
Comprehensive In Silico Functional Prediction Analysis of CDKL5 by Single Amino Acid Substitution in the Catalytic Domain
 ,
,
Abstract
1. Introduction
2. Results
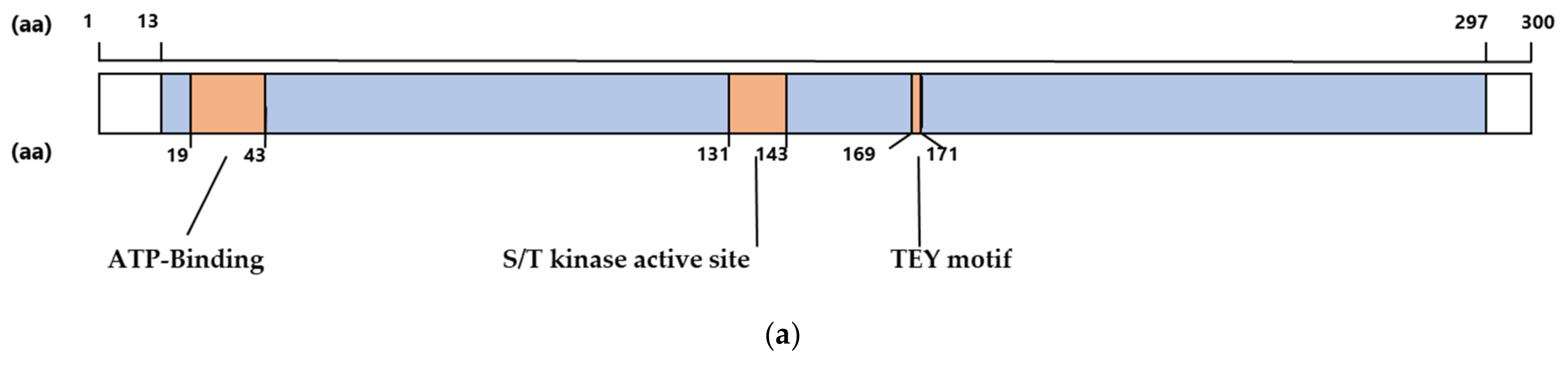
2.1. The CDKL5 Catalytic Domain Is Highly Conserved
2.2. Mutations in the CDKL5 Catalytic Domain Were Predicted to Affect Its Function
2.3. Validation of ClinVar Data Demonstrated the Validity of the In Silico Analysis
3. Discussion
4. Materials and Methods
4.1. Sequence Data
4.2. Bioinformatics Analysis
4.3. Assessment of In Silico Prediction Analysis Data Using Reference Data from ClinVar
4.4. Statistical Processing between Critical Region and Non-Critical Region
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rett, A. On an unusual brain atrophy syndrome in hyperammonemia in childhood. Wien. Med. Wochensch. 1966, 116, 723–726. (In German) [Google Scholar]
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2 encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Weaving, L.S.; Christodoulou, J.; Williamson, S.L.; Friend, K.L.; McKenzie, O.L.; Archer, H.; Evans, J.; Clarke, A.; Pelka, G.J.; Tam, P.P.; et al. Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation. Am. J. Hum. Genet. 2004, 75, 1079–1093. [Google Scholar] [CrossRef] [PubMed]
- Ariani, F.; Hayek, G.; Rondinella, D.; Artuso, R.; Mencarelli, M.A.; Spanhol-Rosseto, A.; Pollazzon, M.; Buoni, S.; Spiga, O.; Ricciardi, S.; et al. FOXG1 is responsible for the congenital variant of Rett syndrome. Am. J. Hum. Genet. 2008, 83, 89–93. [Google Scholar] [CrossRef]
- Montini, E.; Andolfi, G.; Caruso, A.; Buchner, G.; Walpole, S.M.; Mariani, M.; Consalez, G.; Trump, D.; Ballabio, A.; Franco, B. Identification and characterization of a novel serine-threonine kinase gene from the Xp22 region. Genomics 1998, 51, 427–433. [Google Scholar] [CrossRef]
- Kalscheuer, V.M.; Tao, J.; Donnelly, A.; Hollway, G.; Schwinger, E.; Kübart, S.; Menzel, C.; Hoeltzenbein, M.; Tommerup, N.; Eyre, H.; et al. Disruption of the serine/threonine kinase 9 gene causes severe X-linked infantile spasms and mental retardation. Am. J. Hum. Genet. 2003, 72, 1401–1411. [Google Scholar] [CrossRef]
- Tao, J.; Van Esch, H.; Hagedorn-Greiwe, M.; Hoffmann, K.; Moser, B.; Raynaud, M.; Sperner, J.; Fryns, J.P.; Schwinger, E.; Gécz, J.; et al. Mutations in the X-linked cyclin-dependent kinase-like 5 (CDKL5/STK9) gene are associated with severe neurodevelopmental retardation. Am. J. Hum. Genet. 2004, 75, 1149–1154. [Google Scholar] [CrossRef]
- Neul, J.L.; Kaufmann, W.E.; Glaze, D.G.; Christodoulou, J.; Clarke, A.J.; Bahi-Buisson, N.; Leonard, H.; Bailey, M.E.; Schanen, N.C.; Zappella, M.; et al. Rett syndrome: Revised diagnostic criteria and nomenclature. Ann. Neurol. 2010, 68, 944–950. [Google Scholar] [CrossRef]
- Hadzsiev, K.; Polgar, N.; Bene, J.; Komlosi, K.; Karteszi, J.; Hollody, G.; Kosztolanyi, K.; Renieri, A.; Melegh, B. Analysis of Hungarian patients with Rett syndrome phenotype for MECP2, CDKL5 and FOXG1 gene mutations. J. Hum. Genet. 2011, 56, 183–187. [Google Scholar] [CrossRef]
- Vidal, S.; Brandi, N.; Pacheco, P.; Maynou, J.; Fernandez, G.; Xiol, C.; Pascual-Alonso, A.; Pineda, M.; Rett Working Group; Armstrong, J. The most recurrent monogenic disorders that overlap with the phenotype of Rett syndrome. Eur. J. Paediatr. Neurol. 2019, 23, 609–620. [Google Scholar] [CrossRef]
- Symonds, J.D.; Zuberi, S.M.; Stewart, K.; McLellan, A.; O’Regan, M.; MacLeod, S.; Jollands, A.; Joss, S.; Kirkpatrick, M.; Brunklaus, A.; et al. Incidence and phenotypes of childhood-onset genetic epilepsies: A prospective population-based national cohort. Brain 2019, 142, 2303–2318. [Google Scholar] [CrossRef]
- Rusconi, L.; Salvatoni, L.; Giudici, L.; Bertani, I.; Kilstrup-Nielsen, C.; Broccoli, V.; Landsberger, N. CDKL5 expression is modulated during neuronal development and its subcellular distribution is tightly regulated by the C-terminal tail. J. Biol. Chem. 2008, 283, 30101–30111. [Google Scholar] [CrossRef]
- Christianto, A.; Katayama, S.; Kameshita, I.; Inazu, T. A novel CDKL5 mutation in a Japanese patient with atypical Rett syndrome. Clin. Chim. Acta 2016, 459, 132–136. [Google Scholar] [CrossRef]
- Nectoux, J.; Heron, D.; Tallot, M.; Chelly, J.; Bienvenu, T. Maternal origin of a novel C-terminal truncation mutation in CDKL5 causing a severe atypical form of Rett syndrome. Clin. Genet. 2006, 70, 29–33. [Google Scholar] [CrossRef]
- Russo, S.; Marchi, M.; Cogliati, F.; Bonati, M.T.; Pintaudi, M.; Veneselli, E.; Saletti, V.; Balestrini, M.; Ben-Zeev, B.; Larizza, L. Novel mutations in the CDKL5 gene, predicted effects and associated phenotypes. Neurogenetics 2009, 10, 241–250. [Google Scholar] [CrossRef]
- Scala, E.; Ariani, F.; Mari, F.; Caselli, R.; Pescucci, C.; Longo, I.; Meloni, I.; Giachino, D.; Bruttini, M.; Hayek, G.; et al. CDKL5/STK9 is mutated in Rett syndrome variant with infantile spasms. J. Med. Genet. 2005, 42, 103–107. [Google Scholar] [CrossRef]
- Jakimiec, M.; Paprocka, J.; Śmigiel, R. CDKL5 Deficiency Disorder-A Complex Epileptic Encephalopathy. Brain Sci. 2020, 10, 107. [Google Scholar] [CrossRef]
- Bertani, I.; Rusconi, L.; Bolognese, F.; Forlani, G.; Conca, B.; De Monte, L.; Badaracco, G.; Landsberger, N.; Kilstrup-Nielsen, C. Functional consequences of mutations in CDKL5, an X-linked gene involved in infantile spasms and mental retardation. J. Biol. Chem. 2006, 281, 32048–32056. [Google Scholar] [CrossRef]
- Lucariello, M.; Vidal, E.; Vidal, S.; Saez, M.; Roa, L.; Huertas, D.; Pineda, M.; Dalfó, E.; Dopazo, J.; Jurado, P.; et al. Whole exome sequencing of Rett syndrome-like patients reveals the mutational diversity of the clinical phenotype. Hum. Genet. 2016, 135, 1343–1354. [Google Scholar] [CrossRef]
- Vidal, S.; Brandi, N.; Pacheco, P.; Gerotina, E.; Blasco, L.; Trotta, J.R.; Derdak, S.; Del Mar O’Callaghan, M.; Garcia-Cazorla, À.; Pineda, M.; et al. The utility of Next Generation Sequencing for molecular diagnostics in Rett syndrome. Sci. Rep. 2017, 7, 12288. [Google Scholar] [CrossRef]
- Kilstrup-Nielsen, C.; Rusconi, L.; La Montanara, P.; Ciceri, D.; Bergo, A.; Bedogni, F.; Landsberger, N. What we know and would like to know about CDKL5 and its involvement in epileptic encephalopathy. Neural Plast. 2012, 2012, 728267. [Google Scholar] [CrossRef] [PubMed]
- Leong, I.U.; Stuckey, A.; Lai, D.; Skinner, J.R.; Love, D.R. Assessment of the predictive accuracy of five in silico prediction tools, alone or in combination, and two metaservers to classify long QT syndrome gene mutations. BMC Med. Genet. 2015, 16, 1–13, Published online. [Google Scholar] [CrossRef] [PubMed]
- Katayama, S.; Inazu, T. Straightforward and rapid method for detection of cyclin-dependent kinase-like 5 activity. Anal. Biochem. 2019, 566, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Rosas-Vargas, H.; Bahi-Buisson, N.; Philippe, C.; Nectoux, J.; Girard, B.; N’Guyen Morel, A.; Gitiaux, C.; Lazaro, L.; Odent, S.; Jonveaux, P.; et al. Impairment of CDKL5 Nuclear Localisation as a Cause for Severe Infantile Encephalopathy. J. Med. Genet. 2018, 45, 172–178. [Google Scholar] [CrossRef]
- Sekiguchi, M.; Katayama, S.; Hatano, N.; Shigeri, Y.; Sueyoshi, N.; Kameshita, I. Identification of Amphiphysin 1 as an Endogenous Substrate for CDKL5, a Protein Kinase Associated with X-Linked Neurodevelopmental Disorder. Arch. Biochem. Biophys. 2013, 535, 257–267. [Google Scholar] [CrossRef]
- Hicks, S.; Wheeler, D.A.; Plon, S.E.; Kimmel, M. Prediction of missense mutation functionality depends on both the algorithm and sequence alignment employed. Hum. Mutat. 2011, 32, 661–668. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Frazer, J.; Notin, P.; Dias, M.; Gomez, A.; Min, J.K.; Brock, K.; Gal, Y.; Marks, D.S. Disease variant prediction with deep generative models of evolutionary data. Nature 2021, 599, 91–95. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. Predicting deleterious amino acid substitutions. Genome Res. 2001, 11, 863–874. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. Accounting for human polymorphisms predicted to affect protein function. Genome Res. 2002, 12, 436–446. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7–20. [Google Scholar] [CrossRef]
- Baratloo, A.; Hosseini, M.; Negida, A.; El Ashal, G. Part 1: Simple Definition and Calculation of Accuracy, Sensitivity and Specificity. Emerg 2015, 3, 48–49. [Google Scholar]
- Baldi, P.; Brunak, S.; Chauvin, Y.; Andersen, C.A.; Nielsen, H. Assessing the accuracy of prediction algorithms for classification: An overview. Bioinformatics 2000, 16, 412–424. [Google Scholar] [CrossRef]
- Vihinen, M. How to evaluate performance of prediction methods? Measures and their interpretation in variation effect analysis. BMC Genom. 2012, 13 (Suppl. 4), S2. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Prediction Tool | Benign | Pathogenic | Total | |
|---|---|---|---|---|
| PolyPhen-2 HumDiv | prediction | Benign | Probably damaging/ Possibly damaging | |
| frequency | 241 | 5459 | 5700 | |
| rate | (4.2%) | (95.8%) | (100%) | |
| PolyPhen-2 HumVar | prediction | Benign | Probably damaging/ Possibly damaging | |
| frequency | 440 | 5260 | 5700 | |
| rate | (7.7%) | (92.3%) | (100%) | |
| PROVEAN | prediction | Neutral | Deleterious | |
| frequency | 990 | 4710 | 5700 | |
| rate | (17.4%) | (82.6%) | (100%) | |
| SIFT | prediction | Tolerated | Deleterious | |
| frequency | 686 | 5014 | 5700 | |
| rate | (12.0%) | (88.0%) | (100%) |
| Number of In Silico Prediction Tools | SNV Considered as Benign | SNV Considered as Damaging |
|---|---|---|
| 2 tools | Unanimously neutral/tolerated/benign | Unanimously probably damaging/ possibly damaging/deleterious |
| One output is probably damaging/ possibly damaging/deleterious | ||
| 3 tools | Unanimously neutral/tolerated/benign | Unanimously probably damaging/ possibly damaging/deleterious |
| Two outputs are neutral/tolerated/benign | Two outputs are probably damaging/ possibly damaging/deleterious | |
| 4 tools | Unanimously neutral/tolerated/benign | Unanimously probably damaging/ possibly damaging/deleterious |
| Three outputs are neutral/tolerated/benign | Two or more outputs are probably damaging/ possibly damaging/deleterious |
| CDKL5 | |||||
|---|---|---|---|---|---|
| Prediction Tools or Combinations | Accuracy | Sensitivity | Specificity | MCC | |
| (%) | (%) | (%) | |||
| PolyPhen-2 HumDiv | 94.4 | 100 | 33.3 | 0.561 | |
| PolyPhen-2 HumVar | 94.4 | 97.0 | 66.7 | 0.636 | |
| PROVEAN | 94.4 | 97.0 | 66.7 | 0.636 | |
| SIFT | 88.9 | 97.0 | 0.0 | −0.051 | |
| PolyPhen-2 HumDiv & PolyPhen2 HumDiv | 94.4 | 100 | 33.3 | 0.561 | |
| PolyPhen-2 HumDiv & PROVEAN | 94.4 | 100 | 33.3 | 0.561 | |
| PolyPhen-2 HumDiv & SIFT | 91.7 | 100 | 0.0 | ― | |
| PolyPhen-2 HumVar & PROVEAN | 97.2 | 100 | 66.7 | 0.804 | |
| PolyPhen-2 HumVar & SIFT | 91.7 | 100 | 0.0 | ― | |
| PROVEAN & SIFT | 88.9 | 97.0 | 0.0 | −0.051 | |
| ★ | PolyPhen-2 HumDiv & PolyPhen-2 HumVar & PROVEAN | 97.2 | 100 | 66.7 | 0.804 |
| PolyPhen-2 HumDiv & PolyPhen-2 HumVar & SIFT | 94.4 | 100 | 33.3 | 0.561 | |
| PolyPhen-2 HumDiv & PROVEAN & SIFT | 91.7 | 97 | 33.3 | 0.366 | |
| PolyPhen-2 HumVar & PROVEAN & SIFT | 94.4 | 97 | 66.7 | 0.636 | |
| PolyPhen-2 HumDiv & PolyPhen-2 HumVar & PROVEAN & SIFT | 94.4 | 100 | 33.3 | 0.561 | |
| Number of in silico prediction as neutral/tolerated/benign | 0 | 1 | 2 | 3 |
| Number of in silico prediction as probably damaging/possibly damaging/deleterious | 3 | 2 | 1 | 0 |
| Total prediction | P3 | P2 | B2 | B3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoshimura, Y.; Morii, A.; Fujino, Y.; Nagase, M.; Kitano, A.; Ueno, S.; Takeuchi, K.; Yamashita, R.; Inazu, T. Comprehensive In Silico Functional Prediction Analysis of CDKL5 by Single Amino Acid Substitution in the Catalytic Domain. Int. J. Mol. Sci. 2022, 23, 12281. https://doi.org/10.3390/ijms232012281
Yoshimura Y, Morii A, Fujino Y, Nagase M, Kitano A, Ueno S, Takeuchi K, Yamashita R, Inazu T. Comprehensive In Silico Functional Prediction Analysis of CDKL5 by Single Amino Acid Substitution in the Catalytic Domain. International Journal of Molecular Sciences. 2022; 23(20):12281. https://doi.org/10.3390/ijms232012281
Chicago/Turabian StyleYoshimura, Yuri, Atsushi Morii, Yuuki Fujino, Marina Nagase, Arisa Kitano, Shiho Ueno, Kyoka Takeuchi, Riko Yamashita, and Tetsuya Inazu. 2022. "Comprehensive In Silico Functional Prediction Analysis of CDKL5 by Single Amino Acid Substitution in the Catalytic Domain" International Journal of Molecular Sciences 23, no. 20: 12281. https://doi.org/10.3390/ijms232012281
APA StyleYoshimura, Y., Morii, A., Fujino, Y., Nagase, M., Kitano, A., Ueno, S., Takeuchi, K., Yamashita, R., & Inazu, T. (2022). Comprehensive In Silico Functional Prediction Analysis of CDKL5 by Single Amino Acid Substitution in the Catalytic Domain. International Journal of Molecular Sciences, 23(20), 12281. https://doi.org/10.3390/ijms232012281