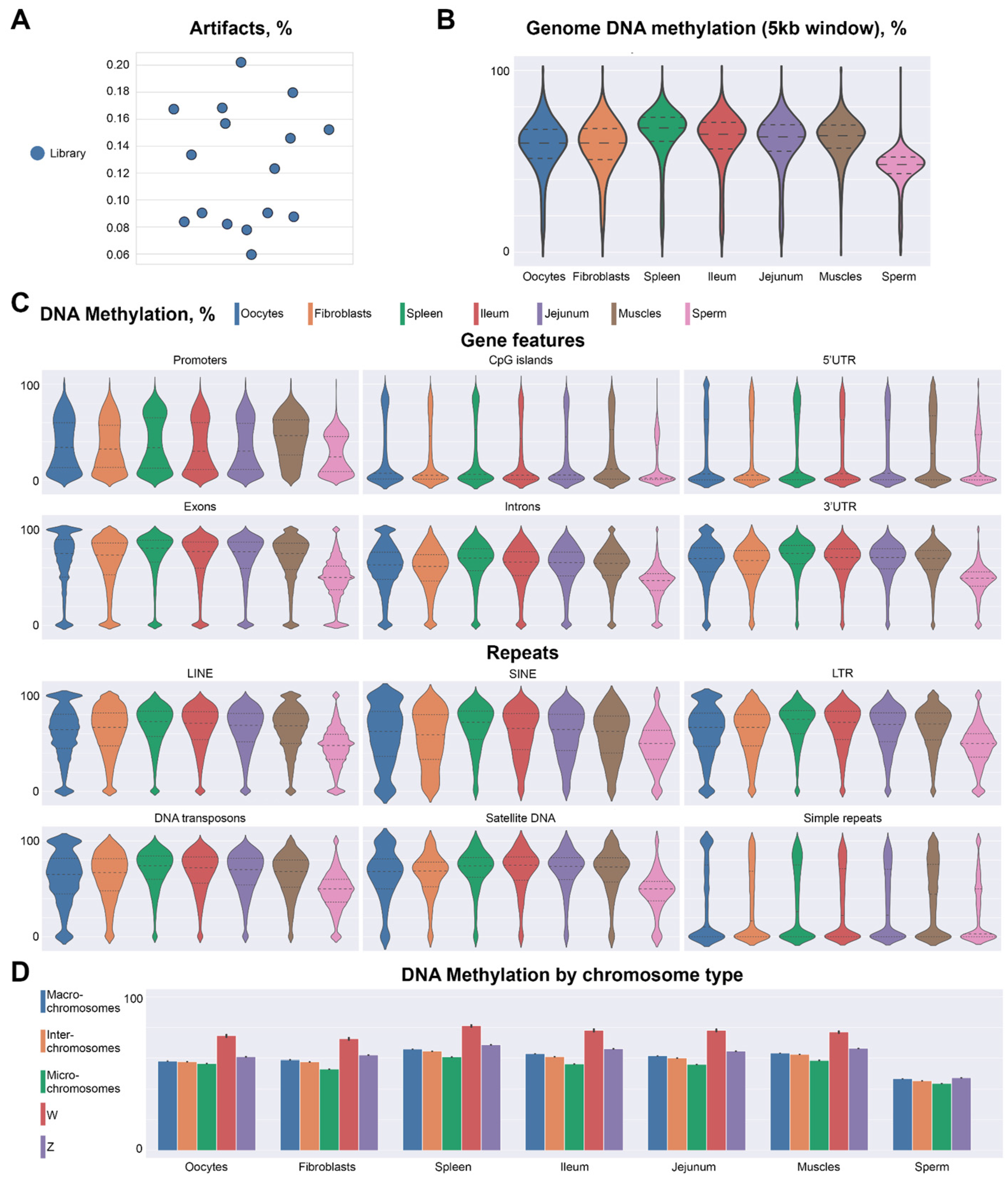
2.2. Methylation Profile of Chicken Oocytes
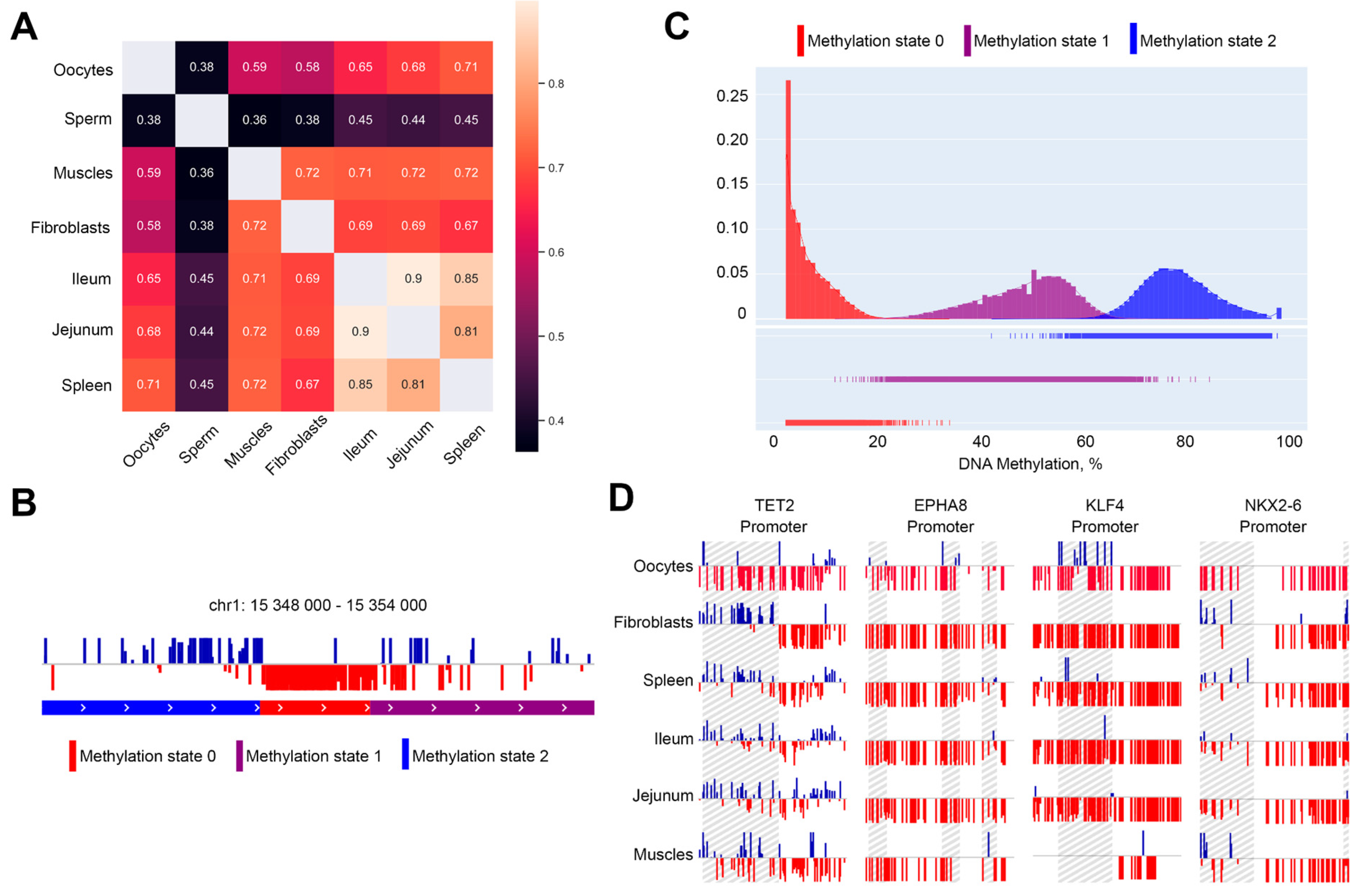
2.3. Methylation-Informed Segmentation of the Chicken Genome Identifies Oocyte-Specific Hypomethylated Regions
3. Discussion
4. Materials and Methods
4.1. Nuclei Isolation
4.2. Staining of Oocyte Nuclei and Confocal Microscopy
4.3. Library Preparation and Sequencing
4.4. Data Analysis
4.4.1. Raw Data Preparation
4.4.2. HMM
4.5. Data Availability
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Greenberg, M.V.C.; Bourc’His, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef] [PubMed]
- Sendžikaitė, G.; Kelsey, G. The role and mechanisms of DNA methylation in the oocyte. Essays Biochem. 2019, 63, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Fishman, V.; Battulin, N.; Nuriddinov, M.; Maslova, A.; Zlotina, A.; Strunov, A.; Chervyakova, D.; Korablev, A.; Serov, O.; Krasikova, A. 3D organization of chicken genome demonstrates evolutionary conservation of topologically associated domains and highlights unique architecture of erythrocytes’ chromatin. Nucleic Acids Res. 2019, 47, 648–665. [Google Scholar] [CrossRef]
- Hwang, Y.S.; Seo, M.; Bang, S.; Kim, H.; Han, J.Y. Transcriptional and translational dynamics during maternal-to-zygotic transition in early chicken development. FASEB J. 2018, 32, 2004–2011. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Sun, C.; Clinton, M.; Yang, N. Dynamic Transcriptional Landscape of the Early Chick Embryo. Front. Cell Dev. Biol. 2019, 7, 196. [Google Scholar] [CrossRef] [PubMed]
- Jhanwar, S.; Malkmus, J.; Stolte, J.; Romashkina, O.; Zuniga, A.; Zeller, R. Conserved and species-specific chromatin remodeling and regulatory dynamics during mouse and chicken limb bud development. Nat. Commun. 2021, 12, 5685. [Google Scholar] [CrossRef]
- Raddatz, G.; Arsenault, R.J.; Aylward, B.; Whelan, R.; Böhl, F.; Lyko, F. A chicken DNA methylation clock for the prediction of broiler health. Commun. Biol. 2021, 4, 76. [Google Scholar] [CrossRef]
- Krasikova, A.V.; Kulikova, T.V. Distribution of heterochromatin markers in lampbrush chromosomes in birds. Russ. J. Genet. 2017, 53, 1022–1029. [Google Scholar] [CrossRef]
- Kulikova, T.; Surkova, A.; Zlotina, A.; Krasikova, A. Mapping epigenetic modifications on chicken lampbrush chromosomes. Mol. Cytogenet. 2020, 13, 32. [Google Scholar] [CrossRef]
- Krasikova, A.; Khodyuchenko, T.; Maslova, A.; Vasilevskaya, E. Three-dimensional organisation of RNA-processing machinery in avian growing oocyte nucleus. Chromosom. Res. 2012, 20, 979–994. [Google Scholar] [CrossRef]
- Maslova, A.; Krasikova, A. Nuclear actin depolymerization in transcriptionally active avian and amphibian oocytes leads to collapse of intranuclear structures. Nucleus 2012, 3, 300–311. [Google Scholar] [CrossRef][Green Version]
- Gridina, M.; Taskina, A.; Lagunov, T.; Nurislamov, A.; Kulikova, T.; Krasikova, A.; Fishman, V. Comparison and Critical Assessment of Single-Cell Hi-C Protocols. bioRxiv 2022, 1–12. [Google Scholar] [CrossRef]
- Gu, C.; Liu, S.; Wu, Q.; Zhang, L.; Guo, F. Integrative single-cell analysis of transcriptome, DNA methylome and chromatin accessibility in mouse oocytes. Cell Res. 2019, 29, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Al Adhami, H.; Bardet, A.F.; Dumas, M.; Cleroux, E.; Guibert, S.; Fauque, P.; Acloque, H.; Weber, M. A comparative methylome analysis reveals conservation and divergence of DNA methylation patterns and functions in vertebrates. BMC Biol. 2022, 20, 70. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Yan, F.-B.; Li, F.; Jiang, K.-R.; Li, D.-H.; Han, R.-L.; Li, Z.-J.; Jiang, R.-R.; Liu, X.-J.; Kang, X.-T.; et al. Genome-wide DNA methylation profiles reveal novel candidate genes associated with meat quality at different age stages in hens. Sci. Rep. 2017, 7, srep45564. [Google Scholar] [CrossRef]
- Mugal, C.F.; Arndt, P.F.; Holm, L.; Ellegren, H. Evolutionary Consequences of DNA Methylation on the GC Content in Vertebrate Genomes. G3 Genes|Genomes|Genetics 2015, 5, 441–447. [Google Scholar] [CrossRef]
- Fudenberg, G.; Imakaev, M.; Lu, C.; Goloborodko, A.; Abdennur, N.; Mirny, L.A. Formation of Chromosomal Domains by Loop Extrusion. Cell Rep. 2016, 15, 2038–2049. [Google Scholar] [CrossRef]
- Hansen, A.S. CTCF as a boundary factor for cohesin-mediated loop extrusion: Evidence for a multi-step mechanism. Nucleus 2020, 11, 132–148. [Google Scholar] [CrossRef]
- Kulikova, T.; Maslova, A.; Starshova, P.; Ramos, J.S.R.; Krasikova, A. Comparison of the somatic TADs and lampbrush chromomere-loop complexes in transcriptionally active prophase I oocytes. Chromosoma 2022. [Google Scholar] [CrossRef]
- Deryusheva, S.; Krasikova, A.; Kulikova, T.; Gaginskaya, E. Tandem 41-bp repeats in chicken and Japanese quail genomes: FISH mapping and transcription analysis on lampbrush chromosomes. Chromosoma 2007, 116, 519–530. [Google Scholar] [CrossRef]
- Krasikova, A.; Fukagawa, T.; Zlotina, A. High-resolution mapping and transcriptional activity analysis of chicken centromere sequences on giant lampbrush chromosomes. Chromosom. Res. 2012, 20, 995–1008. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sommerville, J.; Baird, J.; Turner, B.M. Histone H4 acetylation and transcription in amphibian chromatin. J. Cell Biol. 1993, 120, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Murani, E.; Ponsuksili, S.; Wimmers, K. biomvRhsmm: Genomic Segmentation with Hidden Semi-Markov Model. BioMed Res. Int. 2014, 2014, 910390. [Google Scholar] [CrossRef]
- National Research Council. Guide for the Care and Use of Laboratory Animals, 8th ed.; The National Academies Press: Washington, DC, USA, 2011; p. 12910. [Google Scholar] [CrossRef]
- Pino, F.M.; Sukei, E. Fmorenopino/HeterogeneousHMM: First Stable Release of HeterogenousHMM. Available online: https://zenodo.org/record/3759439 (accessed on 17 September 2022).
- Shokoohi, F.; Stephens, D.A.; Bourque, G.; Pastinen, T.; Greenwood, C.M.T.; Labbe, A. A hidden markov model for identifying differentially methylated sites in bisulfite sequencing data. Biometrics 2019, 75, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Kwok, C.K.; Jiang, H.; Fan, X. Detect differentially methylated regions using non-homogeneous hidden Markov model for bisulfite sequencing data. Methods 2021, 189, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Pino, F.; Sükei, E.; Olmos, P.M.; Artés-Rodríguez, A. PyHHMM: A Python Library for Heterogeneous Hidden Markov Models. arXiv 2022, arXiv:2201.06968. [Google Scholar]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nurislamov, A.; Lagunov, T.; Gridina, M.; Krasikova, A.; Fishman, V. Single-Cell DNA Methylation Analysis of Chicken Lampbrush Chromosomes. Int. J. Mol. Sci. 2022, 23, 12601. https://doi.org/10.3390/ijms232012601
Nurislamov A, Lagunov T, Gridina M, Krasikova A, Fishman V. Single-Cell DNA Methylation Analysis of Chicken Lampbrush Chromosomes. International Journal of Molecular Sciences. 2022; 23(20):12601. https://doi.org/10.3390/ijms232012601
Chicago/Turabian StyleNurislamov, Artem, Timofey Lagunov, Maria Gridina, Alla Krasikova, and Veniamin Fishman. 2022. "Single-Cell DNA Methylation Analysis of Chicken Lampbrush Chromosomes" International Journal of Molecular Sciences 23, no. 20: 12601. https://doi.org/10.3390/ijms232012601
APA StyleNurislamov, A., Lagunov, T., Gridina, M., Krasikova, A., & Fishman, V. (2022). Single-Cell DNA Methylation Analysis of Chicken Lampbrush Chromosomes. International Journal of Molecular Sciences, 23(20), 12601. https://doi.org/10.3390/ijms232012601