
Possible Involvement of the Upregulation of ΔNp63 Expression Mediated by HER2-Activated Aryl Hydrocarbon Receptor in Mammosphere Maintenance
 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
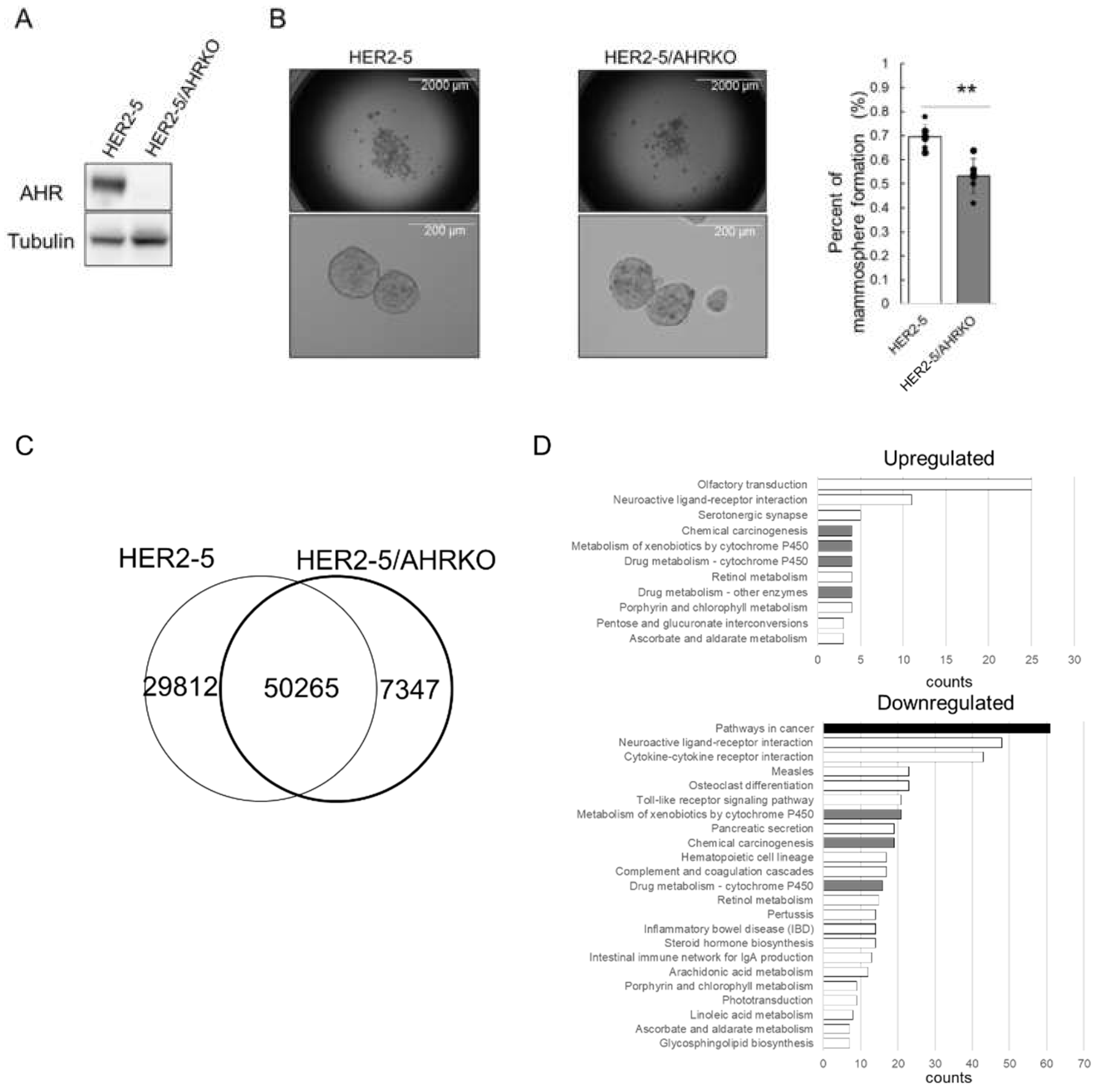
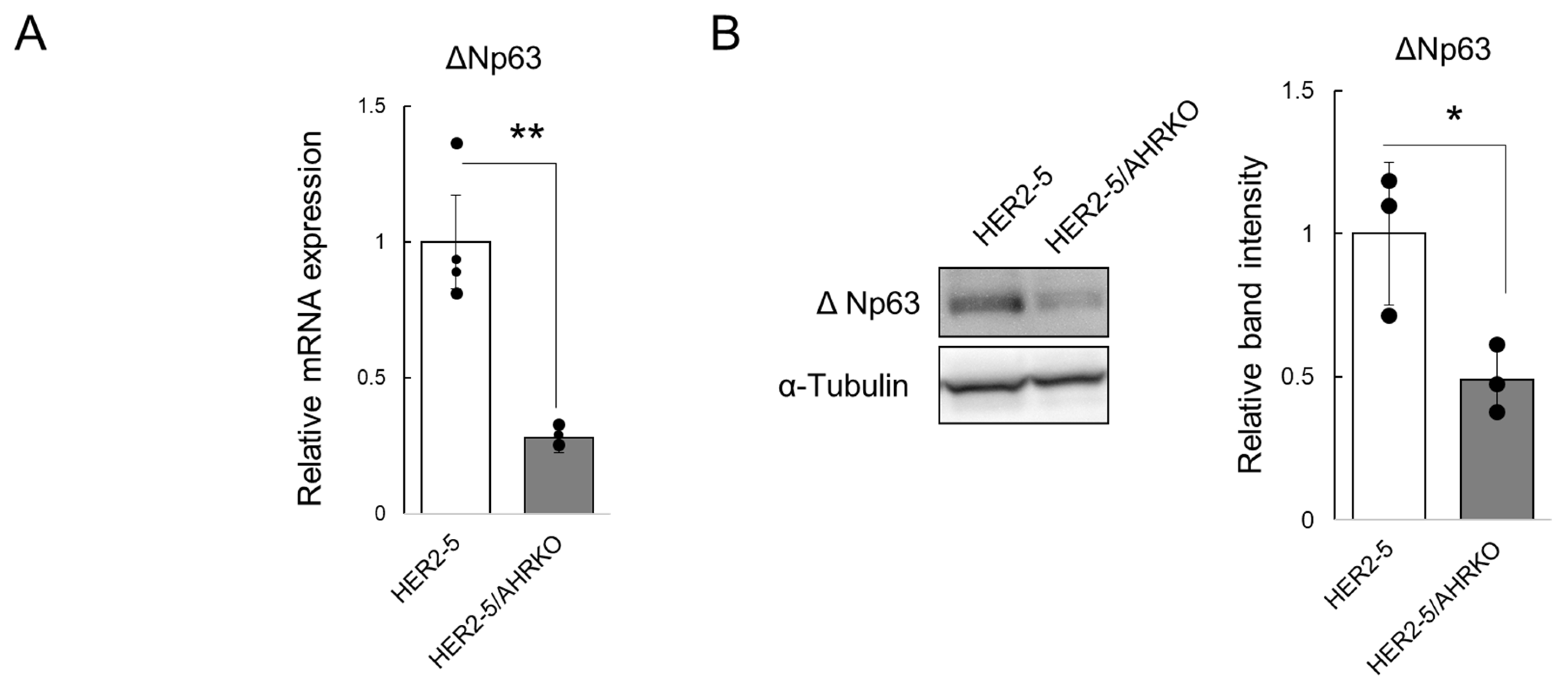
2.1. Knockout of AHR Downregulates Mammosphere Formation and ΔNp63 Expression in HER2-Overexpressing Breast Cancer Cells
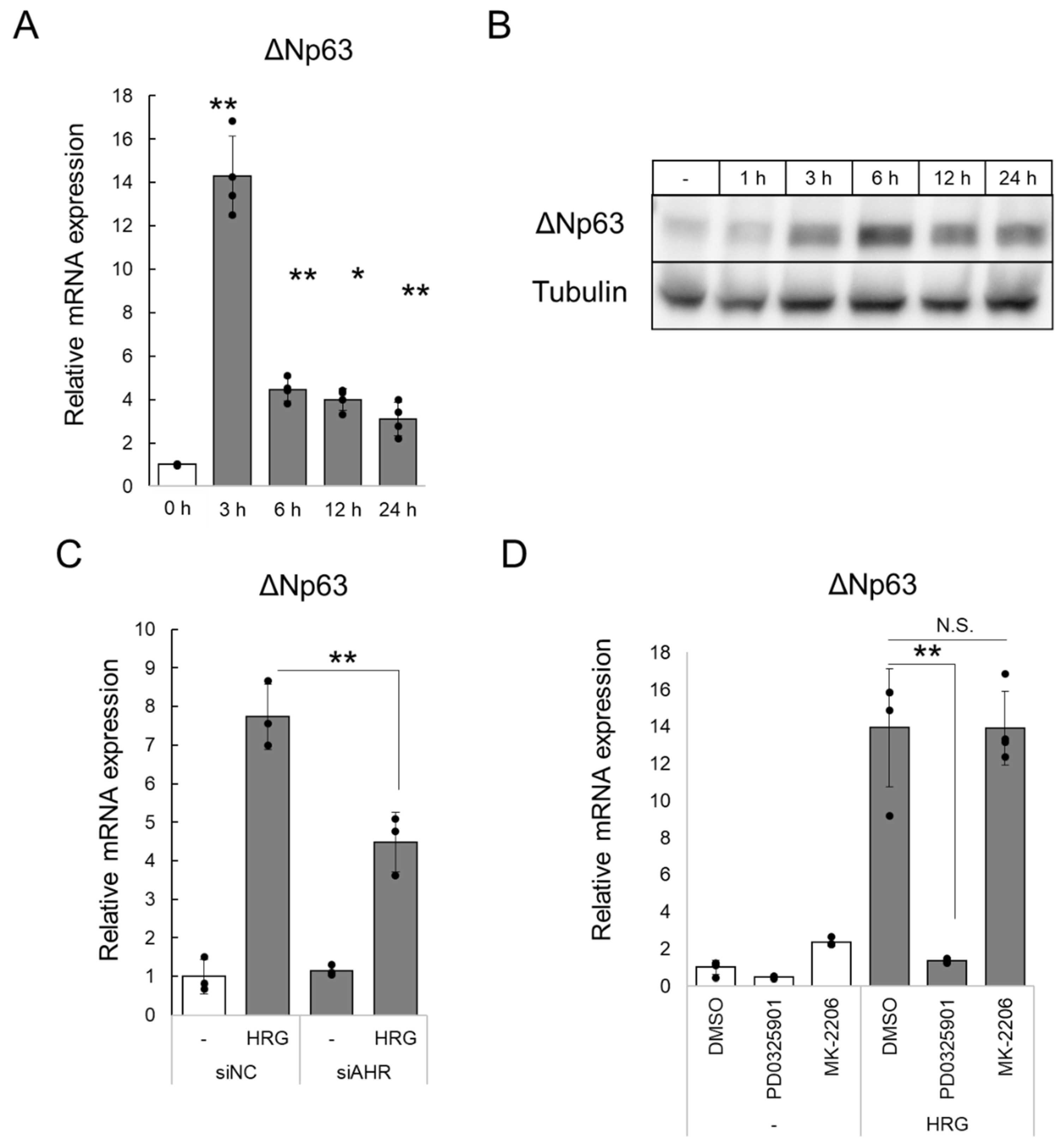
2.2. HRG/HER2 Signaling Induces ΔNp63 Expression via AHR Activation
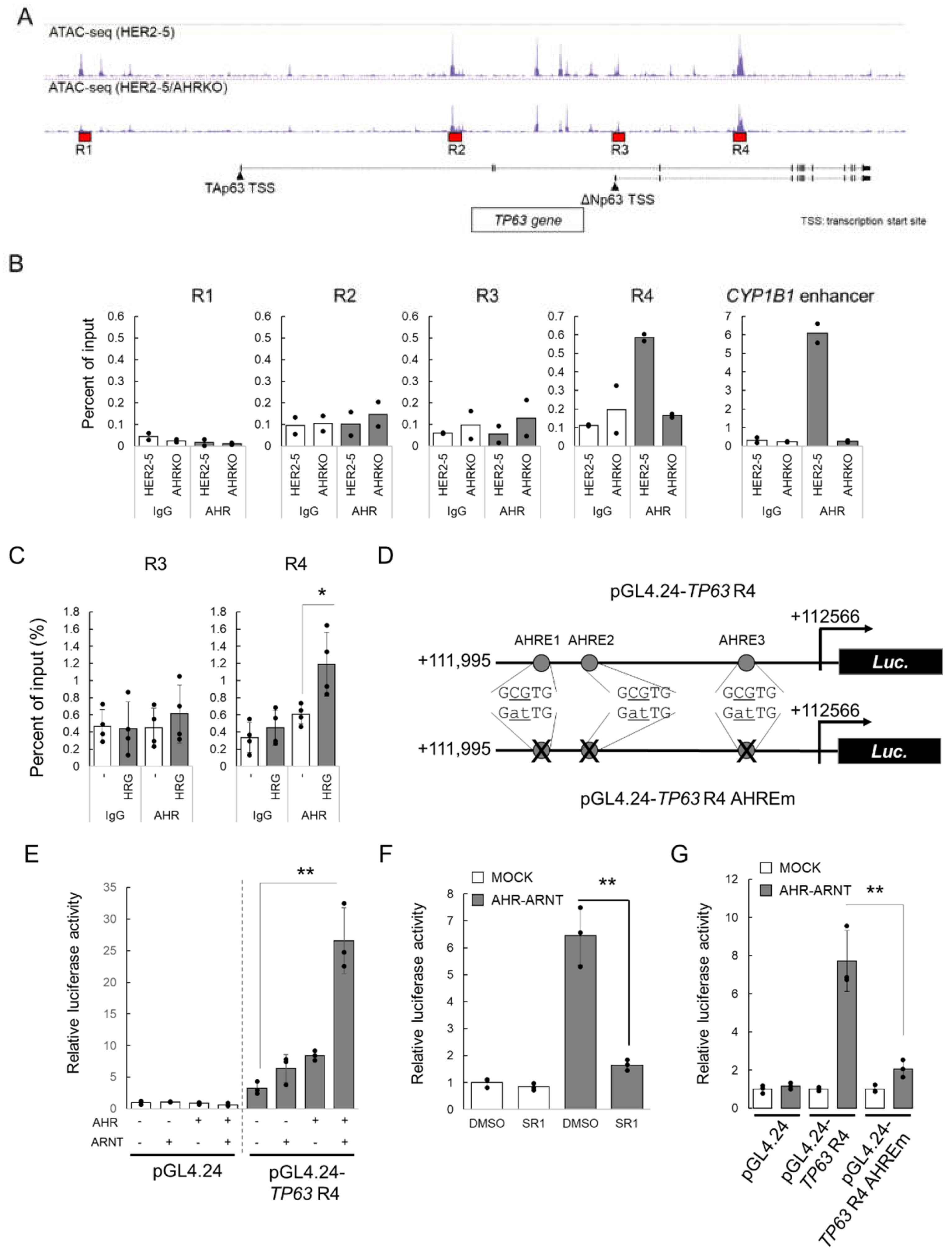
2.3. HRG-Activated AHR Binds to the Enhancer Region of TP63
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Lines and Cell Culture
4.3. Mammosphere Formation Assay
4.4. ATAC-Seq
4.5. Reverse Transcription-Quantitative Polymerase Chain Reaction (RT-qPCR)
4.6. Western Blotting
4.7. ChIP Assay
4.8. Plasmid Construction
4.9. Luciferase Reporter Gene Assay
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fitzmaurice, C.; Allen, C.; Barber, R.M.; Barregard, L.; Bhutta, Z.A.; Brenner, H.; Dicker, D.J.; Chimed-Orchir, O.; Dandona, R.; Dandona, L.; et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: A systematic analysis for the Global Burden of Disease Study Global Burden. JAMA Oncol. 2017, 3, 524–548. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- MWicha, M.S.; Liu, S.; Dontu, G. Cancer Stem Cells: An Old Idea—A Paradigm Shift. Cancer Res. 2006, 66, 1883–1890. [Google Scholar] [CrossRef]
- Duan, H.; Liu, Y.; Gao, Z.; Huang, W. Recent advances in drug delivery systems for targeting cancer stem cells. Acta Pharm. Sin. B 2020, 11, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef]
- Miyoshi, N.; Haraguchi, N.; Mizushima, T.; Ishii, H.; Yamamoto, H.; Mori, M. Targeting cancer stem cells in refractory cancer. Regen. Ther. 2021, 17, 13–19. [Google Scholar] [CrossRef]
- Herreros-Pomares, A. Identification, Culture and Targeting of Cancer Stem Cells. Life 2022, 12, 184. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Pines, G. The ERBB network: At last, cancer therapy meets systems biology. Nat. Cancer 2012, 12, 553–563. [Google Scholar] [CrossRef]
- Peles, E.; Ben-Levy, R.; Tzahar, E.; Liu, N.; Wen, D.; Yarden, Y. Cell-type specific interaction of Neu differentiation factor (NDF/heregulin) with Neu/HER-2 suggests complex ligand-receptor relationships. EMBO J. 1993, 12, 961–971. [Google Scholar] [CrossRef]
- Magnifico, A.; Albano, L.; Campaner, S.; Delia, D.; Castiglioni, F.; Gasparini, P.; Sozzi, G.; Fontanella, E.; Menard, S.; Tagliabue, E. Tumor-Initiating Cells of HER2-Positive Carcinoma Cell Lines Express the Highest Oncoprotein Levels and Are Sensitive to Trastuzumab. Clin. Cancer Res. 2009, 15, 2010–2021. [Google Scholar] [CrossRef]
- Kim, Y.G.; Na Yoon, Y.; Choi, H.S.; Kim, J.-H.; Seol, H.; Lee, J.K.; Seong, M.-K.; Park, I.C.; Kim, K.I.; Kim, H.-A.; et al. Breast cancer stem cells in HER2-negative breast cancer cells contribute to HER2-mediated radioresistance and molecular subtype conversion: Clinical implications for serum HER2 in recurrent HER2-negative breast cancer. Oncotarget 2017, 9, 5811–5822. [Google Scholar] [CrossRef] [PubMed]
- Duru, N.; Fan, M.; Candas, D.; Menaa, C.; Liu, H.-C.; Nantajit, D.; Wen, Y.; Xiao, K.; Eldridge, A.; Chromy, B.A.; et al. HER2-Associated Radioresistance of Breast Cancer Stem Cells Isolated from HER2-Negative Breast Cancer Cells. Clin. Cancer Res. 2012, 18, 6634–6647. [Google Scholar] [CrossRef] [PubMed]
- Korkaya, H.; Paulson, A.; Iovino, F.; Wicha, M.S. HER2 regulates the mammary stem/progenitor cell population driving tumorigenesis and invasion. Oncogene 2008, 27, 6120–6130. [Google Scholar] [CrossRef]
- Gu, Y.-Z.; Hogenesch, J.B.; Bradfield, C.A. The PAS Superfamily: Sensors of Environmental and Developmental Signals. Annu. Rev. Pharmacol. Toxicol. 2000, 40, 519–561. [Google Scholar] [CrossRef]
- Hoffman, E.C.; Reyes, H.; Chu, F.-F.; Sander, F.; Conley, L.H.; Brooks, B.A.; Hankinson, O. Cloning of a Factor Required for Activity of the Ah (Dioxin) Receptor. Science 1991, 252, 954–958. [Google Scholar] [CrossRef]
- Fujii-Kuriyama, Y.; Mimura, J. Molecular mechanisms of AhR functions in the regulation of cytochrome P450 genes. Biochem. Biophys. Res. Commun. 2005, 338, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, N.; Saito, N.; Zhao, S.; Terai, K.; Hiruta, N.; Park, Y.; Bujo, H.; Nemoto, K.; Kanno, Y. Heregulin-induced cell migration is promoted by aryl hydrocarbon receptor in HER2-overexpressing breast cancer cells. Exp. Cell Res. 2018, 366, 34–40. [Google Scholar] [CrossRef]
- Zhao, S.; Ohara, S.; Kanno, Y.; Midorikawa, Y.; Nakayama, M.; Makimura, M.; Park, Y.; Inouye, Y. HER2 overexpression-mediated inflammatory signaling enhances mammosphere formation through up-regulation of aryl hydrocarbon receptor transcription. Cancer Lett. 2013, 330, 41–48. [Google Scholar] [CrossRef]
- Yallowitz, A.R.; Alexandrova, E.M.; Talos, F.; Xu, S.; Marchenko, N.D.; Moll, U.M. p63 is a prosurvival factor in the adult mammary gland during post-lactational involution, affecting PI-MECs and ErbB2 tumorigenesis. Cell Death Differ. 2014, 21, 645–654. [Google Scholar] [CrossRef]
- Memmi, E.M.; Sanarico, A.G.; Giacobbe, A.; Peschiaroli, A.; Frezza, V.; Cicalese, A.; Pisati, F.; Tosoni, D.; Zhou, H.; Tonon, G.; et al. p63 sustains self-renewal of mammary cancer stem cells through regulation of Sonic Hedgehog signaling. Proc. Natl. Acad. Sci. USA 2015, 112, 3499–3504. [Google Scholar] [CrossRef]
- Du, Z.; Li, J.; Wang, L.; Bian, C.; Wang, Q.; Liao, L.; Dou, X.; Bian, X.; Zhao, R.C. Overexpression of ΔNp63α induces a stem cell phenotype in MCF7 breast carcinoma cell line through the Notch pathway. Cancer Sci. 2010, 101, 2417–2424. [Google Scholar] [CrossRef]
- Saito, N.; Kanno, Y.; Yamashita, N.; Degawa, M.; Yoshinari, K.; Nemoto, K. The differential selectivity of aryl hydrocarbon receptor (AHR) agonists towards AHR-dependent suppression of mammosphere formation and gene transcription in human breast cancer cells. Biol. Pharm. Bull. 2021, 44, 571–578. [Google Scholar] [CrossRef]
- Holbro, T.; Beerli, R.R.; Maurer, F.; Koziczak, M.; Barbas, C.F.; Hynes, N.E. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc. Natl. Acad. Sci. USA 2003, 100, 8933–8938. [Google Scholar] [CrossRef] [PubMed]
- Fornes, O.; Castro-Mondragon, J.A.; Khan, A.; Van Der Lee, R.; Zhang, X.; Richmond, P.A.; Modi, B.P.; Correard, S.; Gheorghe, M.; Baranašić, D.; et al. JASPAR 2020: Update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2020, 48, D87–D92. [Google Scholar] [CrossRef]
- Yang, X.; Solomon, S.; Fraser, L.R.; Trombino, A.F.; Liu, D.; Sonenshein, G.E.; Hestermann, E.V.; Sherr, D.H. Constitutive regulation ofCYP1B1 by the aryl hydrocarbon receptor (AhR) in pre-malignant and malignant mammary tissue. J. Cell. Biochem. 2008, 104, 402–417. [Google Scholar] [CrossRef]
- Vacher, S.; Castagnet, P.; Chemlali, W.; Lallemand, F.; Meseure, D.; Pocard, M.; Bieche, I.; Perrot-Applanat, M. High AHR expression in breast tumors correlates with expression of genes from several signaling pathways namely inflammation and endogenous tryptophan metabolism. PLoS ONE 2018, 13, e0190619. [Google Scholar] [CrossRef]
- Mohamed, H.T.; Gadalla, R.; El-Husseiny, N.; Hassan, H.; Wang, Z.; Ibrahim, S.A.; El-Shinawi, M.; Sherr, D.H.; Mohamed, M.M. Inflammatory breast cancer: Activation of the aryl hydrocarbon receptor and its target CYP1B1 correlates closely with Wnt5a/b-β-catenin signalling, the stem cell phenotype and disease progression. J. Adv. Res. 2019, 16, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, U.; Zhang, X.; Kuhn, C.; Jalaguier, S.; Colinge, J.; Pfender, K.; Mayr, D.; Ditsch, N.; Harbeck, N.; Mahner, S.; et al. The Prognostic Impact of the Aryl Hydrocarbon Receptor (AhR) in Primary Breast Cancer Depends on the Lymph Node Status. Int. J. Mol. Sci. 2019, 20, 1016. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-D.; Wang, K.; Yang, X.-W.; Zhuang, Z.-G.; Wang, J.-J.; Tong, X.-W. Expression of aryl hydrocarbon receptor in relation to p53 status and clinicopathological parameters in breast cancer. Int. J. Clin. Exp. Pathol. 2014, 7, 7931–7937. [Google Scholar]
- Novikov, O.; Wang, Z.; Stanford, E.A.; Parks, A.J.; Ramirez-Cardenas, A.; Landesman, E.; Laklouk, I.; Sarita-Reyes, C.; Gusenleitner, D.; Li, A.; et al. An Aryl Hydrocarbon Receptor-Mediated Amplification Loop That Enforces Cell Migration in ER−/PR−/Her2−Human Breast Cancer Cells. Mol. Pharmacol. 2016, 90, 674–688. [Google Scholar] [CrossRef]
- Goode, G.D.; Ballard, B.R.; Manning, H.C.; Freeman, M.L.; Kang, Y.; Eltom, S.E. Knockdown of aberrantly upregulated aryl hydrocarbon receptor reduces tumor growth and metastasis of MDA-MB-231 human breast cancer cell line. Int. J. Cancer 2013, 133, 2769–2780. [Google Scholar] [CrossRef]
- Mengoni, M.; Braun, A.; Gaffal, E.; Tüting, T. The aryl hydrocarbon receptor promotes inflammation-induced dedifferentiation and systemic metastatic spread of melanoma cells. Int. J. Cancer 2020, 147, 2902–2913. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Niu, Y.; Leng, J.; Xu, J.; Chen, H.; Li, H.; Wang, L.; Hu, J.; Xia, D.; Wu, Y. Benzo(a)pyrene regulated A549 cell migration, invasion and epithelial-mesenchymal transition by up-regulating long non-coding RNA linc00673. Toxicol. Lett. 2020, 320, 37–45. [Google Scholar] [CrossRef]
- Nagarajan, S.; Bedi, U.; Budida, A.; Hamdan, F.; Mishra, V.K.; Najafova, Z.; Xie, W.; Alawi, M.; Indenbirken, D.; Knapp, S.; et al. BRD4 promotes p63 and GRHL3 expression downstream of FOXO in mammary epithelial cells. Nucleic Acids Res. 2017, 45, 3130–3145. [Google Scholar] [CrossRef] [PubMed]
- Swedenborg, E.; Pongratz, I. AhR and ARNT modulate ER signaling. Toxicology 2010, 268, 132–138. [Google Scholar] [CrossRef]
- Ohtake, F.; Fujii-Kuriyama, Y.; Kawajiri, K.; Kato, S. Cross-talk of dioxin and estrogen receptor signals through the ubiquitin system. J. Steroid Biochem. Mol. Biol. 2011, 127, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Labrecque, M.P.; Takhar, M.K.; Hollingshead, B.D.; Prefontaine, G.G.; Perdew, G.H.; Beischlag, T.V. Distinct Roles for Aryl Hydrocarbon Receptor Nuclear Translocator and Ah Receptor in Estrogen-Mediated Signaling in Human Cancer Cell Lines. PLoS ONE 2012, 7, e29545. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, F.; Takeyama, K.-I.; Matsumoto, T.; Kitagawa, H.; Yamamoto, Y.; Nohara, K.; Tohyama, C.; Krust, A.; Mimura, J.; Chambon, P.; et al. Modulation of oestrogen receptor signalling by association with the activated dioxin receptor. Nature 2003, 423, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Henry, M.D.; Triplett, A.A.; Oh, K.B.; Smith, G.H.; Wagner, K.-U. Parity-induced mammary epithelial cells facilitate tumorigenesis in MMTV-neu transgenic mice. Oncogene 2004, 23, 6980–6985. [Google Scholar] [CrossRef]
- Eyermann, C.E.; Li, J.; Alexandrova, E.M. p63 suppresses the ability of pregnancy-identified mammary epithelial cells (PIMECs) to drive HER2-positive breast cancer. Cell Death Dis. 2021, 12, 525. [Google Scholar] [CrossRef]
- Chakrabarti, R.; Wei, Y.; Hwang, J.; Hang, X.; Blanco, M.A.; Choudhury, A.; Tiede, B.; Romano, R.-A.; DeCoste, C.; Mercatali, L.; et al. ΔNp63 promotes stem cell activity in mammary gland development and basal-like breast cancer by enhancing Fzd7 expression and Wnt signalling. Nat. Cell Biol. 2014, 16, 1004–1015. [Google Scholar] [CrossRef] [PubMed]
- Stanford, E.A.; Wang, Z.; Novikov, O.; Mulas, F.; Landesman-Bollag, E.; Monti, S.; Smith, B.W.; Seldin, D.C.; Murphy, G.J.; Sherr, D.H. The role of the aryl hydrocarbon receptor in the development of cells with the molecular and functional characteristics of cancer stem-like cells. BMC Biol. 2016, 14, 20. [Google Scholar] [CrossRef] [PubMed]
- Al-Dhfyan, A.; Alhoshani, A.; Korashy, H.M. Aryl hydrocarbon receptor/cytochrome P450 1A1 pathway mediates breast cancer stem cells expansion through PTEN inhibition and β-Catenin and Akt activation. Mol. Cancer 2017, 16, 14. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, N.; Kanno, Y.; Saito, N.; Terai, K.; Sanada, N.; Kizu, R.; Hiruta, N.; Park, Y.; Bujo, H.; Nemoto, K. Aryl hydrocarbon receptor counteracts pharmacological efficacy of doxorubicin via enhanced AKR1C3 expression in triple negative breast cancer cells. Biochem. Biophys. Res. Commun. 2019, 516, 693–698. [Google Scholar] [CrossRef]
- Gao, W.-Q.; Ma, J.; Sun, L.-L.; Li, Q.; Zhu, R.-Y.; Jin, J. Paclitaxel-mediated human aryl hydrocarbon receptor mRNA translation by an internal ribosomal entry site-dependent mechanism. Oncol. Rep. 2017, 38, 3211–3219. [Google Scholar] [CrossRef][Green Version]
- Kolluri, S.K.; Jin, U.H.; Safe, S. Role of the aryl hydrocarbon receptor in carcinogenesis and potential as an anti-cancer drug target. Arch. Toxicol. 2017, 91, 2497–2513. [Google Scholar] [CrossRef]
- Zhao, S.; Kanno, Y.; Nakayama, M.; Makimura, M.; Ohara, S.; Inouye, Y. Activation of the aryl hydrocarbon receptor represses mammosphere formation in MCF-7 cells. Cancer Lett. 2012, 317, 192–198. [Google Scholar] [CrossRef]
- Prud’Homme, G.J.; Glinka, Y.; Toulina, A.; Ace, O.; Subramaniam, V.; Jothy, S. Breast Cancer Stem-Like Cells Are Inhibited by a Non-Toxic Aryl Hydrocarbon Receptor Agonist. PLoS ONE 2010, 5, e13831. [Google Scholar] [CrossRef]
- Yamashita, N.; Taga, C.; Ozawa, M.; Kanno, Y.; Sanada, N.; Kizu, R. Camalexin, an indole phytoalexin, inhibits cell proliferation, migration, and mammosphere formation in breast cancer cells via the aryl hydrocarbon receptor. J. Nat. Med. 2021, 76, 110–118. [Google Scholar] [CrossRef]
- Brantley, E.; Callero, M.A.; Berardi, D.E.; Campbell, P.; Rowland, L.; Zylstra, D.; Amis, L.; Yee, M.; Simian, M.; Todaro, L.; et al. AhR ligand Aminoflavone inhibits α6-integrin expression and breast cancer sphere-initiating capacity. Cancer Lett. 2016, 376, 53–61. [Google Scholar] [CrossRef]
- Yamashita, N.; Yoshizuka, A.; Kase, A.; Ozawa, M.; Taga, C.; Sanada, N.; Kanno, Y.; Nemoto, K.; Kizu, R. Activation of the aryl hydrocarbon receptor by 3-methylcholanthrene, but not by indirubin, suppresses mammosphere formation via downregulation of CDC20 expression in breast cancer cells. Biochem. Biophys. Res. Commun. 2021, 570, 131–136. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanno, Y.; Saito, N.; Yamashita, N.; Ota, K.; Shizu, R.; Hosaka, T.; Nemoto, K.; Yoshinari, K. Possible Involvement of the Upregulation of ΔNp63 Expression Mediated by HER2-Activated Aryl Hydrocarbon Receptor in Mammosphere Maintenance. Int. J. Mol. Sci. 2022, 23, 12095. https://doi.org/10.3390/ijms232012095
Kanno Y, Saito N, Yamashita N, Ota K, Shizu R, Hosaka T, Nemoto K, Yoshinari K. Possible Involvement of the Upregulation of ΔNp63 Expression Mediated by HER2-Activated Aryl Hydrocarbon Receptor in Mammosphere Maintenance. International Journal of Molecular Sciences. 2022; 23(20):12095. https://doi.org/10.3390/ijms232012095
Chicago/Turabian StyleKanno, Yuichiro, Nao Saito, Naoya Yamashita, Kazuki Ota, Ryota Shizu, Takuomi Hosaka, Kiyomitsu Nemoto, and Kouichi Yoshinari. 2022. "Possible Involvement of the Upregulation of ΔNp63 Expression Mediated by HER2-Activated Aryl Hydrocarbon Receptor in Mammosphere Maintenance" International Journal of Molecular Sciences 23, no. 20: 12095. https://doi.org/10.3390/ijms232012095
APA StyleKanno, Y., Saito, N., Yamashita, N., Ota, K., Shizu, R., Hosaka, T., Nemoto, K., & Yoshinari, K. (2022). Possible Involvement of the Upregulation of ΔNp63 Expression Mediated by HER2-Activated Aryl Hydrocarbon Receptor in Mammosphere Maintenance. International Journal of Molecular Sciences, 23(20), 12095. https://doi.org/10.3390/ijms232012095