
Regulation of Molecular Targets in Osteosarcoma Treatment




{kind=link}
{kind=link}
Abstract
1. Introduction
2. Molecular Targets in Osteosarcoma Treatment
2.1. Ubiquitin-Specific Protease 1
2.2. ErbB Receptor Family
2.3. Lysophosphatidic Acid Acyltransferase ß (LPAATβ)
2.4. Notch Signaling Pathway
2.5. Extracellular Matrix Molecules
2.6. MicroRNAs and Protein Interactions
2.6.1. NOB1
2.6.2. HMGB1
2.6.3. MIF
2.7. DNA-PKcs
2.8. GREM1
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mutsaers, A.J.; Walkley, C.R. Cells of Origin in Osteosarcoma: Mesenchymal Stem Cells or Osteoblast Committed Cells? Bone 2014, 62, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Ottaviani, G.; Jaffe, N. The Epidemiology of Osteosarcoma. Cancer Treat. Res. 2009, 152, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-M.; Wang, W.; Qui, E.-D. Osteosarcoma Cells Induce Differentiation of Mesenchymal Stem Cells into Cancer Associated Fibroblasts through Notch and Akt Signaling Pathways. Int. J. Clin. Exp. Pathol. 2017, 10, 8479–8486. [Google Scholar] [PubMed]
- Chen, X.; Bahrami, A.; Pappo, A.; Easton, J.; Dalton, J.; Hedlund, E.; Ellison, D.; Shurtleff, S.; Wu, G.; Wei, L.; et al. Recurrent Somatic Structural Variations Contribute to Tumorigenesis in Pediatric Osteosarcoma. Cell Rep. 2014, 7, 104–112. [Google Scholar] [CrossRef]
- Sayles, L.C.; Breese, M.R.; Koehne, A.L.; Leung, S.G.; Lee, A.G.; Liu, H.Y.; Spillinger, A.; Shah, A.T.; Tanasa, B.; Straessler, K.; et al. Genome-Informed Targeted Therapy for Osteosarcoma. Cancer Discov. 2019, 9, 43–46. [Google Scholar] [CrossRef]
- Zhang, J.; Edmonson, M.N.; Hedges, D.; Yergeau, D.A.; Chen, X.; Nuccio, R.; Rusch, M.; Wang, S.; Mardis, E.R.; Ellison, D.W.; et al. Germline Mutations in Predisposition Genes in Pediatric Cancer. N. Engl. J. Med. 2015, 373, 2336–2382. [Google Scholar] [CrossRef]
- Saraf, A.J.; Fenger, J.M.; Roberts, R.D. Osteosarcoma: Accelerating Progress Makes for a Hopeful Future. Front. Oncol. 2018, 8, 26. [Google Scholar] [CrossRef]
- Bosma, S.E.; Wong, K.C.; Paul, L.; Gerbers, J.G.; Jutte, P.C. A Cadaveric Comparative Study on the Surgical Accuracy of Freehand, Computer Navigation, and Patient-Specific Instruments in Joint-Preserving Bone Tumor Resections. Sarcoma 2018, 2018, 4065846. [Google Scholar] [CrossRef]
- Liu, J.; Zhu, H.; Zhong, N.; Jiang, Z.; Xu, L.; Deng, Y.; Jiang, Z.; Wang, H.W.; Wang, J. Gene Silencing of USP1 by Lentivirus Effectively Inhibits Proliferation and Invasion of Human Osteosarcoma Cells. Int. J. Oncol. 2016, 49, 2549–2557. [Google Scholar] [CrossRef]
- Jullien, N.; Dieudonné, F.; Habel, N.; Marty, C.; Modrowski, D.; Patino, A.; Lecanda, F.; Sévère, N.; Marie, P.J. ErbB3 Silencing Reduces Osteosarcoma Cell Proliferation and Tumor Growth in Vivo. Gene 2013, 521, 55–61. [Google Scholar] [CrossRef]
- Guimarães, G.M.; Tesser-Gamba, F.; Petrilli, A.S.; Donato-Macedo, C.R.P.; Alves, M.T.S.; de Lima, F.T.; Garcia-Filho, R.J.; Oliveira, R.; Toledo, S.R.C. Molecular Profiling of Osteosarcoma in Children and Adolescents from Different Age Groups Using a Next-Generation Sequencing Panel. Cancer Genet. 2021, 258–259, 85–92. [Google Scholar] [CrossRef] [PubMed]
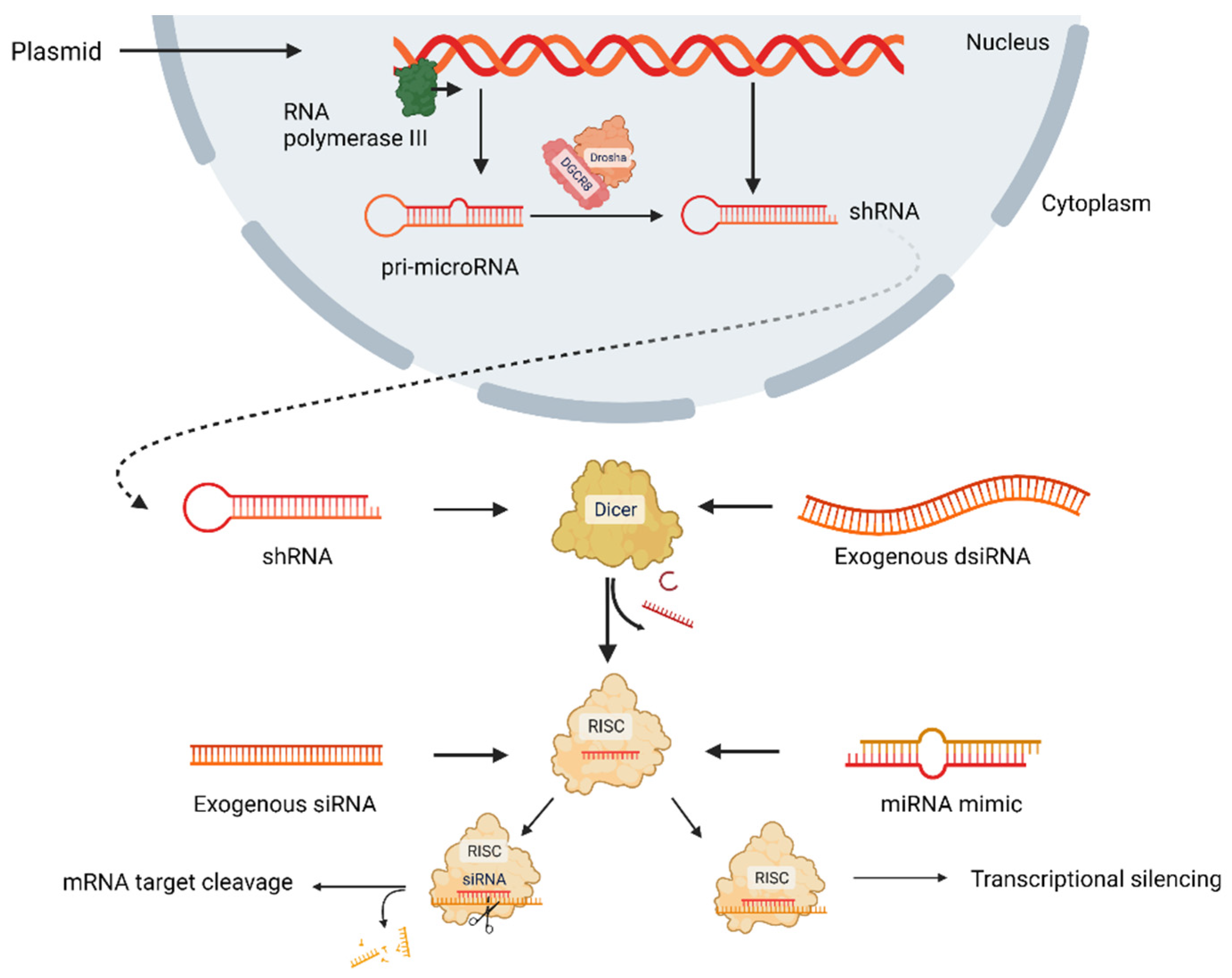
- Bobbin, M.L.; Rossi, J.J. RNA Interference (RNAi)-Based Therapeutics: Delivering on the Promise? Annu. Rev. Pharmacol. Toxicol. 2016, 56, 103–122. [Google Scholar] [CrossRef] [PubMed]
- Mahmoodi Chalbatani, G.; Dana, H.; Gharagouzloo, E.; Grijalvo, S.; Eritja, R.; Logsdon, C.D.; Memari, F.; Miri, S.R.; Rad, M.R.; Marmari, V. Small Interfering RNAs (SiRNAs) in Cancer Therapy: A Nano-Based Approach. Int. J. Nanomed. 2019, 14, 3111–3128. [Google Scholar] [CrossRef]
- Hammond, S.M. Dicing and Slicing: The Core Machinery of the RNA Interference Pathway. FEBS Lett. 2005, 579, 5822–5829. [Google Scholar] [CrossRef] [PubMed]
- Łabno, A.; Tomecki, R.; Dziembowski, A. Cytoplasmic RNA Decay Pathways—Enzymes and Mechanisms. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 3125–3147. [Google Scholar] [CrossRef]
- Tian, Z.; Liang, G.; Cui, K.; Liang, Y.; Wang, Q.; Lv, S.; Cheng, X.; Zhang, L. Insight Into the Prospects for RNAi Therapy of Cancer. Front. Pharmacol. 2021, 12, 644718. [Google Scholar] [CrossRef]
- Lu, T.X.; Rothenberg, M.E. MicroRNA. J. Allergy Clin. Immunol. 2018, 141, 1202–1207. [Google Scholar] [CrossRef]
- Davidson, B.L.; McCray, P.B. Current Prospects for RNA Interference-Based Therapies. Nat. Rev. Genet. 2011, 12, 329–340. [Google Scholar] [CrossRef]
- Ou, L.; Przybilla, M.J.; Koniar, B.L.; Whitley, C.B. Elements of Lentiviral Vector Design toward Gene Therapy for Treating Mucopolysaccharidosis I. Mol. Genet. Metab. Rep. 2016, 8, 87–93. [Google Scholar] [CrossRef]
- Gu, Q.; Luo, Y.; Chen, C.; Jiang, D.; Huang, Q.; Wang, X. GREM1 Overexpression Inhibits Proliferation, Migration and Angiogenesis of Osteosarcoma. Exp. Cell Res. 2019, 384, 111619. [Google Scholar] [CrossRef]
- Jullien, N.; Maudinet, A.; Leloutre, B.; Ringe, J.; Marie, P.J. Downregulation of ErbB3 by Wnt3a contributes to wnt-induced osteoblast differentiation in mesenchymal cells. J. Cell. Biochem. 2012, 113, 2047–2056. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Schmitt, S.; Buac, D.; Dou, Q.P. Targeting the Ubiquitin-Proteasome System for Cancer Therapy. Expert Opin. Targets 2013, 17, 1091–1108. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.; Feng, Z.; Huang, H.; Wang, G.; Chen, Z.; Xu, G.; Xie, Z.; Jie, Z.; Zhao, X.; Ma, Q.; et al. USP1 Inhibition Suppresses the Progression of Osteosarcoma via Destabilizing TAZ. Int. J. Biol. Sci. 2022, 18, 3122–3136. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Liu, Z.; Yang, Q. The Role of Ubiquitination and Deubiquitination in Cancer Metabolism. Signal Transduct. Target. Ther. 2020, 5, 11. [Google Scholar] [CrossRef]
- Liu, Y.; Luo, X.; Hu, H.; Wang, R.; Sun, Y.; Zeng, R.; Chen, H. Integrative Proteomics and Tissue Microarray Profiling Indicate the Association between Overexpressed Serum Proteins and Non-Small Cell Lung Cancer. PLoS ONE 2012, 7, e0051748. [Google Scholar] [CrossRef]
- Xu, X.; Li, S.; Cui, X.; Han, K.; Wang, J.; Hou, X.; Cui, L.; He, S.; Xiao, J.; Yang, Y. Inhibition of Ubiquitin Specific Protease 1 Sensitizes Colorectal Cancer Cells to DNA-Damaging Chemotherapeutics. Front. Oncol. 2019, 9, 1406. [Google Scholar] [CrossRef]
- Chen, F.; Chen, L.; Qin, Q.; Sun, X. Salt-Inducible Kinase 2: An Oncogenic Signal Transmitter and Potential Target for Cancer Therapy. Front. Oncol. 2019, 9, 18. [Google Scholar] [CrossRef]
- Bon, H.; Wadhwa, K.; Schreiner, A.; Osborne, M.; Carroll, T.; Ramos-Montoya, A.; Ross-Adams, H.; Visser, M.; Hoffmann, R.; Ahmed, A.A.; et al. Salt-Inducible Kinase 2 Regulates Mitotic Progression and Transcription in Prostate Cancer. Mol. Cancer Res. 2015, 13, 620–635. [Google Scholar] [CrossRef]
- Williams, S.A.; Maecker, H.L.; French, D.M.; Liu, J.; Gregg, A.; Silverstein, L.B.; Cao, T.C.; Carano, R.A.D.; Dixit, V.M. USP1 Deubiquitinates ID Proteins to Preserve a Mesenchymal Stem Cell Program in Osteosarcoma. Cell 2011, 146, 918–930. [Google Scholar] [CrossRef]
- Wrighton, K.H. Tumorigenesis: USP1 Keeps ID Proteins Stable. Nat. Rev. Cancer 2011, 11, 757. [Google Scholar] [CrossRef]
- Zheng, S.; Qiao, G.; Min, D.; Zhang, Z.; Lin, F.; Yang, Q.; Feng, T.; Tang, L.; Sun, Y.; Zhao, H.; et al. Heterogeneous Expression and Biological Function of Ubiquitin Carboxy-Terminal Hydrolase-L1 in Osteosarcoma. Cancer Lett. 2014, 359, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Sun, H.; Cheng, C.; Wang, G. BRCA1-Associated Protein-1 Suppresses Osteosarcoma Cell Proliferation and Migration Through Regulation PI3K/Akt Pathway. DNA Cell Biol. 2017, 36, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Li, Z.; Zhao, X.; Guo, L.; Yu, C.; Qin, J.; Zhang, S.; Zhang, Y.; Yang, X. Ubiquitin-Specific Protease 7 Promotes Osteosarcoma Cell Metastasis by Inducing Epithelial-Mesenchymal Transition. Oncol. Rep. 2019, 41, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Bhanumathy, K.K.; Balagopal, A.; Vizeacoumar, F.S.; Vizeacoumar, F.J.; Freywald, A.; Giambra, V.; Bhanumathy, K.; Balagopal, K.; Vizeacoumar, A.; Vizeacoumar, F.S.; et al. Protein Tyrosine Kinases: Their Roles and Their Targeting in Leukemia. Cancers 2021, 13, 184. [Google Scholar] [CrossRef]
- Wang, S.; Huang, X.; Lee, C.K.; Liu, B. Elevated Expression of ErbB3 Confers Paclitaxel Resistance in ErbB2-Overexpressing Breast Cancer Cells via Upregulation of Survivin. Oncogene 2010, 29, 4225–4236. [Google Scholar] [CrossRef]
- Wieduwilt, M.J.; Moasser, M.M. The Epidermal Growth Factor Receptor Family: Biology Driving Targeted Therapeutics. Cell Mol. Life Sci. 2011, 65, 1566–1584. [Google Scholar] [CrossRef]
- Schaefer, K.L.; Brachwitz, K.; Braun, Y.; Diallo, R.; Wai, D.H.; Zahn, S.; Schneider, D.T.; Kuhnen, C.; Vollmann, A.; Brockhoff, G.; et al. Constitutive Activation of Neuregulin/ERBB3 Signaling Pathway in Clear Cell Sarcoma of Soft Tissue. Neoplasia 2006, 8, 613–622. [Google Scholar] [CrossRef]
- Huang, Z.; Wang, S.L.; Chen, H.; Shen, R.K.; Li, X.D.; Huang, Q.S.; Wu, C.Y.; Weng, D.F.; Lin, J.H. Clinicopathological and Prognostic Values of ErbB Receptor Family Amplification in Primary Osteosarcoma. Scand. J. Clin. Lab. Investig. 2019, 79, 601–612. [Google Scholar] [CrossRef]
- Mu, X.; Isaac, C.; Schott, T.; Huard, J.; Weiss, K. Rapamycin Inhibits ALDH Activity, Resistance to Oxidative Stress, and Metastatic Potential in Murine Osteosarcoma Cells. Sarcoma 2013, 2013, 11. [Google Scholar] [CrossRef]
- Knight, M.N.; Hankenson, K.D. Mesenchymal Stem Cells in Bone Regeneration. Adv. Wound Care (New Rochelle) 2013, 2, 306–316. [Google Scholar] [CrossRef]
- Niesporek, S.; Denkert, C.; Weichert, W.; Kö Bel, M.; Noske, A.; Sehouli, J.; Singer, J.W.; Dietel, M.; Hauptmann, S. Expression of Lysophosphatidic Acid Acyltransferase Beta (LPAAT-b) in Ovarian Carcinoma: Correlation with Tumour Grading and Prognosis. Br. J. Cancer 2005, 92, 1729–1736. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Duan, P.; Gan, Y.; Li, P.; Zhao, C.; Xu, J.; Zhang, Z.; Zhou, Q. Silencing LPAATβ Inhibits Tumor Growth of Cisplatin-Resistant Human Osteosarcoma in Vivo and in Vitro. Int. J. Oncol. 2017, 50, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Chang, Z.; Wang, W.; Wang, B. Rapamycin-Mediated MTOR Inhibition Reverses Drug Resistance to Adriamycin in Colon Cancer Cells. Available online: https://pubmed.ncbi.nlm.nih.gov/26902021/ (accessed on 7 December 2021).
- Randle, R.A.; Raguz, S.; Higgins, C.F.; Yag¨ue, E.; Yag¨ue, Y. Role of the Highly Structured 5-End Region of MDR1 MRNA in P-Glycoprotein Expression. Biochem. J. 2007, 406, 445–455. [Google Scholar] [CrossRef]
- Marklein, D.; Graab, U.; Naumann, I.; Yan, T.; Ridzewski, R.; Nitzki, F.; Rosenberger, A.; Dittmann, K.; Wienands, J.; Wojnowski, L.; et al. PI3K Inhibition Enhances Doxorubicin-Induced Apoptosis in Sarcoma Cells. PLoS ONE 2012, 7, e0052898. [Google Scholar] [CrossRef]
- Gan, Y.; Wang, Y.; Tan, Z.; Zhou, J.; Kitazawa, R.; Jiang, X.; Tang, Y.; Yang, J. TDRG1 Regulates Chemosensitivity of Seminoma TCam-2 Cells to Cisplatin via PI3K/Akt/MTOR Signaling Pathway and Mitochondria-Mediated Apoptotic Pathway. Cancer Biol. 2016, 17, 741–750. [Google Scholar] [CrossRef]
- Diefenbach, C.S.M.; Soslow, R.A.; Iasonos, A.; Linkov, I.; Hedvat, C.; Bonham, L.; Singer, J.; Barakat, R.R.; Aghajanian, C.; Dupont, J. Lysophosphatidic Acid Acyltransferase-Beta (LPAAT-Beta) Is Highly Expressed in Advanced Ovarian Cancer and Is Associated with Aggressive Histology and Poor Survival. Cancer 2006, 107, 1511–1519. [Google Scholar] [CrossRef]
- Springett, G.M.; Bonham, L.; Hummer, A.; Linkov, I.; Misra, D.; Ma, C.; Pezzoni, G.; di Giovine, S.; Singer, J.; Kawasaki, H.; et al. Lysophosphatidic Acid Acyltransferase-B Is a Prognostic Marker and Therapeutic Target in Gynecologic Malignancies. Am. Assoc. Cancer Res. 2005, 65, 9415–9425. [Google Scholar] [CrossRef]
- Colombo, M.; Galletti, S.; Garavelli, S.; Platonova, N.; Paoli, A.; Basile, A.; Taiana, E.; Neri, A.; Chiaramonte, R. Notch Signaling Deregulation in Multiple Myeloma: A Rational Molecular Target. Oncotarget 2015, 6, 26826–26840. [Google Scholar] [CrossRef]
- Wolter, J. The Notch Signaling Pathway in Embryogenesis | The Embryo Project Encyclopedia. Available online: https://embryo.asu.edu/pages/notch-signaling-pathway-embryogenesis (accessed on 7 December 2021).
- Cao, J.; Wei, Y.; Lian, J.; Yang, L.; Zhang, X.; Xie, J.; Liu, Q.; Luo, J.; He, B.; Tang, M. Notch Signaling Pathway Promotes Osteogenic Differentiation of Mesenchymal Stem Cells by Enhancing BMP9/Smad Signaling. Int. J. Mol. Med. 2017, 40, 378–388. [Google Scholar] [CrossRef]
- Zanotti, S.; Canalis, E. Notch Signaling and the Skeleton. Endocr. Rev. 2016, 37, 223. [Google Scholar] [CrossRef]
- Bazzoni, R.; Bentivegna, A. Role of Notch Signaling Pathway in Glioblastoma Pathogenesis. Cancers 2019, 11, 292. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Shang, X.; Zhang, H.; Wang, G.; Massey, P.A.; Barton, S.R.; Kevil, C.G.; Dong, Y. Notch Signaling in Osteogenesis, Osteoclastogenesis, and Angiogenesis. Am. J. Pathol. 2019, 189, 1495–1500. [Google Scholar] [CrossRef]
- Si, J.; Wang, C.; Zhang, D.; Wang, B.; Hou, W.; Zhou, Y. Osteopontin in Bone Metabolism and Bone Diseases. Med. Sci. Monit. 2020, 26, e919159. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Zhang, C. The Role of Bone Morphogenetic Proteins in Liver Fibrosis. Gastroenterol. Hepatol. Open Access 2021, 12, 17–20. [Google Scholar] [CrossRef]
- Parrow, N.L.; Fleming, R.E. Bone Morphogenetic Proteins as Regulators of Iron Metabolism. Annu. Rev. Nutr. 2014, 34, 77–94. [Google Scholar] [CrossRef]
- Deng, Z.H.; Li, Y.S.; Gao, X.; Lei, G.H.; Huard, J. Bone Morphogenetic Proteins for Articular Cartilage Regeneration. Osteoarthr. Cartil. 2018, 26, 1153–1161. [Google Scholar] [CrossRef]
- Nikolic, I.; Yung, L.M.; Yang, P.; Malhotra, R.; Paskin-Flerlage, S.D.; Dinter, T.; Bocobo, G.A.; Tumelty, K.E.; Faugno, A.J.; Troncone, L.; et al. Bone Morphogenetic Protein 9 Is a Mechanistic Biomarker of Portopulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2019, 199, 891–902. [Google Scholar] [CrossRef]
- Desroches-Castan, A.; Tillet, E.; Bouvard, C.; Bailly, S. BMP9 and BMP10: Two Close Vascular Quiescence Partners That Stand Out. Dev. Dyn. 2021, 251, 178–197. [Google Scholar] [CrossRef]
- Majumdar, M.K.; Wang, E.; Morris, E.A. BMP-2 and BMP-9 Promote Chondrogenic Differentiation of Human Multipotential Mesenchymal Cells and Overcome the Inhibitory Effect of IL-1. J. Cell. Physiol. 2001, 189, 275–284. [Google Scholar] [CrossRef]
- Hughes, D.P.M. How the NOTCH Pathway Contributes to the Ability of Osteosarcoma Cells to Metastasize. Cancer Treat. Res. 2009, 152, 479–496. [Google Scholar] [CrossRef]
- Tao, J.; Jiang, M.M.; Jiang, L.; Salvo, J.S.; Zeng, H.C.; Dawson, B.; Bertin, T.K.; Rao, P.H.; Chen, R.; Donehower, L.A.; et al. Notch Activation as a Driver of Osteogenic Sarcoma. Cancer Cell 2014, 26, 390–401. [Google Scholar] [CrossRef] [PubMed]
- di Martino, J.S.; Akhter, T.; Bravo-Cordero, J.J. Remodeling the ECM: Implications for Metastasis and Tumor Dormancy. Cancers 2021, 13, 4916. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Dean, D.; Hornicek, F.J.; Chen, Z.; Duan, Z. The Role of Extracelluar Matrix in Osteosarcoma Progression and Metastasis. J. Exp. Clin. Cancer Res. 2020, 39, 178. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental Regulation of Tumor Progression and Metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Bergamaschi, A.; Tagliabue, E.; Sørlie, T.; Naume, B.; Triulzi, T.; Orlandi, R.; Russnes, H.G.; Nesland, J.M.; Tammi, R.; Auvinen, P.; et al. Extracellular Matrix Signature Identifies Breast Cancer Subgroups with Different Clinical Outcome. J. Pathol. 2008, 214, 357–367. [Google Scholar] [CrossRef]
- Theocharis, A.D.; Manou, D.; Karamanos, N.K. The Extracellular Matrix as a Multitasking Player in Disease. FEBS J. 2019, 286, 2830–2869. [Google Scholar] [CrossRef]
- Casale, J.; Crane, J.S. Biochemistry, Glycosaminoglycans. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Sasisekharan, R.; Shriver, Z.; Venkataraman, G.; Narayanasami, U. Roles of Heparan-Sulphate Glycosaminoglycans in Cancer. Nat. Rev. Cancer 2002, 2, 521–528. [Google Scholar] [CrossRef]
- Afratis, N.; Gialeli, C.; Nikitovic, D.; Tsegenidis, T.; Karousou, E.; Theocharis, A.D.; Pavão, M.S.; Tzanakakis, G.N.; Karamanos, N.K. Glycosaminoglycans: Key Players in Cancer Cell Biology and Treatment. FEBS J. 2012, 279, 1177–1197. [Google Scholar] [CrossRef]
- Asano, K.; Nelson, C.M.; Nandadasa, S.; Aramaki-Hattori, N.; Lindner, D.J.; Alban, T.; Inagaki, J.; Ohtsuki, T.; Oohashi, T.; Apte, S.S.; et al. Stromal Versican Regulates Tumor Growth by Promoting Angiogenesis. Sci. Rep. 2017, 7, 17225. [Google Scholar] [CrossRef]
- Bi, X.L.; Yang, W. Biological Functions of Decorin in. Cancer. Chin. J. Cancer 2013, 32, 266. [Google Scholar] [CrossRef]
- Skandalis, S.S.; Labropoulou, V.T.; Ravazoula, P.; Likaki-Karatza, E.; Dobra, K.; Kalofonos, H.P.; Karamanos, N.K.; Theocharis, A.D. Versican but Not Decorin Accumulation Is Related to Malignancy in Mammographically Detected High Density and Malignant-Appearing Microcalcifications in Non-Palpable Breast Carcinomas. BMC Cancer 2011, 11, 314. [Google Scholar] [CrossRef] [PubMed]
- Zafiropoulos, A.; Nikitovic, D.; Katonis, P.; Tsatsakis, A.; Karamanos, N.K.; Tzanakakis, G.N. Decorin-Induced Growth Inhibition Is Overcome through Protracted Expression and Activation of Epidermal Growth Factor Receptors in Osteosarcoma Cells. Mol. Cancer Res. 2008, 6, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Theocharis, A.D.; Tsolakis, I.; Tzanakakis, G.N.; Karamanos, N.K. Chondroitin Sulfate as a Key Molecule in the Development of Atherosclerosis and Cancer Progression. Adv. Pharm. 2006, 53, 281–295. [Google Scholar] [CrossRef]
- Nikitovic, D.; Zafiropoulos, A.; Katonis, P.; Tsatsakis, A.; Theocharis, A.D.; Karamanos, N.K.; Tzanakakis, G.N. Transforming Growth Factor-β as a Key Molecule Triggering the Expression of Versican Isoforms V0 and V1, Hyaluronan Synthase-2 and Synthesis of Hyaluronan in Malignant Osteosarcoma Cells. IUBMB Life 2006, 58, 47–53. [Google Scholar] [CrossRef]
- Nikitovic, D.; Zafiropoulos, A.; Tzanakakis, G.N.; Karamanos, N.K.; Tsatsakis, A.M. Effects of Glycosaminoglycans on Cell Proliferation of Normal Osteoblasts and Human Osteosarcoma Cells Depend on Their Type and Fine Chemical Compositions. Anticancer Res. 2005, 25, 2851–2856. [Google Scholar]
- Song, B.; Yan, J.; Liu, C.; Zhou, H.; Zheng, Y. Tumor Suppressor Role of Mir-363-3p in Gastric Cancer. Med. Sci. Monit. 2015, 21, 4074–4080. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, T.; Huang, H.; Jiang, Y.; Yang, L.; Lin, Z.; He, H.; Liu, T.; Wu, B.; Chen, J.; et al. MiR-363-3p Inhibits Tumor Growth by Targeting PCNA in Lung Adenocarcinoma. Oncotarget 2013, 8, 20133–20144. [Google Scholar] [CrossRef]
- Hu, F.; Min, J.; Cao, X.; Liu, L.; Ge, Z.; Hu, J.; Li, X. MiR-363-3p Inhibits the Epithelial-to-Mesenchymal Transition and Suppresses Metastasis in Colorectal Cancer by Targeting Sox4. Biochem. Biophys. Res. Commun. 2016, 474, 35–42. [Google Scholar] [CrossRef]
- He, F.; Fang, L.; Yin, Q. MiR-363 Acts as a Tumor Suppressor in Osteosarcoma Cells by Inhibiting PDZD2. Oncol. Rep. 2019, 41, 2729–2738. [Google Scholar] [CrossRef]
- Fatica, A.; Tollervey, D.; Dlakic´3, M.; Dlakic´3, D. PIN Domain of Nob1p Is Required for D-Site Cleavage in 20S Pre-RRNA. RNA 2004, 10, 1698–1701. [Google Scholar] [CrossRef]
- Wang, H.; Li, P.; Zhao, B. Knockdown of NOB1 Expression by RNAi Inhibits Cellular Proliferation and Migration in Human Gliomas. Gene 2013, 528, 146–153. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, F.; Wang, L.; Zhang, Q. MiR-363 Suppresses Cell Migration, Invasion, and Epithelial-Mesenchymal Transition of Osteosarcoma by Binding to NOB1. World J. Surg Oncol. 2020, 18, 83. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Liu, J.; Wu, D.; Qin, Y.; Peng, C.; Li, C.; Wang, J. Gene Silencing of NOB1 by Lentivirus Suppresses Growth and Migration of Human Osteosarcoma Cells. Mol. Med. Rep. 2014, 9, 2173. [Google Scholar] [CrossRef]
- Pandit, B.; Gartel, A.L. Thiazole Antibiotic Thiostrepton Synergize with Bortezomib to Induce Apoptosis in Cancer Cells. PLoS ONE 2011, 6, e0017110. [Google Scholar] [CrossRef]
- Shapovalov, Y.; Benavidez, D.; Zuch, D.; Eliseev, R.A. Proteasome Inhibition with Bortezomib Suppresses Growth and Induces Apoptosis in Osteosarcoma. Int. J. Cancer 2010, 127, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Pandit, B.; Bhat, U.; Gartel, A.L. Proteasome Inhibitory Activity of Thiazole Antibiotics. Cancer Biol. 2011, 11, 43–47. [Google Scholar] [CrossRef] [PubMed]
- HMGB1 High Mobility Group Box 1 [Homo Sapiens (Human)]-Gene-NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene/3146 (accessed on 7 December 2021).
- Kang, R.; Livesey, K.M.; Zeh, H.J.; Lotze, M.T.; Tang, D. HMGB1: A Novel Beclin 1-Binding Protein Active in Autophagy. Autophagy 2010, 6, 1209–1211. [Google Scholar] [CrossRef]
- Wu, L.; Yang, L. The Function and Mechanism of HMGB1 in Lung Cancer and Its Potential Therapeutic Implications. Oncol. Lett. 2018, 15, 6799–6805. [Google Scholar] [CrossRef]
- Huang, J.; Ni, J.; Liu, K.; Yu, Y.; Xie, M.; Kang, R.; Vernon, P.; Cao, L.; Tang, D. HMGB1 Promotes Drug Resistance in Osteosarcoma. Cancer Res. 2012, 72, 230–238. [Google Scholar] [CrossRef]
- Huang, J.; Liu, K.; Yu, Y.; Xie, M.; Kang, R.; Vernon, P.; Cao, L.; Tang, D.; Ni, J. Targeting HMGB1-Mediated Autophagy as a Novel Therapeutic Strategy for Osteosarcoma. Autophagy 2012, 8, 275–277. [Google Scholar] [CrossRef]
- Charoonpatrapong, K.; Shah, R.; Robling, A.G.; Alvarez, M.; Clapp, D.W.; Chen, S.; Kopp, R.P.; Pavalko, F.M.; Yu, J.; Bidwell, J.P. HMGB1 Expression and Release by Bone Cells. J. Cell. Physiol. 2006, 207, 480–490. [Google Scholar] [CrossRef]
- Liu, Y.J.; Li, W.; Chang, F.; Liu, J.N.; Lin, J.X.; Chen, D.X. MicroRNA-505 Is Downregulated in Human Osteosarcoma and Regulates Cell Proliferation, Migration and Invasion. Oncol. Rep. 2018, 39, 491–500. [Google Scholar] [CrossRef]
- NCBI MIF Macrophage Migration Inhibitory Factor [Homo Sapiens (Human)]. Available online: https://www.ncbi.nlm.nih.gov/gene/4282 (accessed on 7 December 2021).
- Sumaiya, K.; Langford, D.; Natarajaseenivasan, K.; Shanmughapriya, S. Macrophage Migration Inhibitory Factor (MIF): A Multifaceted Cytokine Regulated by Genetic and Physiological Strategies. Pharmacol. Ther. 2021, 233, 108024. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Jiang, N.; Guo, R.; Jiang, W.; He, Q.M.; Xu, Y.F.; Li, Y.Q.; Tang, L.L.; Mao, Y.P.; Sun, Y.; et al. MiR-451 Inhibits Cell Growth and Invasion by Targeting MIF and Is Associated with Survival in Nasopharyngeal Carcinoma. Mol. Cancer 2013, 12, 123. [Google Scholar] [CrossRef]
- Tian, Y.; Nan, Y.; Han, L.; Zhang, A.; Wang, G.; Jia, Z.; Hao, J.; Pu, P.; Zhong, Y.; Kang, C. MicroRNA MiR-451 Downregulates the PI3K/AKT Pathway through CAB39 in Human Glioma. Int. J. Oncol. 2012, 40, 1105–1112. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Wang, Z.X.; Yang, J.S.; Pan, X.; De, W.; Chen, L.B. MicroRNA-451 Functions as a Tumor Suppressor in Human Non-Small Cell Lung Cancer by Targeting Ras-Related Protein 14 (RAB14). Oncogene 2011, 30, 2644–2658. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Liu, S.Y.; He, Y.b.; Huang, R.L.; Deng, S.Y.; Ni, G.X.; Yu, B. MiR-451 Suppresses Proliferation, Migration and Promotes Apoptosis of the Human Osteosarcoma by Targeting Macrophage Migration Inhibitory Factor. Biomed. Pharmacother. 2017, 87, 621–627. [Google Scholar] [CrossRef] [PubMed]
- le Romencer, M.; Cosulich, S.C.; Jackson, S.P.; Clarke, P.R. Cleavage and Inactivation of DNA-Dependent Protein Kinase Catalytic Subunit. J. Cell Sci. 1996, 109, 3121–3127. [Google Scholar] [CrossRef]
- Fang, X.; Huang, Z.; Zhai, K.; Huang, Q.; Tao, W.; Kim, L.; Wu, Q.; Almasan, A.; Yu, J.S.; Li, X.; et al. Inhibiting DNA-PK Induces Glioma Stem Cell Differentiation and Sensitizes Glioblastoma to Radiation in Mice. Sci. Transl. Med. 2021, 13, eabc7275. [Google Scholar] [CrossRef]
- Jin, P.Y.; Lu, H.J.; Tang, Y.; Fan, S.H.; Zhang, Z.F.; Wang, Y.; Li, X.N.; Wu, D.M.; Lu, J.; Zheng, Y.L. The Effect of DNA-PKcs Gene Silencing on Proliferation, Migration, Invasion and Apoptosis, and in Vivo Tumorigenicity of Human Osteosarcoma MG-63 Cells. Biomed. Pharmacother. 2017, 96, 1324–1334. [Google Scholar] [CrossRef]
- Karagiannis, G.S.; Berk, A.; Dimitromanolakis, A.; Diamandis, E.P. Enrichment Map Profiling of the Cancer Invasion Front Suggests Regulation of Colorectal Cancer Progression by the Bone Morphogenetic Protein Antagonist, Gremlin-1. Mol. Oncol. 2013, 7, 826–839. [Google Scholar] [CrossRef] [PubMed]
- Karagiannis, G.S.; Musrap, N.; Saraon, P.; Treacy, A.; Schaeffer, D.F.; Kirsch, R.; Riddell, R.H.; Diamandis, E.P. Bone Morphogenetic Protein Antagonist Gremlin-1 Regulates Colon Cancer Progression. Biol. Chem. 2015, 396, 163–183. [Google Scholar] [CrossRef]
- Ren, J.; Smid, M.; Iaria, J.; Salvatori, D.C.F.; van Dam, H.; Zhu, H.J.; Martens, J.W.M.; ten Dijke, P. Cancer-Associated Fibroblast-Derived Gremlin 1 Promotes Breast Cancer Progression. Breast Cancer Res. 2019, 21, 109. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Tissari, M.; Tamminen, J.; Ylivinkka, I.; Rönty, M.; von Nandelstadh, P.; Lehti, K.; Hyytiäinen, M.; Myllärniemi, M.; Koli, K. Gremlin-1 Is a Key Regulator of the Invasive Cell Phenotype in Mesothelioma. Oncotarget 2017, 8, 98280. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Cheng, W.; Zou, C.; Wang, T.; Cao, Z. Gremlin1 Promotes Carcinogenesis of Glioma in Vitro. Clin. Exp. Pharm. Physiol. 2017, 44, 244–256. [Google Scholar] [CrossRef]
- Cho, W.H.; Lee, S.Y.; Song, W.S.; Park, J.H. Osteosarcoma in Pre-Adolescent Patients. J. Int. Med. Res. 2006, 34, 676–681. [Google Scholar] [CrossRef]
- Nair, J.K.; Willoughby, J.L.; Chan, A. Multivalent N-Acetylgalactosamine-Conjugated SiRNA Localizes in Hepatocytes and Elicits Robust RNAi-Mediated Gene Silencing. Publication Date. J. Am. Chem. Soc. 2014, 136, 16958–16961. [Google Scholar] [CrossRef]
- Watts, J.K.; Deleavey, G.F.; Damha, M.J. Chemically Modified SiRNA: Tools and Applications. Drug Discov. Today 2008, 13, 842–855. [Google Scholar] [CrossRef]
- Dong, Y.; Siegwart, D.J.; Anderson, D.G. Strategies, Design, and Chemistry in SiRNA Delivery Systems. Adv. Drug Deliv. Rev. 2019, 144, 133–147. [Google Scholar] [CrossRef]
- Shim, M.S.; Kwon, Y.J. Efficient and Targeted Delivery of SiRNA in Vivo. FEBS J. 2010, 277, 4814–4827. [Google Scholar] [CrossRef]
- Goguen, R.P.; del Corpo, O.; Malard, C.M.G.; Daher, A.; Alpuche-Lazcano, S.P.; Chen, M.J.; Scarborough, R.J.; Gatignol, A. Efficacy, Accumulation, and Transcriptional Profile of Anti-HIV ShRNAs Expressed from Human U6, 7SK, and H1 Promoters. Mol. Nucleic Acids 2021, 23, 1020–1034. [Google Scholar] [CrossRef] [PubMed]
- Dubey, P.K.; Singodia, D.; Vyas, S.P. Liposomes Modified with YIGSR Peptide for Tumor Targeting. J. Drug Target. 2010, 18, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Cheung, C.S.F.; Lui, J.C.; Baron, J. Identification of Chondrocyte-Binding Peptides by Phage Display. J. Orthop. Res. 2013, 31, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Shan, Z.; Zhou, X.; Peng, L.; Zhi, C.; Chai, J.; Liu, H.; Yang, J.; Zhang, Z. Knockdown of LncRNA GHET1 Inhibits Osteosarcoma Cells Proliferation, Invasion, Migration and EMT in Vitro and in Vivo. Cancer Biomark. 2018, 23, 589–601. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Celik, B.; Cicek, K.; Leal, A.F.; Tomatsu, S. Regulation of Molecular Targets in Osteosarcoma Treatment. Int. J. Mol. Sci. 2022, 23, 12583. https://doi.org/10.3390/ijms232012583
Celik B, Cicek K, Leal AF, Tomatsu S. Regulation of Molecular Targets in Osteosarcoma Treatment. International Journal of Molecular Sciences. 2022; 23(20):12583. https://doi.org/10.3390/ijms232012583
Chicago/Turabian StyleCelik, Betul, Kader Cicek, Andrés Felipe Leal, and Shunji Tomatsu. 2022. "Regulation of Molecular Targets in Osteosarcoma Treatment" International Journal of Molecular Sciences 23, no. 20: 12583. https://doi.org/10.3390/ijms232012583
APA StyleCelik, B., Cicek, K., Leal, A. F., & Tomatsu, S. (2022). Regulation of Molecular Targets in Osteosarcoma Treatment. International Journal of Molecular Sciences, 23(20), 12583. https://doi.org/10.3390/ijms232012583