
Targeting CXCR4 and CD47 Receptors: An Overview of New and Old Molecules for a Biological Personalized Anticancer Therapy


Abstract
1. Introduction
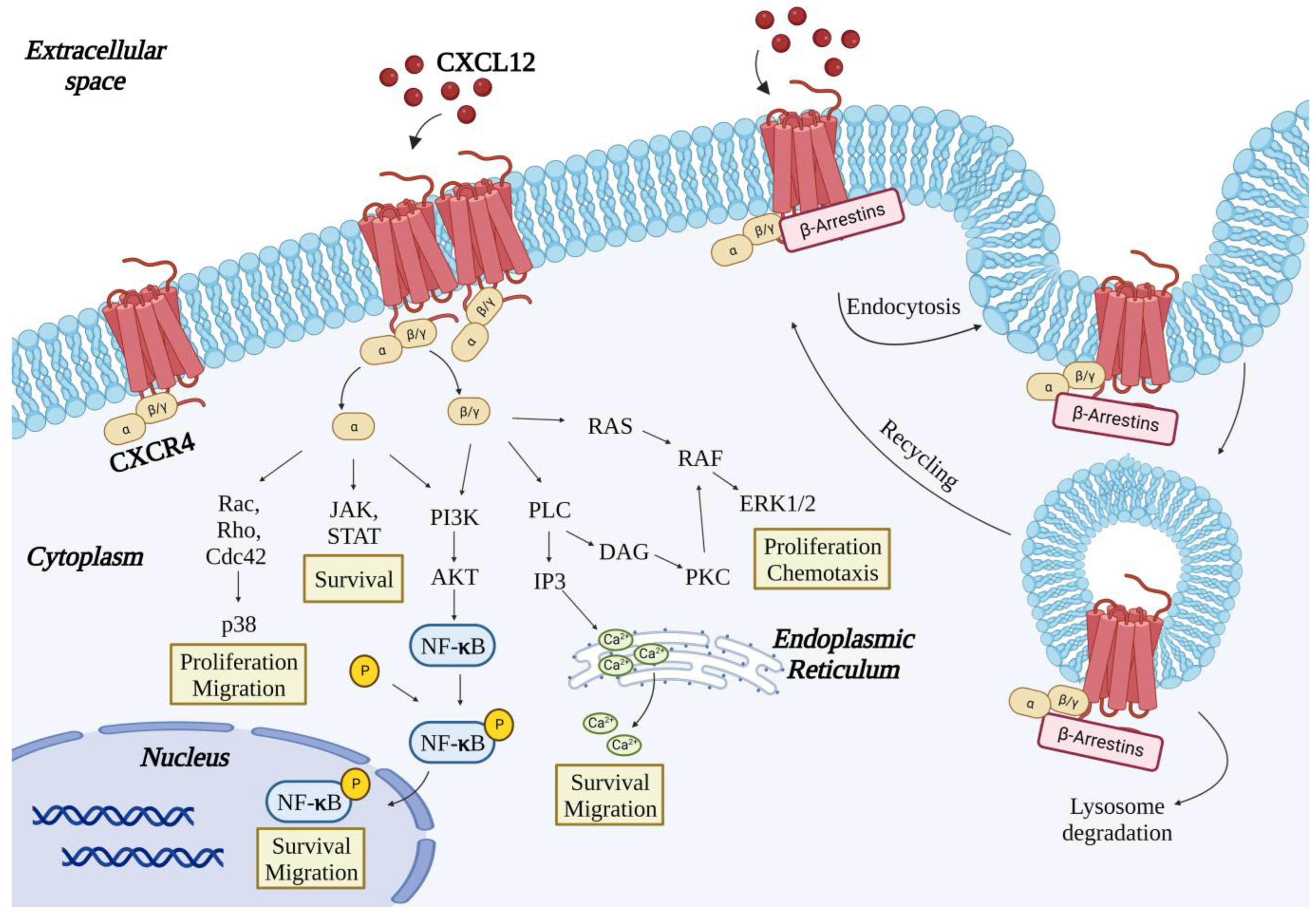
2. The CXCR4 Receptor
3. Binding of CXCL12 to CXCR4
4. The CXCR7 Receptor
5. CXCR4 Antagonists
- (1)
- nonpeptide CXCR4 antagonists;
- (2)
- small-peptide CXCR4 antagonists;
- (3)
- antibodies to CXCR4.
5.1. Nonpeptide CXCR4 Antagonists
5.2. Small-Peptide CXCR4 Antagonists
5.3. Antibodies to CXCR4
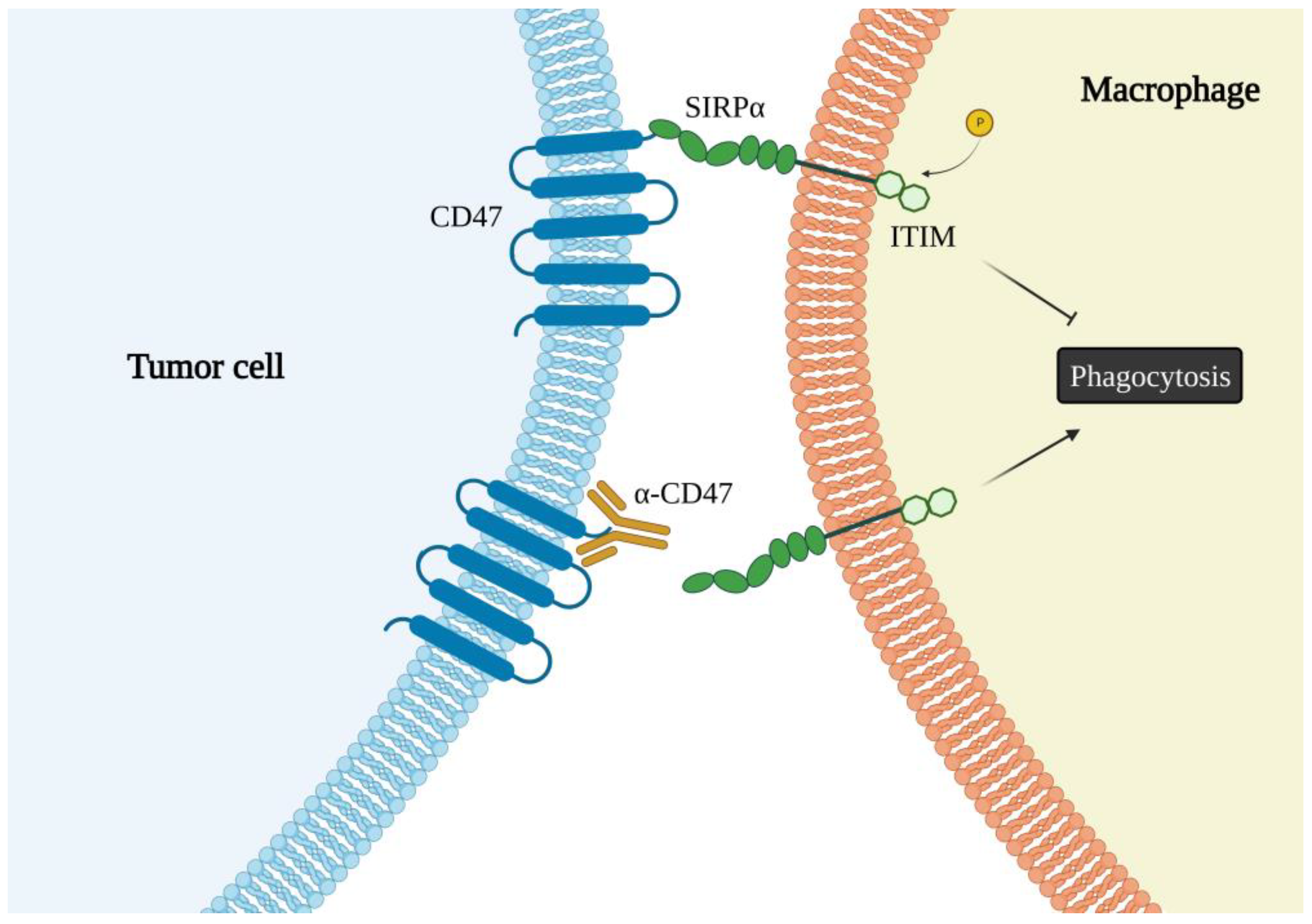
6. The CD47 Receptor
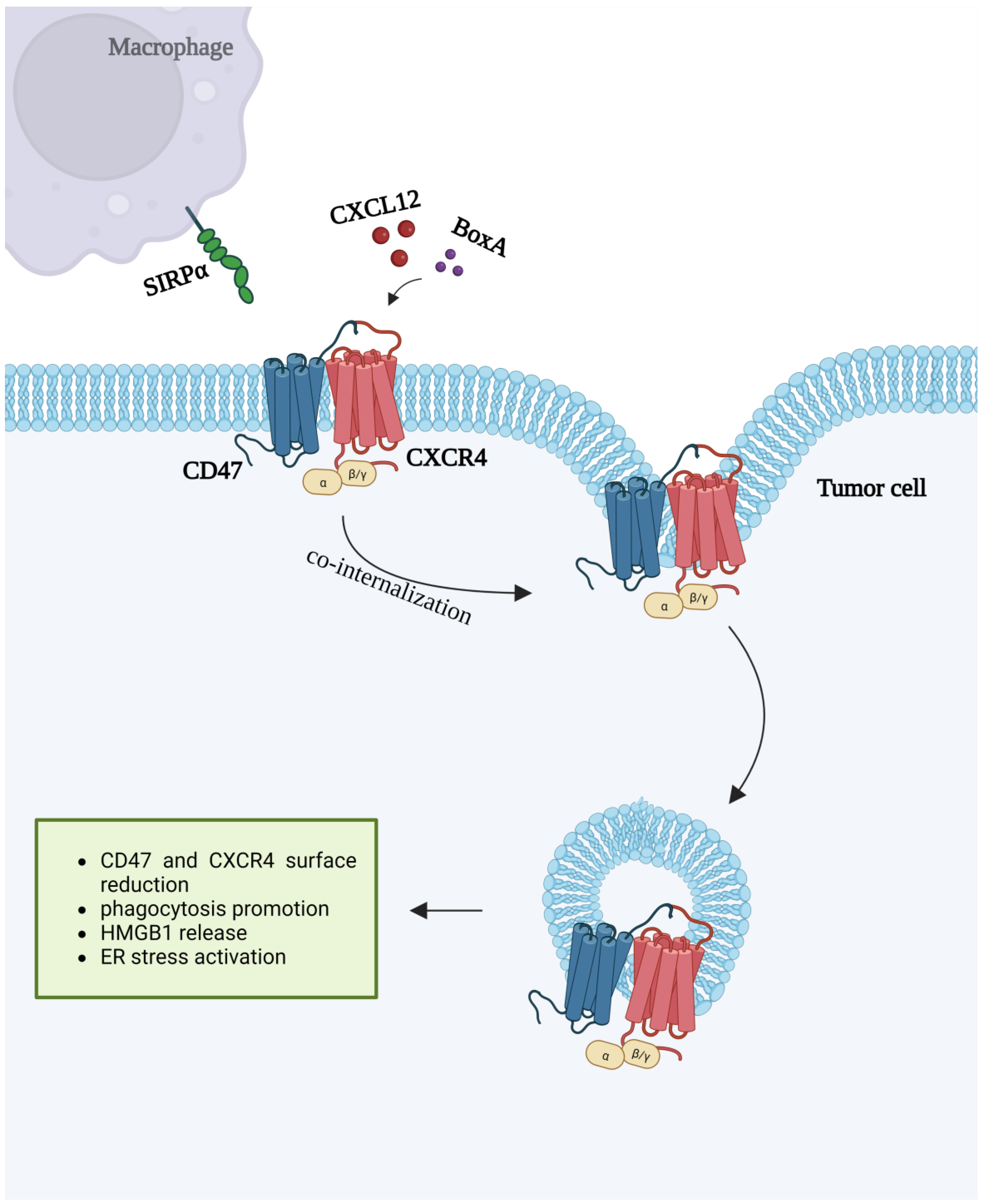
7. CXCR4 and CD47 Interactions
8. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Papież, M.A.; Krzyściak, W. Biological therapies in the treatment of cancer—Update and new directions. Int. J. Mol. Sci. 2021, 22, 1694. [Google Scholar] [CrossRef] [PubMed]
- Han, X.J.; Ma, X.L.; Yang, L.; Wei, Y.Q.; Peng, Y.; Wei, X.W. Progress in Neoantigen Targeted Cancer Immunotherapies. Front. Cell Dev. Biol. 2020, 8, 728. [Google Scholar] [CrossRef] [PubMed]
- Luker, G.D.; Yang, J.; Richmond, A.; Scala, S.; Festuccia, C.; Schottelius, M.; Wester, H.J.; Zimmermann, J. At the Bench: Pre-clinical evidence for multiple functions of CXCR4 in cancer. J. Leukoc. Biol. 2021, 109, 969–989. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M.E.; McClanahan, T.; Murphy, E.; Yuan, W.; Wagner, S.N.; et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 410, 5016. [Google Scholar] [CrossRef]
- Subramanian, S.; Parthasarathy, R.; Sen, S.; Boder, E.T.; Discher, D.E. Species- and cell type-specific interactions between CD47 and human SIRPα. Blood 2006, 107, 1463. [Google Scholar] [CrossRef]
- Adams, S.; van der Laan, L.J.; Vernon-Wilson, E.; Renardel de Lavalette, C.; Döpp, E.A.; Dijkstra, C.D.; Simmons, D.L.; van den Berg, T.K. Signal-regulatory protein is selectively expressed by myeloid and neuronal cells. J. Immunol. 1998, 161, 1853–1859. [Google Scholar]
- Logtenberg, M.E.W.; Scheeren, F.A.; Schumacher, T.N. The CD47-SIRPα Immune Checkpoint. Immunity 2020, 52, 11. [Google Scholar] [CrossRef]
- Kharitonenkov, A.; Chen, Z.; Sures, I.; Wang, H.; Schilling, J.; Ullrich, A. A family of proteins that inhibit signalling through tyrosine kinase receptors. Nature 1997, 386, 181–186. [Google Scholar] [CrossRef]
- Chao, M.P.; Jaiswal, S.; Weissman-Tsukamoto, R.; Alizadeh, A.A.; Gentles, A.J.; Volkmer, J.; Weiskopf, K.; Willingham, S.B.; Raveh, T.; Park, C.Y.; et al. Calreticulin is the dominant pro-phagocytic signal on multiple human cancers and is counterbalanced by CD47. Sci. Transl. Med. 2010, 2, 1375. [Google Scholar] [CrossRef]
- Martínez-Muñoz, L.; Rodríguez-Frade, J.M.; Barroso, R.; Sorzano, C.Ó.S.; Torreño-Pina, J.A.; Santiago, C.A.; Manzo, C.; Lucas, P.; García-Cuesta, E.M.; Gutierrez, E.; et al. Separating Actin-Dependent Chemokine Receptor Nanoclustering from Dimerization Indicates a Role for Clustering in CXCR4 Signaling and Function. Mol. Cell 2018, 70, 34. [Google Scholar] [CrossRef]
- Qing, M.; Jones, D.; Springer, T.A. The chemokine receptor CXCR4 is required for the retention of B lineage and granulocytic precursors within the bone marrow microenvironment. Immunity 1999, 10, 463–471. [Google Scholar] [CrossRef]
- Ödemis, V.; Lamp, E.; Pezeshki, G.; Moepps, B.; Schilling, K.; Gierschik, P.; Littman, D.R.; Engele, J. Mice deficient in the chemokine receptor CXCR4 exhibit impaired limb innervation and myogenesis. Mol. Cell. Neurosci. 2005, 30, 19. [Google Scholar] [CrossRef] [PubMed]
- Takabatake, Y.; Sugiyama, T.; Kohara, H.; Matsusaka, T.; Kurihara, H.; Koni, P.A.; Nagasawa, Y.; Hamano, T.; Matsui, I.; Kawada, N.; et al. The CXCL12 (SDF-1)/CXCR4 axis is essential for the development of renal vasculature. J. Am. Soc. Nephrol. 2009, 20, 640. [Google Scholar] [CrossRef] [PubMed]
- Bleul, C.C.; Wu, L.; Hoxie, J.A.; Springer, T.A.; Mackay, C.R. The HIV coreceptors CXCR4 and CCR5 are differentially expressed and regulated on human T lymphocytes. Proc. Natl. Acad. Sci. USA 1997, 94, 1925–1930. [Google Scholar] [CrossRef] [PubMed]
- Döring, Y.; Pawig, L.; Weber, C.; Noels, H. The CXCL12/CXCR4 chemokine ligand/receptor axis in cardiovascular disease. Front. Physiol. 2014, 5, 212. [Google Scholar] [CrossRef]
- Li, H.; Wang, R. A focus on CXCR4 in Alzheimer’s disease. Brain Circ. 2017, 3, 199. [Google Scholar] [CrossRef]
- Teixidó, J.; Martínez-Moreno, M.; Díaz-Martínez, M.; Sevilla-Movilla, S. The good and bad faces of the CXCR4 chemokine receptor. Int. J. Biochem. Cell Biol. 2018, 95, 18. [Google Scholar] [CrossRef]
- Chatterjee, S.; Behnam Azad, B.; Nimmagadda, S. The intricate role of CXCR4 in cancer. Adv. Cancer Res. 2014, 124, 31–82. [Google Scholar] [CrossRef]
- Clarke, M.F.; Dick, J.E.; Dirks, P.B.; Eaves, C.J.; Jamieson, C.H.M.; Jones, D.L.; Visvader, J.; Weissman, I.L.; Wahl, G.M. Cancer stem cells—Perspectives on current status and future directions: AACR workshop on cancer stem cells. Cancer Res. 2006, 66, 9339–9344. [Google Scholar] [CrossRef]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct Populations of Cancer Stem Cells Determine Tumor Growth and Metastatic Activity in Human Pancreatic Cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef]
- Allinen, M.; Beroukhim, R.; Cai, L.; Brennan, C.; Lahti-Domenici, J.; Huang, H.; Porter, D.; Hu, M.; Chin, L.; Richardson, A.; et al. Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell 2004, 6, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Kojima, Y.; Acar, A.; Eaton, E.N.; Mellody, K.T.; Scheel, C.; Ben-Porath, I.; Onder, T.T.; Wang, Z.C.; Richardson, A.L.; Weinberg, R.A.; et al. Autocrine TGF-β and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc. Natl. Acad. Sci. USA 2010, 107, 5107. [Google Scholar] [CrossRef] [PubMed]
- Santagata, S.; Ieranò, C.; Trotta, A.M.; Capiluongo, A.; Auletta, F.; Guardascione, G.; Scala, S. CXCR4 and CXCR7 Signaling Pathways: A Focus on the Cross-Talk Between Cancer Cells and Tumor Microenvironment. Front. Oncol. 2021, 11, 591386. [Google Scholar] [CrossRef]
- Schioppa, T.; Uranchimeg, B.; Saccani, A.; Biswas, S.K.; Doni, A.; Rapisarda, A.; Bernasconi, S.; Saccani, S.; Nebuloni, M.; Vago, L.; et al. Regulation of the Chemokine Receptor CXCR4 by Hypoxia. J. Exp. Med. 2003, 198, 30267. [Google Scholar] [CrossRef]
- Jin, F.; Brockmeier, U.; Otterbach, F.; Metzen, E. New insight into the SDF-1/CXCR4 axis in a breast carcinoma model: Hypoxia-induced endothelial SDF-1 and tumor cell CXCR4 are required for tumor cell intravasation. Mol. Cancer Res. 2012, 10, 498. [Google Scholar] [CrossRef]
- Liang, Z.; Brooks, J.; Willard, M.; Liang, K.; Yoon, Y.; Kang, S.; Shim, H. CXCR4/CXCL12 axis promotes VEGF-mediated tumor angiogenesis through Akt signaling pathway. Biochem. Biophys. Res. Commun. 2007, 359, 182. [Google Scholar] [CrossRef]
- Zhou, W.; Guo, S.; Liu, M.; Burow, M.E.; Wang, G. Targeting CXCL12/CXCR4 Axis in Tumor Immunotherapy. Curr. Med. Chem. 2017, 26, 3026–3041. [Google Scholar] [CrossRef]
- López-Gil, J.C.; Martin-Hijano, L.; Hermann, P.C.; Sainz, B. The CXCL12 crossroads in cancer stem cells and their niche. Cancers 2021, 13, 469. [Google Scholar] [CrossRef]
- Bianchi, M.E.; Mezzapelle, R. The Chemokine Receptor CXCR4 in Cell Proliferation and Tissue Regeneration. Front. Immunol. 2020, 11, 2109. [Google Scholar] [CrossRef]
- Sun, Y.; Cheng, Z.; Ma, L.; Pei, G. β-arrestin2 is critically involved in CXCR4-mediated chemotaxis, and this is mediated by its enhancement of p38 MAPK activation. J. Biol. Chem. 2002, 277, 49212–49219. [Google Scholar] [CrossRef]
- Busillo, J.M.; Armando, S.; Sengupta, R.; Meucci, O.; Bouvier, M.; Benovic, J.L. Site-specific phosphorylation of CXCR4 is dynamically regulated by multiple kinases and results in differential modulation of CXCR4 signaling. J. Biol. Chem. 2010, 285, 91173. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; He, L.; Combs, C.A.; Roderiquez, G.; Norcross, M.A. Dimerization of CXCR4 in living malignant cells: Control of cell migration by a synthetic peptide that reduces homologous CXCR4 interactions. Mol. Cancer Ther. 2006, 5, 2474–2488. [Google Scholar] [CrossRef] [PubMed]
- Ge, B.; Lao, J.; Li, J.; Chen, Y.; Song, Y.; Huang, F. Single-molecule imaging reveals dimerization/oligomerization of CXCR4 on plasma membrane closely related to its function. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef]
- Lao, J.; He, H.; Wang, X.; Wang, Z.; Song, Y.; Yang, B.; Ullahkhan, N.; Ge, B.; Huang, F. Single-Molecule Imaging Demonstrates Ligand Regulation of the Oligomeric Status of CXCR4 in Living Cells. J. Phys. Chem. B 2017, 121, 1466–1474. [Google Scholar] [CrossRef]
- Vila-Coro, A.J.; Rodríguez-Frade, J.M.; De Ana, A.M.; Moreno-Ortíz, M.C.; Martínez-A, C.; Mellado, M. The chemokine SDF-lα triggers CXCR4 receptor dimerization and activates the JAK/STAT pathway. FASEB J. 1999, 13, 1699–1710. [Google Scholar] [CrossRef]
- Percherancier, Y.; Berchiche, Y.A.; Slight, I.; Volkmer-Engert, R.; Tamamura, H.; Fujii, N.; Bouvier, M.; Heveker, N. Bioluminescence resonance energy transfer reveals ligand-induced conformational changes in CXCR4 homo- and heterodimers. J. Biol. Chem. 2005, 280, 9895–9903. [Google Scholar] [CrossRef]
- Sohy, D.; Parmentier, M.; Springael, J.Y. Allosteric transinhibition by specific antagonists in CCR2/CXCR4 heterodimers. J. Biol. Chem. 2007, 282, 2200. [Google Scholar] [CrossRef]
- Hayasaka, H.; Kobayashi, D.; Yoshimura, H.; Nakayama, E.E.; Shioda, T.; Miyasaka, M. The HIV-1 Gp120/CXCR4 axis promotes CCR7 ligand-dependent CD4 T cell migration: CCR7 homo- and CCR7/CXCR4 hetero-oligomer formation as a possible mechanism for up-regulation of functional CCR7. PLoS ONE 2015, 10, e117454. [Google Scholar] [CrossRef]
- Patrussi, L.; Ulivieri, C.; Lucherini, O.M.; Paccani, S.R.; Gamberucci, A.; Lanfrancone, L.; Pelicci, P.G.; Baldari, C.T. p52Shc is required for CXCR4-dependent signaling and chemotaxis in T cells. Blood 2007, 110, 68411. [Google Scholar] [CrossRef]
- Wu, C.Y.; Tsai, Y.Y.; Chen, S.Y.; Lin, Y.P.; Shin, J.W.; Wu, C.C.; Yang, B.C. Interaction of Zap70 and CXCR4 receptor at lamellipodia that determines the directionality during Jurkat T cells chemotaxis. Mol. Immunol. 2017, 90, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Cascio, G.; Martín-Cófreces, N.B.; Rodríguez-Frade, J.M.; López-Cotarelo, P.; Criado, G.; Pablos, J.L.; Rodríguez-Fernández, J.L.; Sánchez-Madrid, F.; Mellado, M. CXCL12 Regulates through JAK1 and JAK2 Formation of Productive Immunological Synapses. J. Immunol. 2015, 194, 1402491. [Google Scholar] [CrossRef] [PubMed]
- Felce, J.H.; Parolini, L.; Sezgin, E.; Céspedes, P.F.; Korobchevskaya, K.; Jones, M.; Peng, Y.; Dong, T.; Fritzsche, M.; Aarts, D.; et al. Single-Molecule, Super-Resolution, and Functional Analysis of G Protein-Coupled Receptor Behavior within the T Cell Immunological Synapse. Front. Cell Dev. Biol. 2021, 8, 608484. [Google Scholar] [CrossRef] [PubMed]
- Becker, M.; Hobeika, E.; Jumaa, H.; Reth, M.; Maity, P.C. CXCR4 signaling and function require the expression of the IgD-class B-cell antigen receptor. Proc. Natl. Acad. Sci. USA 2017, 114, 5231–5236. [Google Scholar] [CrossRef]
- Gustavsson, M.; Dyer, D.P.; Zhao, C.; Handel, T.M. Kinetics of CXCL12 binding to atypical chemokine receptor 3 reveal a role for the receptor N terminus in chemokine binding. Sci. Signal. 2019, 12, aaw3657. [Google Scholar] [CrossRef]
- Sierro, F.; Biben, C.; Martínez-Muñoz, L.; Mellado, M.; Ransohoff, R.M.; Li, M.; Woehl, B.; Leung, H.; Groom, J.; Batten, M.; et al. Disrupted cardiac development but normal hematopoiesis in mice deficient in the second CXCL12/SDF-1 receptor, CXCR7. Proc. Natl. Acad. Sci. USA 2007, 104, 14759–14764. [Google Scholar] [CrossRef]
- Koenen, J.; Bachelerie, F.; Balabanian, K.; Schlecht-Louf, G.; Gallego, C. Atypical chemokine receptor 3 (ACKR3): A comprehensive overview of its expression and potential roles in the immune system. Mol. Pharmacol. 2019, 96, 115329. [Google Scholar] [CrossRef]
- Kawai, T.; Choi, U.; Cardwell, L.; DeRavin, S.S.; Naumann, N.; Whiting-Theobald, N.L.; Linton, G.F.; Moon, J.; Murphy, P.M.; Malech, H.L. WHIM syndrome myelokathexis reproduced in the NOD/SCID mouse xenotransplant model engrafted with healthy human stem cells transduced with C-terminus-truncated CXCR4. Blood 2007, 109, 25296. [Google Scholar] [CrossRef][Green Version]
- Naumann, U.; Cameroni, E.; Pruenster, M.; Mahabaleshwar, H.; Raz, E.; Zerwes, H.G.; Rot, A.; Thelen, M. CXCR7 functions as a scavenger for CXCL12 and CXCL11. PLoS ONE 2010, 5, e9175. [Google Scholar] [CrossRef]
- Rajagopal, S.; Kim, J.; Ahn, S.; Craig, S.; Lam, C.M.; Gerard, N.P.; Gerard, C.; Lefkowitz, R.J. β-Arrestin- But not G protein-mediated signaling by the “decoy” receptor CXCR7. Proc. Natl. Acad. Sci. USA 2010, 107, 628–632. [Google Scholar] [CrossRef]
- Boldajipour, B.; Mahabaleshwar, H.; Kardash, E.; Reichman-Fried, M.; Blaser, H.; Minina, S.; Wilson, D.; Xu, Q.; Raz, E. Control of Chemokine-Guided Cell Migration by Ligand Sequestration. Cell 2008, 132, 34. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.; Feitzinger, A.; Venkiteswaran, G.; Wang, J.; Lewellis, S.W.; Koplinski, C.A.; Peterson, F.C.; Volkman, B.F.; Meier-Schellersheim, M.; Knaut, H. A negative-feedback loop maintains optimal chemokine concentrations for directional cell migration. Nat. Cell Biol. 2020, 22, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Levoye, A.; Balabanian, K.; Baleux, F.; Bachelerie, F.; Lagane, B. CXCR7 heterodimerizes with CXCR4 and regulates CXCL12-mediated G protein signaling. Blood 2009, 11, 1966183. [Google Scholar] [CrossRef] [PubMed]
- Luker, K.E.; Gupta, M.; Luker, G.D. Imaging chemokine receptor dimerization with firefly luciferase complementation. FASEB J. 2009, 23, 116749. [Google Scholar] [CrossRef]
- Décaillot, F.M.; Kazmi, M.A.; Lin, Y.; Ray-Saha, S.; Sakmar, T.P.; Sachdev, P. CXCR7/CXCR4 heterodimer constitutively recruits β-arrestin to enhance cell migration. J. Biol. Chem. 2011, 286, 277038. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A.; Stewart, D.J.; Wald, O.; Peled, A. Potential of CXCR4 antagonists for the treatment of metastatic lung cancer. Expert Rev. Anticancer Ther. 2011, 11, 621–630. [Google Scholar] [CrossRef]
- Hatse, S.; Princen, K.; Bridger, G.; De Clercq, E.; Schols, D. Chemokine receptor inhibition by AMD3100 is strictly confined to CXCR4. FEBS Lett. 2002, 527, 255–262. [Google Scholar] [CrossRef]
- Fricker, S.P.; Anastassov, V.; Cox, J.; Darkes, M.C.; Grujic, O.; Idzan, S.R.; Labrecque, J.; Lau, G.; Mosi, R.M.; Nelson, K.L.; et al. Characterization of the molecular pharmacology of AMD3100: A specific antagonist of the G-protein coupled chemokine receptor, CXCR4. Biochem. Pharmacol. 2006, 72, 588–596. [Google Scholar] [CrossRef]
- De Clercq, E. The bicyclam AMD3100 story. Nat. Rev. Drug Discov. 2003, 2, 1134. [Google Scholar] [CrossRef]
- Hendrix, C.W.; Flexner, C.; Macfarland, R.T.; Giandomenico, C.; Fuchs, E.J.; Redpath, E.; Bridger, G.; Henson, G.W. Pharmacokinetics and safety of AMD-3100, a novel antagonist of the CXCR- 4 chemokine receptor, in human volunteers. Antimicrob. Agents Chemother. 2000, 44, 1667–1673. [Google Scholar] [CrossRef]
- Hendrix, C.W.; Collier, A.C.; Lederman, M.M.; Schols, D.; Pollard, R.B.; Brown, S.; Jackson, J.B.; Coombs, R.W.; Glesby, M.J.; Flexner, C.W.; et al. Safety, pharmacokinetics, and antiviral activity of AMD3100, a selective CXCR4 receptor inhibitor, in HIV-1 infection. J. Acquir. Immune Defic. Syndr. 2004, 37, 1253–1262. [Google Scholar] [CrossRef] [PubMed]
- Fruehauf, S.; Ehninger, G.; Hübel, K.; Topaly, J.; Goldschmidt, H.; Ho, A.D.; Müller, S.; Moos, M.; Badel, K.; Calandra, G. Mobilization of peripheral blood stem cells for autologous transplant in non-Hodgkin’s lymphoma and multiple myeloma patients by plerixafor and G-CSF and detection of tumor cell mobilization by PCR in multiple myeloma patients. Bone Marrow Transplant. 2010, 45, 142. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ghobrial, I.M.; Liu, C.J.; Zavidij, O.; Azab, A.K.; Baz, R.; Laubach, J.P.; Mishima, Y.; Armand, P.; Munshi, N.C.; Basile, F.; et al. Phase I/II trial of the CXCR4 inhibitor plerixafor in combination with bortezomib as a chemosensitization strategy in relapsed/refractory multiple myeloma. Am. J. Hematol. 2019, 94, 25627. [Google Scholar] [CrossRef]
- Devine, S.M.; Vij, R.; Rettig, M.; Todt, L.; McGlauchlen, K.; Fisher, N.; Devine, H.; Link, D.C.; Calandra, G.; Bridger, G.; et al. Rapid mobilization of functional donor hematopoietic cells without G-CSF using AMD3100, an antagonist of the CXCR4/SDF-1 interaction. Blood 2008, 112, 990–998. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.Q.; Duda, D.G.; Muzikansky, A.; Gerstner, E.R.; Kuhn, J.G.; Reardon, D.A.; Nayak, L.; Norden, A.D.; Doherty, L.; LaFrankie, D.; et al. Phase I and biomarker study of plerixafor and bevacizumab in recurrent high-grade glioma. Clin. Cancer Res. 2018, 24, 1025. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 43330. [Google Scholar] [CrossRef] [PubMed]
- Uchida, D.; Kuribayashi, N.; Kinouchi, M.; Sawatani, Y.; Shimura, M.; Mori, T.; Hasegawa, T.; Miyamoto, Y.; Kawamata, H. Effect of a novel orally bioavailable CXCR4 inhibitor, AMD070, on the metastasis of oral cancer cells. Oncol. Rep. 2018, 40, 6400. [Google Scholar] [CrossRef] [PubMed]
- Stone, N.D.; Dunaway, S.B.; Flexner, C.; Tierney, C.; Calandra, G.B.; Becker, S.; Cao, Y.J.; Wiggins, I.P.; Conley, J.; MacFarland, R.T.; et al. Multiple-dose escalation study of the safety, pharmacokinetics, and biologic activity of oral AMD070, a selective CXCR4 receptor inhibitor, in human subjects. Antimicrob. Agents Chemother. 2007, 51, 2351–2358. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Atkins, M.B.; Rose, T.L.; Alter, R.S.; Ju, Y.; Niland, K.; Wang, Y.; Arbeit, R.; Parasuraman, S.; Gan, L.; et al. A phase 1b trial of the CXCR4 inhibitor mavorixafor and nivolumab in advanced renal cell carcinoma patients with no prior response to nivolumab monotherapy. Investig. New Drugs 2021, 39, 1019–1027. [Google Scholar] [CrossRef]
- Ichiyama, K.; Yokoyama-Kumakura, S.; Tanaka, Y.; Tanaka, R.; Hirose, K.; Bannai, K.; Edamatsu, T.; Yanaka, M.; Niitani, Y.; Miyano-Kurosaki, N.; et al. A duodenally absorbable CXC chemokine receptor 4 antagonist, KRH-1636, exhibits a potent and selective anti-HIV-1 activity. Proc. Natl. Acad. Sci. USA 2003, 100, 4185–4190. [Google Scholar] [CrossRef]
- Zachariassen, Z.G.; Karlshøj, S.; Haug, B.E.; Rosenkilde, M.M.; Våbenø, J. Probing the Molecular Interactions between CXC Chemokine Receptor 4 (CXCR4) and an Arginine-Based Tripeptidomimetic Antagonist (KRH-1636). J. Med. Chem. 2015, 58, 987. [Google Scholar] [CrossRef]
- Martin, M.; Mayer, I.A.; Walenkamp, A.M.E.; Lapa, C.; Andreeff, M.; Bobirca, A. At the Bedside: Profiling and treating patients with CXCR4-expressing cancers. J. Leukoc. Biol. 2021, 109, 953–967. [Google Scholar] [CrossRef]
- Van Hout, A.; D’Huys, T.; Oeyen, M.; Schols, D.; Van Loy, T. Comparison of cell-based assays for the identification and evaluation of competitive CXCR4 inhibitors. PLoS ONE 2017, 12, 176057. [Google Scholar] [CrossRef]
- Iyer, C.V.; Evans, R.J.; Lou, Q.; Lin, D.; Wang, J.; Kohn, W.; Yan, L.Z.; Pulley, S.; Peng, S. Bin Rapid and recurrent neutrophil mobilization regulated by T134, a CXCR4 peptide antagonist. Exp. Hematol. 2008, 36, 21. [Google Scholar] [CrossRef] [PubMed]
- Tamamura, H.; Xu, Y.; Hattori, T.; Zhang, X.; Arakaki, R.; Kanbara, K.; Omagari, A.; Otaka, A.; Ibuka, T.; Yamamoto, N.; et al. A low-molecular-weight inhibitor against the chemokine receptor CXCR4: A strong anti-HIV peptide T140. Biochem. Biophys. Res. Commun. 1998, 253, 9871. [Google Scholar] [CrossRef] [PubMed]
- Tamamura, H.; Murakami, T.; Masuda, M.; Otaka, A.; Takada, W.; Ibuka, T.; Nakashima, H.; Waki, M.; Matsumoto, A.; Yamamoto, N.; et al. Structure-activity relationships of an anti-HIV peptide, T22. Biochem. Biophys. Res. Commun. 1994, 205, 2868. [Google Scholar] [CrossRef] [PubMed]
- Serna, N.; Carratalá, J.V.; Conchillo-Solé, O.; Martínez-Torró, C.; Unzueta, U.; Mangues, R.; Ferrer-Miralles, N.; Daura, X.; Vázquez, E.; Villaverde, A. Antibacterial activity of t22, a specific peptidic ligand of the tumoral marker cxcr4. Pharmaceutics 2021, 13, 1922. [Google Scholar] [CrossRef] [PubMed]
- Tamamura, H.; Fujii, N. The therapeutic potential of CXCR4 antagonists in the treatment of HIV infection, cancer metastasis and rheumatoid arthritis. Expert Opin. Ther. Targets 2005, 9, 1267. [Google Scholar] [CrossRef]
- Abraham, M.; Biyder, K.; Begin, M.; Wald, H.; Weiss, I.D.; Galun, E.; Nagler, A.; Peled, A. Enhanced Unique Pattern of Hematopoietic Cell Mobilization Induced by the CXCR4 Antagonist 4F-Benzoyl-TN14003. Stem Cells 2007, 25, 161. [Google Scholar] [CrossRef]
- Borthakur, G.; Ofran, Y.; Tallman, M.S.; Foran, J.; Uy, G.L.; DiPersio, J.F.; Showel, M.M.; Shimoni, A.; Nagler, A.; Rowe, J.M.; et al. BL-8040 CXCR4 antagonist is safe and demonstrates antileukemic activity in combination with cytarabine for the treatment of relapsed/refractory acute myelogenous leukemia: An open-label safety and efficacy phase 2a study. Cancer 2021, 127, 33338. [Google Scholar] [CrossRef]
- Crees, Z.D.; Stockerl-Goldstein, K.; Vainstein, A.; Chen, H.; Dipersio, J.F. GENESIS: Phase III trial evaluating BL-8040 + G-CSF to mobilize hematopoietic cells for autologous transplant in myeloma. Futur. Oncol. 2019, 15, 380. [Google Scholar] [CrossRef] [PubMed]
- Results of COMBAT/KEYNOTE-202 Study for Metastatic Pancreatic Cancer. Oncology Times, 20 January 2020; Volume 42, p. 29. [CrossRef]
- Bockorny, B.; Semenisty, V.; Macarulla, T.; Borazanci, E.; Wolpin, B.M.; Stemmer, S.M.; Golan, T.; Geva, R.; Borad, M.J.; Pedersen, K.S.; et al. BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: The COMBAT trial. Nat. Med. 2020, 26, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zeng, Z.; Shi, Y.; Jacamo, R.; Ludin, C.; Dembowsky, K.; Frenette, P.S.; Konopleva, M.; Andreeff, M. Targeting CXCR4, SDF1 and Beta-Adrenergic Receptors in the AML Microenvironment by Novel Antagonist POL6326, G-CSF and Isoproterenol. Blood 2010, 116, 2179. [Google Scholar] [CrossRef]
- Schmitt, S.; Weinhold, N.; Dembowsky, K.; Neben, K.; Witzens-Harig, M.; Braun, M.; Klemmer, J.; Wuchter, P.; Ludin, C.; Ho, A.D.; et al. First Results of a Phase-II Study with the New CXCR4 Antagonist POL6326 to Mobilize Hematopoietic Stem Cells (HSC) In Multiple Myeloma (MM). Blood 2010, 116, 824. [Google Scholar] [CrossRef]
- Pernas, S.; Martin, M.; Kaufman, P.A.; Gil-Martin, M.; Gomez Pardo, P.; Lopez-Tarruella, S.; Manso, L.; Ciruelos, E.; Perez-Fidalgo, J.A.; Hernando, C.; et al. Balixafortide plus eribulin in HER2-negative metastatic breast cancer: A phase 1, single-arm, dose-escalation trial. Lancet Oncol. 2018, 19, 812–824. [Google Scholar] [CrossRef]
- Peng, S.B.; Zhang, X.; Paul, D.; Kays, L.M.; Gough, W.; Stewart, J.; Uhlik, M.T.; Chen, Q.; Hui, Y.H.; Zamek-Gliszczynski, M.J.; et al. Identification of LY2510924, a novel cyclic peptide CXCR4 antagonist that exhibits antitumor activities in solid tumor and breast cancer metastatic models. Mol. Cancer Ther. 2015, 14, 850. [Google Scholar] [CrossRef]
- Cho, B.S.; Zeng, Z.; Mu, H.; Wang, Z.; Konoplev, S.; McQueen, T.; Protopopova, M.; Cortes, J.; Marszalek, J.R.; Peng, S.-B.; et al. Antileukemia activity of the novel peptidic CXCR4 antagonist LY2510924 as monotherapy and in combination with chemotherapy. Blood 2015, 126, 628677. [Google Scholar] [CrossRef]
- Boddu, P.; Borthakur, G.; Koneru, M.; Huang, X.; Naqvi, K.; Wierda, W.; Bose, P.; Jabbour, E.; Estrov, Z.; Burger, J.; et al. Initial Report of a Phase I Study of LY2510924, Idarubicin, and Cytarabine in Relapsed/Refractory Acute Myeloid Leukemia. Front. Oncol. 2018, 8, 369. [Google Scholar] [CrossRef]
- O’Hara, M.H.; Messersmith, W.; Kindler, H.; Zhang, W.; Pitou, C.; Szpurka, A.M.; Wang, D.; Peng, S.-B.; Vangerow, B.; Khan, A.A.; et al. Safety and Pharmacokinetics of CXCR4 Peptide Antagonist, LY2510924, in Combination with Durvalumab in Advanced Refractory Solid Tumors. J. Pancreat. Cancer 2020, 6, 18. [Google Scholar] [CrossRef]
- Portella, L.; Vitale, R.; De Luca, S.; D’Alterio, C.; Ieranò, C.; Napolitano, M.; Riccio, A.; Polimeno, M.N.; Monfregola, L.; Barbieri, A.; et al. Preclinical Development of a Novel Class of CXCR4 Antagonist Impairing Solid Tumors Growth and Metastases. PLoS ONE 2013, 8, 74578. [Google Scholar] [CrossRef]
- Fontanella, R.; Pelagalli, A.; Nardelli, A.; D’Alterio, C.; Ieranò, C.; Cerchia, L.; Lucarelli, E.; Scala, S.; Zannetti, A. A novel antagonist of CXCR4 prevents bone marrow-derived mesenchymal stem cell-mediated osteosarcoma and hepatocellular carcinoma cell migration and invasion. Cancer Lett. 2016, 370, 18. [Google Scholar] [CrossRef] [PubMed]
- D’Alterio, C.; Zannetti, A.; Trotta, A.M.; Ieranò, C.; Napolitano, M.; Rea, G.; Greco, A.; Maiolino, P.; Albanese, S.; Scognamiglio, G.; et al. New CXCR4 antagonist peptide R (Pep R) improves standard therapy in colorectal cancer. Cancers 2020, 12, 1952. [Google Scholar] [CrossRef]
- Baribaud, F.; Edwards, T.G.; Sharron, M.; Brelot, A.; Heveker, N.; Price, K.; Mortari, F.; Alizon, M.; Tsang, M.; Doms, R.W. Antigenically Distinct Conformations of CXCR4. J. Virol. 2001, 75, 2001. [Google Scholar] [CrossRef] [PubMed]
- Strizki, J.M.; Turner, J.D.; Collman, R.G.; Hoxie, J.; González-Scarano, F. A monoclonal antibody (12G5) directed against CXCR-4 inhibits infection with the dual-tropic human immunodeficiency virus type 1 isolate HIV-1(89.6) but not the T-tropic isolate HIV-1(HxB). J. Virol. 1997, 71, 5678–5683. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Lan, C.; Xiao, Y.; Chen, Y.H. Antibody to CD14 like CXCR4-specific antibody 12G5 could inhibit CXCR4-dependent chemotaxis and HIV Env-mediated cell fusion. Immunol. Lett. 2003, 88, 27–30. [Google Scholar] [CrossRef]
- Wei, L.; Kong, P.Y.; Chen, X.H.; Peng, X.G.; Zeng, D.F.; Chang, C.; Yang, W.B.; Liu, H.; Liu, L.; Wang, Q.Y.; et al. Effects of anti-CXCR4 monoclonal antibody on adhesion and proliferation of human acute myelocytic leukemia cell line HL-60. Ai Zheng 2004, 23, 1273–1277. [Google Scholar]
- Brennecke, P.; Arlt, M.J.E.; Campanile, C.; Husmann, K.; Gvozdenovic, A.; Apuzzo, T.; Thelen, M.; Born, W.; Fuchs, B. CXCR4 antibody treatment suppresses metastatic spread to the lung of intratibial human osteosarcoma xenografts in mice. Clin. Exp. Metastasis 2014, 31, 339–349. [Google Scholar] [CrossRef]
- Kashyap, M.K.; Kumar, D.; Jones, H.; Amaya-Chanaga, C.I.; Choi, M.Y.; Melo-Cardenas, J.; Ale-Ali, A.; Kuhne, M.R.; Sabbatini, P.; Cohen, L.J.; et al. Ulocuplumab (BMS-936564/MDX1338): A fully human anti- CXCR4 antibody induces cell death in chronic lymphocytic leukemia mediated through a reactive oxygen speciesdependent pathway. Oncotarget 2016, 7, 6465. [Google Scholar] [CrossRef]
- Treon, S.P.; Meid, K.; Hunter, Z.R.; Flynn, C.A.; Sarosiek, S.R.; Leventoff, C.R.; White, T.P.; Cao, Y.; Roccaro, A.M.; Sacco, A.; et al. Phase 1 study of ibrutinib and the CXCR4 antagonist ulocuplumab in CXCR4-mutated Waldenström macroglobulinemia. Blood 2021, 138, 2953. [Google Scholar] [CrossRef]
- Ghobrial, I.M.; Liu, C.J.; Redd, R.A.; Perez, R.P.; Baz, R.; Zavidij, O.; Sklavenitis-Pistofidis, R.; Richardson, P.G.; Anderson, K.C.; Laubach, J.; et al. A phase Ib/II trial of the first-in-class anti-CXCR4 antibody ulocuplumab in combination with lenalidomide or bortezomib plus dexamethasone in relapsed multiple myeloma. Clin. Cancer Res. 2020, 26, 647. [Google Scholar] [CrossRef]
- Becker, P.S.; Foran, J.M.; Altman, J.K.; Yacoub, A.; Castro, J.E.; Sabbatini, P.; Dilea, C.; Wade, M.; Xing, G.; Gutierrez, A.; et al. Targeting the CXCR4 Pathway: Safety, Tolerability and Clinical Activity of Ulocuplumab (BMS-936564), an Anti-CXCR4 Antibody, in Relapsed/Refractory Acute Myeloid Leukemia. Blood 2014, 124, 386. [Google Scholar] [CrossRef]
- Liu, S.H.; Gu, Y.; Pascual, B.; Yan, Z.; Hallin, M.; Zhang, C.; Fan, C.; Wang, W.; Lam, J.; Spilker, M.E.; et al. A novel CXCR4 antagonist IgG1 antibody (PF-06747143) for the treatment of hematologic malignancies. Blood Adv. 2017, 1, 3921. [Google Scholar] [CrossRef] [PubMed]
- Murata, Y.; Saito, Y.; Kotani, T.; Matozaki, T. CD47-signal regulatory protein α signaling system and its application to cancer immunotherapy. Cancer Sci. 2018, 109, 13663. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, F.P.; Gresham, H.D.; Schwarz, E.; Brown, E.J. Molecular cloning of integrin-associated protein: An immunoglobulin family member with multiple membrane-spanning domains implicated in α(v)β3- dependent ligand binding. J. Cell Biol. 1993, 123, 485. [Google Scholar] [CrossRef]
- Majeti, R.; Chao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaiswal, S.; Gibbs, K.D.; van Rooijen, N.; Weissman, I.L. CD47 Is an Adverse Prognostic Factor and Therapeutic Antibody Target on Human Acute Myeloid Leukemia Stem Cells. Cell 2009, 138, 45. [Google Scholar] [CrossRef]
- Chao, M.P.; Alizadeh, A.A.; Tang, C.; Myklebust, J.H.; Varghese, B.; Gill, S.; Jan, M.; Cha, A.C.; Chan, C.K.; Tan, B.T.; et al. Anti-CD47 Antibody Synergizes with Rituximab to Promote Phagocytosis and Eradicate Non-Hodgkin Lymphoma. Cell 2010, 142, 44. [Google Scholar] [CrossRef]
- Brown, E.; Hooper, L.; Ho, T.; Gresham, H. Integrin-associated protein: A 50-kD plasma membrane antigen physically and functionally associated with integrins. J. Cell Biol. 1990, 111, 2785. [Google Scholar] [CrossRef]
- Seiffert, M.; Cant, C.; Chen, Z.; Rappold, I.; Brugger, W.; Kanz, L.; Brown, E.J.; Ullrich, A.; Bühring, H.J. Human signal-regulatory protein is expressed on normal, but not on subsets of leukemic myeloid cells and mediates cellular adhesion involving its counterreceptor CD47. Blood 1999, 94, 3633. [Google Scholar] [CrossRef]
- Kaur, S.; Martin-Manso, G.; Pendrak, M.L.; Garfield, S.H.; Isenberg, J.S.; Roberts, D.D. Thrombospondin-1 inhibits VEGF receptor-2 signaling by disrupting its association with CD47. J. Biol. Chem. 2010, 285, 172304. [Google Scholar] [CrossRef]
- Manna, P.P.; Dimitry, J.; Oldenborg, P.A.; Frazier, W.A. CD47 augments fas/CD95-mediated apoptosis. J. Biol. Chem. 2005, 280, 2200. [Google Scholar] [CrossRef]
- Liu, M.; O’Connor, R.S.; Trefely, S.; Graham, K.; Snyder, N.W.; Beatty, G.L. Metabolic rewiring of macrophages by CpG potentiates clearance of cancer cells and overcomes tumor-expressed CD47−mediated ‘don’t-eat-me’ signal. Nat. Immunol. 2019, 20, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Willingham, S.B.; Volkmer, J.P.; Gentles, A.J.; Sahoo, D.; Dalerba, P.; Mitra, S.S.; Wang, J.; Contreras-Trujillo, H.; Martin, R.; Cohen, J.D.; et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 6662–6667. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Sun, C.; Li, M.; Xia, B.; Wang, Y.; Zhang, L.; Zhang, Y.; Wang, J.; Sun, F.; Lu, S.; et al. Novel fully human anti-CD47 antibodies stimulate phagocytosis and promote elimination of AML cells. J. Cell. Physiol. 2021, 236, 30163. [Google Scholar] [CrossRef]
- Schürch, C.M.; Roelli, M.A.; Forster, S.; Wasmer, M.H.; Brühl, F.; Maire, R.S.; Di Pancrazio, S.; Ruepp, M.D.; Giger, R.; Perren, A.; et al. Targeting CD47 in Anaplastic Thyroid Carcinoma Enhances Tumor Phagocytosis by Macrophages and Is a Promising Therapeutic Strategy. Thyroid 2019, 29, 555. [Google Scholar] [CrossRef] [PubMed]
- Upton, R.; Banuelos, A.; Feng, D.; Biswas, T.; Kao, K.; McKenna, K.; Willingham, S.; Ho, P.Y.; Rosental, B.; Tal, M.C.; et al. Combining CD47 blockade with trastuzumab eliminates HER2-positive breast cancer cells and overcomes trastuzumab tolerance. Proc. Natl. Acad. Sci. USA 2021, 118, e2026849118. [Google Scholar] [CrossRef] [PubMed]
- Maute, R.; Xu, J.; Weissman, I.L. CD47–SIRPα-targeted therapeutics: Status and prospects. Immuno-Oncology Technol. 2022, 13, 100070. [Google Scholar] [CrossRef]
- Liu, J.; Wang, L.; Zhao, F.; Tseng, S.; Narayanan, C.; Shura, L.; Willingham, S.; Howard, M.; Prohaska, S.; Volkmer, J.; et al. Pre-clinical development of a humanized anti-CD47 antibody with anti-cancer therapeutic potential. PLoS ONE 2015, 10, e137345. [Google Scholar] [CrossRef]
- Naval, D.; Vyas, P.; Kambhampati, S.; Al Malki, M.; Larson, R.; Asch, A.; Mannis, G.; Chai-Ho, W.; Tanaka, T.; Bradley, T.; et al. AML-464 Tolerability and Efficacy of the First-In-Class Anti-CD47 Antibody Magrolimab Combined With Azacitidine in Frontline Patients With TP53-Mutated Acute Myeloid Leukemia (AML): Phase 1b Results. Clin. Lymphoma Myeloma Leuk. 2022, 22, S253–S254. [Google Scholar] [CrossRef]
- Sikic, B.I.; Lakhani, N.; Patnaik, A.; Shah, S.A.; Chandana, S.R.; Rasco, D.; Colevas, A.D.; O’Rourke, T.; Narayanan, S.; Papadopoulos, K.; et al. First-in-human, first-in-class phase i trial of the anti-CD47 antibody Hu5F9-G4 in patients with advanced cancers. J. Clin. Oncol. 2019, 37, 946. [Google Scholar] [CrossRef]
- Sallman, D.; Al Malki, M.; Asch, A.; Wang, E.; Jurcic, J.; Bradley, T.; Flinn, I.; Pollyea, D.; Kambhampati, S.; Tanaka, T.; et al. MDS-445 Magrolimab In Combination With Azacitidine for Patients With Untreated Higher-Risk Myelodysplastic Syndromes (HR MDS): 5F9005 Phase 1b Study Results. Clin. Lymphoma, Myeloma Leuk. 2022, 22, S314–S315. [Google Scholar] [CrossRef]
- Berlin, J.; Harb, W.; Adjei, A.; Xing, Y.; Swiecicki, P.; Seetharam, M.; Nandagopal, L.; Gopal, A.; Xu, C.; Xu, C.; et al. 385 A first-in-human study of lemzoparlimab, a differentiated anti-CD47 antibody, in subjects with relapsed/refractory malignancy: Initial monotherapy results. J. ImmunoTher. Cancer 2020, 8 (Suppl. 3), A233. [Google Scholar] [CrossRef]
- Andrejeva, G.; Capoccia, B.J.; Hiebsch, R.R.; Donio, M.J.; Darwech, I.M.; Puro, R.J.; Pereira, D.S. Novel SIRPα Antibodies That Induce Single-Agent Phagocytosis of Tumor Cells while Preserving T Cells. J. Immunol. 2021, 206, 2001019. [Google Scholar] [CrossRef] [PubMed]
- Burris III, H.A.; Spira, A.I.; Taylor, M.H.; Yeku, O.O.; Liu, J.F.; Munster, P.N.; Hamilton, E.P.; Thomas, J.S.; Gatlin, F.; Penson, R.T.; et al. A first-in-human study of AO-176, a highly differentiated anti-CD47 antibody, in patients with advanced solid tumors. J. Clin. Oncol. 2021, 39, 2516. [Google Scholar] [CrossRef]
- Peluso, M.O.; Adam, A.; Armet, C.M.; Zhang, L.; O’Connor, R.W.; Lee, B.H.; Lake, A.C.; Normant, E.; Chappel, S.C.; Hill, J.A.; et al. The Fully human anti-CD47 antibody SRF231 exerts dual-mechanism antitumor activity via engagement of the activating receptor CD32a. J. Immunother. Cancer 2020, 8, 413. [Google Scholar] [CrossRef]
- Patnaik, A.; Spreafico, A.; Paterson, A.M.; Peluso, M.; Chung, J.-K.; Bowers, B.; Niforos, D.; O’Neill, A.M.; Beeram, M.; Iafolla, M.; et al. Results of a first-in-human phase I study of SRF231, a fully human, high-affinity anti-CD47 antibody. J. Clin. Oncol. 2020, 38 (Suppl. 15), 3064. [Google Scholar] [CrossRef]
- Yang, C.P.; Gao, S.F.; Zhang, H.Z.; Xu, L.; Liu, J.G.; Wang, M.; Zhang, S.R. CD47 is a Potential Target for the Treatment of Laryngeal Squamous Cell Carcinoma. Cell. Physiol. Biochem. 2016, 40, 452530. [Google Scholar] [CrossRef]
- Zeidan, A.M.; DeAngelo, D.J.; Palmer, J.; Seet, C.S.; Tallman, M.S.; Wei, X.; Raymon, H.; Sriraman, P.; Kopytek, S.; Bewersdorf, J.P.; et al. Phase 1 study of anti-CD47 monoclonal antibody CC-90002 in patients with relapsed/refractory acute myeloid leukemia and high-risk myelodysplastic syndromes. Ann. Hematol. 2022, 101, 557–569. [Google Scholar] [CrossRef]
- Lentz, R.W.; Colton, M.D.; Mitra, S.S.; Messersmith, W.A. Innate immune checkpoint inhibitors: The next breakthrough in medical oncology? Mol. Cancer Ther. 2021, 20, 41. [Google Scholar] [CrossRef]
- Lakhani, N.J.; Chow, L.Q.M.; Gainor, J.F.; LoRusso, P.; Lee, K.W.; Chung, H.C.; Lee, J.; Bang, Y.J.; Hodi, F.S.; Kim, W.S.; et al. Evorpacept alone and in combination with pembrolizumab or trastuzumab in patients with advanced solid tumours (ASPEN-01): A first-in-human, open-label, multicentre, phase 1 dose-escalation and dose-expansion study. Lancet Oncol. 2021, 22, 1740–1751. [Google Scholar] [CrossRef]
- Petrova, P.S.; Viller, N.N.; Wong, M.; Pang, X.; Lin, G.H.Y.; Dodge, K.; Chai, V.; Chen, H.; Lee, V.; House, V.; et al. TTI-621 (SIRPαFc): A CD47-blocking innate immune checkpoint inhibitor with broad antitumor activity and minimal erythrocyte binding. Clin. Cancer Res. 2017, 23, 1700. [Google Scholar] [CrossRef]
- Ansell, S.M.; Maris, M.B.; Lesokhin, A.M.; Chen, R.W.; Flinn, I.W.; Sawas, A.; Minden, M.D.; Villa, D.; Percival, M.E.M.; Advani, A.S.; et al. Phase I study of the CD47 blocker TTI-621 in patients with relapsed or refractory hematologic malignancies. Clin. Cancer Res. 2021, 27, 3706. [Google Scholar] [CrossRef] [PubMed]
- Mezzapelle, R.; De Marchis, F.; Passera, C.; Leo, M.; Brambilla, F.; Colombo, F.; Casalgrandi, M.; Preti, A.; Zambrano, S.; Castellani, P.; et al. CXCR4 engagement triggers CD47 internalization and antitumor immunization in a mouse model of mesothelioma. EMBO Mol. Med. 2021, 13, 12344. [Google Scholar] [CrossRef]
- Mezzapelle, R.; Leo, M.; Caprioglio, F.; Colley, L.S.; Lamarca, A.; Sabatino, L.; Colantuoni, V.; Crippa, M.P.; Bianchi, M.E. CXCR4/CXCL12 Activities in the Tumor Microenvironment and Implications for Tumor Immunotherapy. Cancers 2022, 14, 92314. [Google Scholar] [CrossRef] [PubMed]
- Penzo, M.; Molteni, R.; Suda, T.; Samaniego, S.; Raucci, A.; Habiel, D.M.; Miller, F.; Jiang, H.; Li, J.; Pardi, R.; et al. Inhibitor of NF-κB Kinases α and β Are Both Essential for High Mobility Group Box 1-Mediated Chemotaxis. J. Immunol. 2010, 184, 903131. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Class | Name | Clinical Trial | Tumor | References |
|---|---|---|---|---|
| Nonpeptide | AMD3100, plerixafor | NCT01455025 completed | r/r AML | - |
| NCT00322842 completed | non-Hodgkin’s lymphoma, MM | [62] | ||
| NCT00903968 completed | r/r MM | [63] | ||
| NCT02179970 completed | pancreatic, ovarian, CRC | - | ||
| NCT04177810 in progress | pancreatic | - | ||
| NCT01339039 terminated | high-grade glioma | [65] | ||
| NCT01977677 completed | high-grade glioma | [66] | ||
| NCT03746080 recruiting | glioblastoma | - | ||
| AMD070, mavorixafor, X4P-001 | NCT02823405 completed | advanced melanoma | - | |
| NCT02667886 active, not recruiting | RCC | - | ||
| NCT02923531 completed | RCC | [69] | ||
| Small-peptide | BL-8040, motixafortide | NCT01838395 completed | r/r AML | [80] |
| NCT03154827 terminated | AML | - | ||
| NCT03246529 active, not recruiting | MM | [81] | ||
| NCT02826486 active, not recruiting | metastatic pancreatic adenocarcinoma | [82] | ||
| NCT02907099 active, not recruiting | metastatic pancreatic | [83] | ||
| POL6326, balixafortide | NCT01837095 completed | metastatic BC | [86] | |
| LY2510924 | NCT02652871 completed | r/r AML | [89] | |
| NCT02737072 terminated | solid tumor | [90] | ||
| Antibody | LY2624587 | NCT01139788 completed | advanced or metastatic cancer | - |
| MDX-1338, ulocuplumab | NCT03225716 active, not recruiting | Waldenström macroglobulinemia | [100] | |
| NCT01359657 completed | MM | [101] | ||
| NCT01120457 completed | acute myelogeneus leukemia, B-cell cancers | - | ||
| NCT02472977 terminated | solid tumor | - | ||
| PF-06747143 | NCT02954653 terminated | AML | [103] |
| Name | Clinical Trial | Tumor | References |
|---|---|---|---|
| Hu5F9-G4, ONO-7913, magrolimab | NCT03248479 active, not recruiting | hematologic, AML | [119] |
| NCT02216409 completed | solid tumor | [120] | |
| NCT04313881 recruiting | myelodysplastic Syndromes | [121] | |
| TJC4, lemzoparlimab | NCT03934814 active, not recruiting | advanced r/r solid tumor and lymphoma | [122] |
| AO-176 | NCT03834948 active, not recruiting | multiple solid | [123,124] |
| SRF231 | NCT03512340 completed | advanced solid tumor and hematologic | [126] |
| CC-90002, INBRX 103 | NCT02641002 terminated | myelodysplastic and r/r AML | [128] |
| ALX148, evorpacept | NCT03013218 active, not recruiting | solid tumor and lymphoma | [130] |
| TTI-621, trillium | NCT04996004 recruiting | leiomyosarcoma | - |
| NCT02663518 active, not recruiting | hematologic malignancies and solid tumor | [132] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leo, M.; Sabatino, L. Targeting CXCR4 and CD47 Receptors: An Overview of New and Old Molecules for a Biological Personalized Anticancer Therapy. Int. J. Mol. Sci. 2022, 23, 12499. https://doi.org/10.3390/ijms232012499
Leo M, Sabatino L. Targeting CXCR4 and CD47 Receptors: An Overview of New and Old Molecules for a Biological Personalized Anticancer Therapy. International Journal of Molecular Sciences. 2022; 23(20):12499. https://doi.org/10.3390/ijms232012499
Chicago/Turabian StyleLeo, Manuela, and Lina Sabatino. 2022. "Targeting CXCR4 and CD47 Receptors: An Overview of New and Old Molecules for a Biological Personalized Anticancer Therapy" International Journal of Molecular Sciences 23, no. 20: 12499. https://doi.org/10.3390/ijms232012499
APA StyleLeo, M., & Sabatino, L. (2022). Targeting CXCR4 and CD47 Receptors: An Overview of New and Old Molecules for a Biological Personalized Anticancer Therapy. International Journal of Molecular Sciences, 23(20), 12499. https://doi.org/10.3390/ijms232012499