
CRLF1 and CLCF1 in Development, Health and Disease



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Cytokines and Specific Receptors
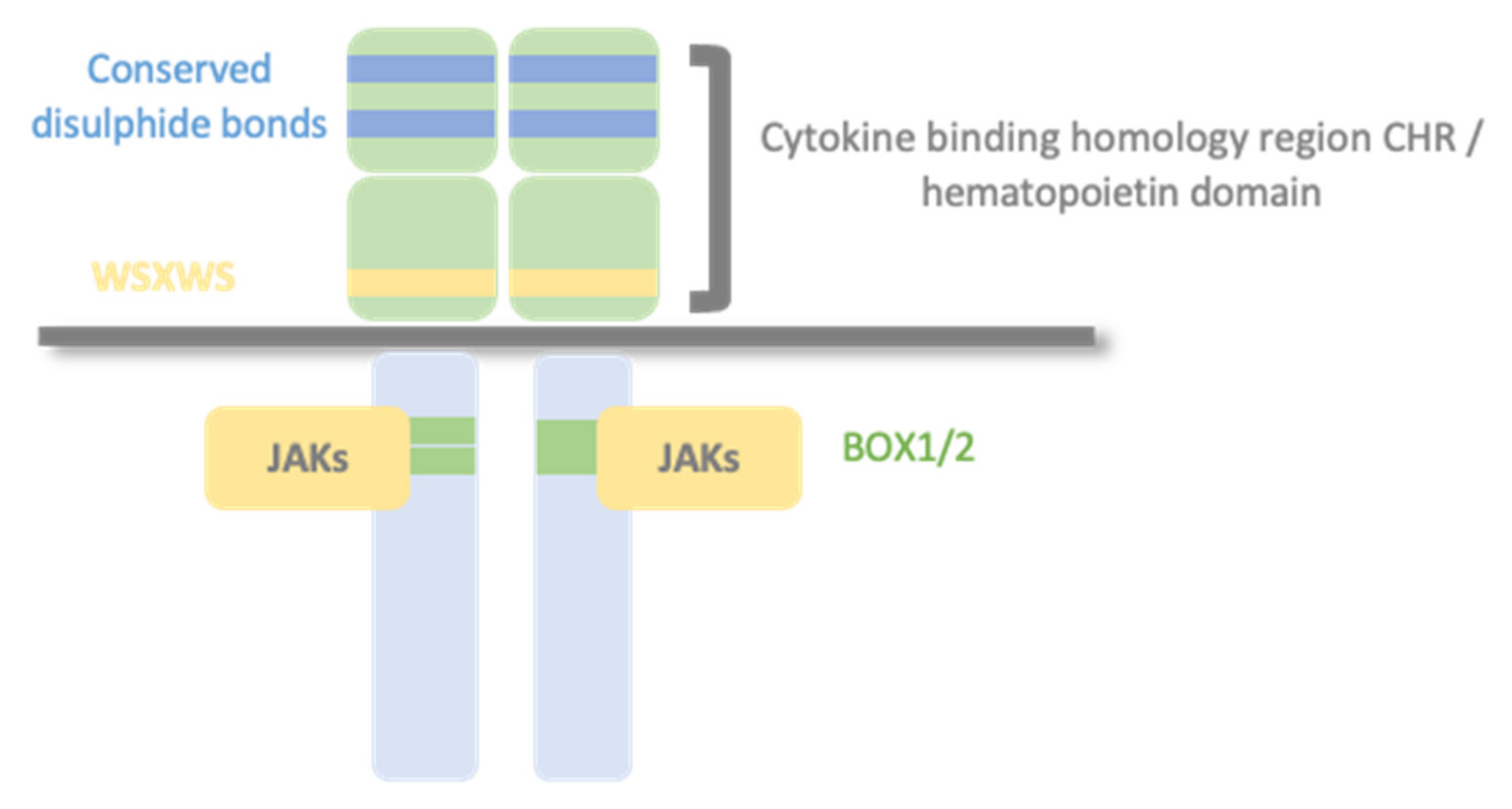
1.2. Type I Cytokine Receptors
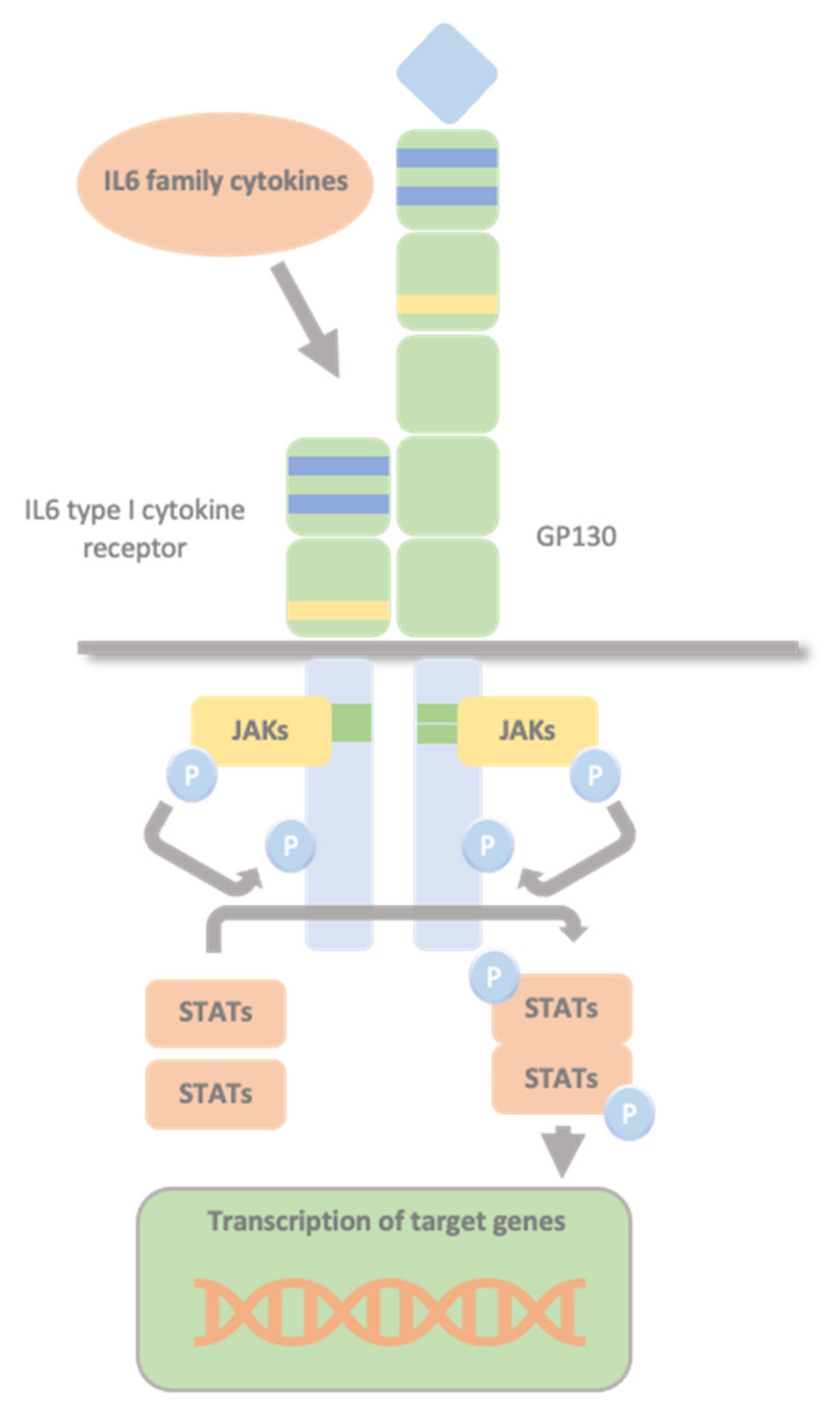
1.3. Glycoprotein 130 Cytokine Receptor Family and IL-6 Cytokines
1.4. The JAK/STAT Pathway
2. Overview of Cardiotrophin-like Cytokine Factor 1/Cytokine Receptor-like Factor-1
2.1. CLCF1 Key Concepts
2.2. CRLF1 Key Concepts
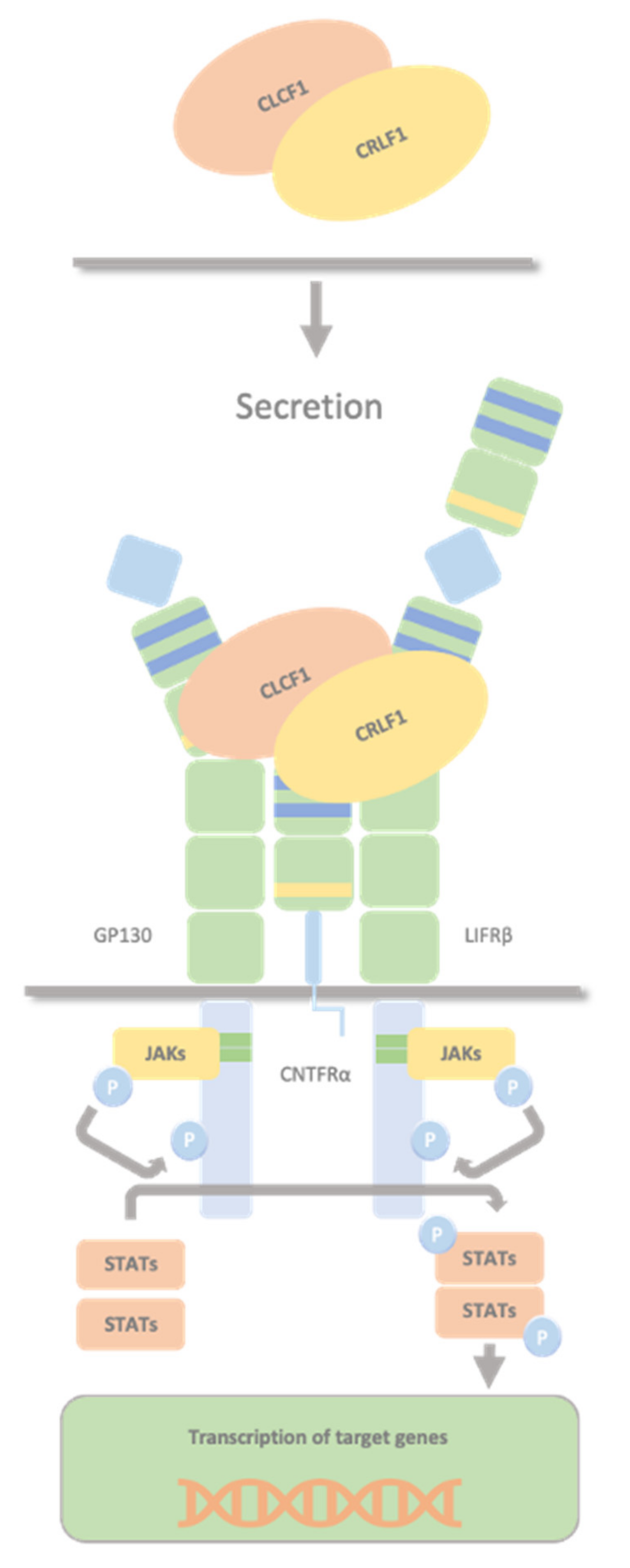
2.3. CLCF1/sCNTFRα Complex and CRLF1/CLCF1/CNTFRα Complex
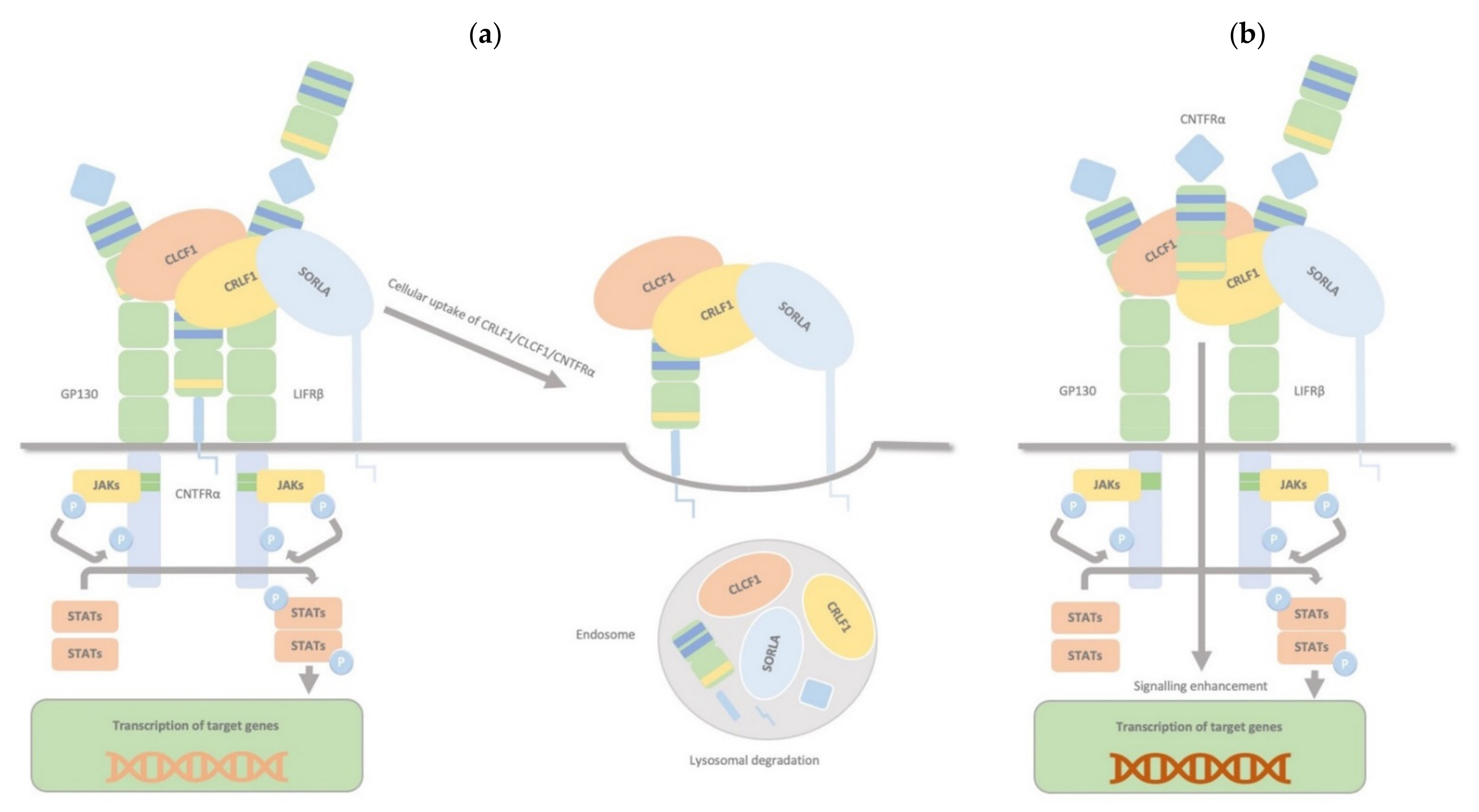
2.4. CRLF1/CLCF1/CNTFRα/SorLA Complex
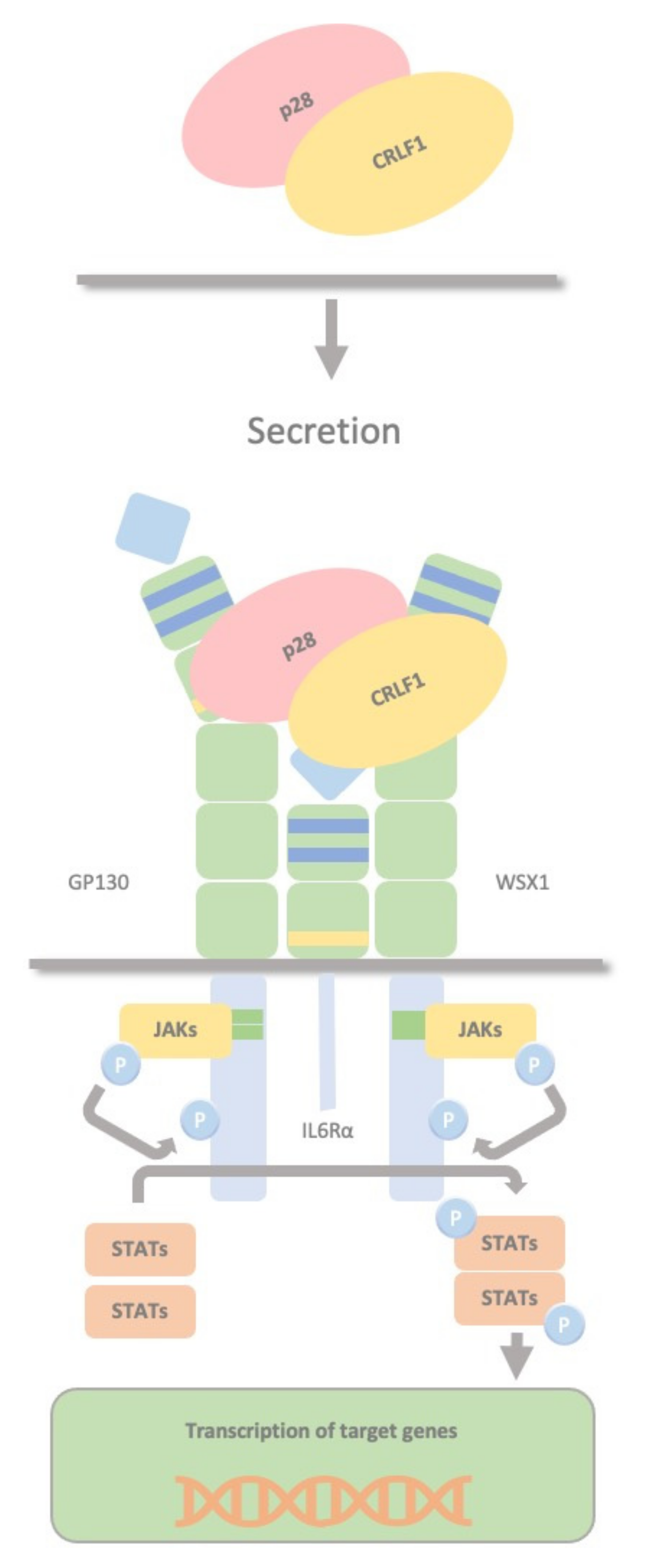
2.5. CRLF1/p28 Complex
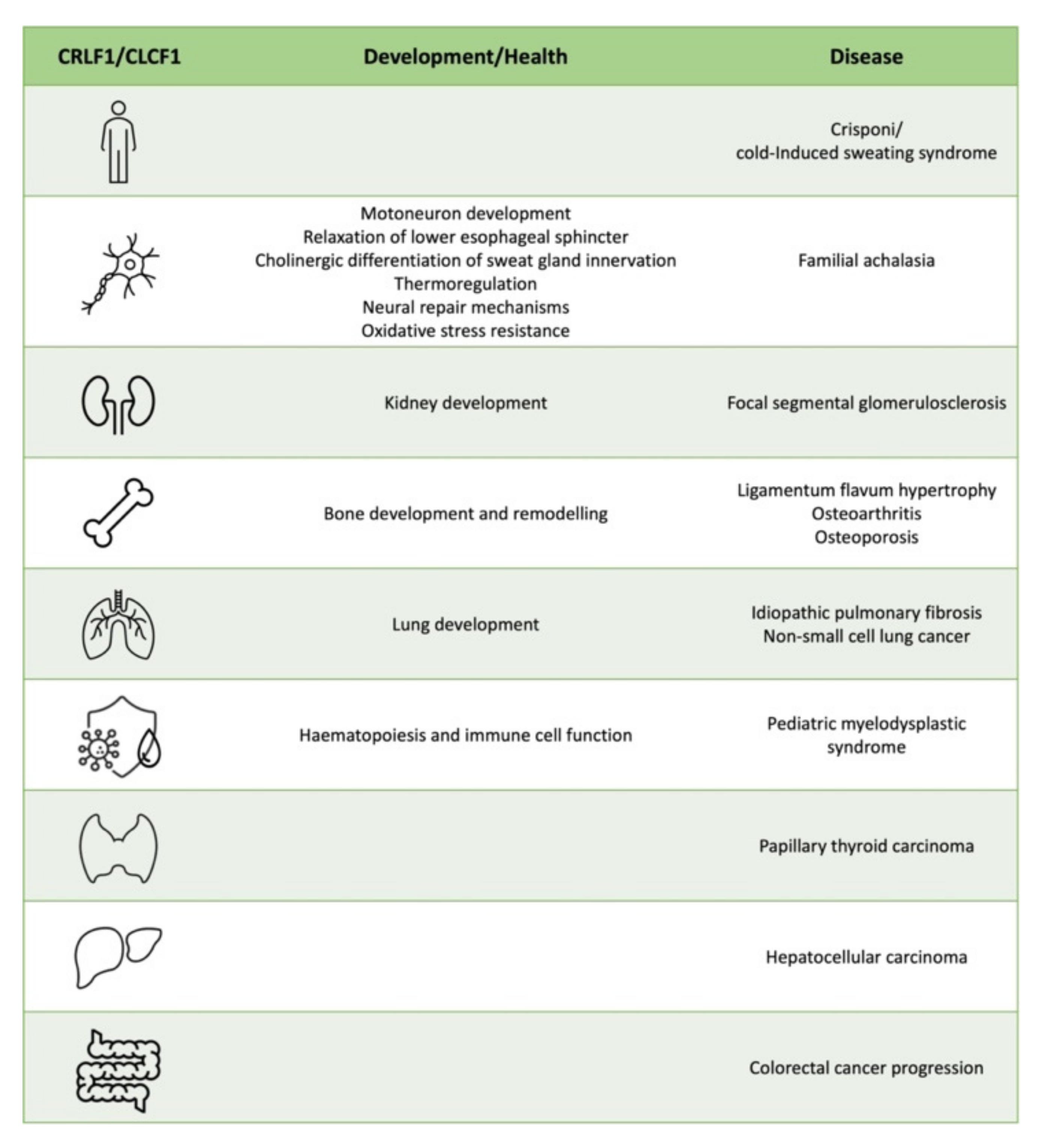
3. CRLF1 and CLCF1 in Development, Health and Disease
3.1. CS/CISS1 and CS/CISS2
3.2. Nervous System
3.3. Kidney
3.4. Bone and Cartilage
3.5. Lung
3.6. Haematopoiesis and Immune cell Function
3.7. Cancer
4. Therapeutic Opportunities
5. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Khan, M.M. Immunopharmacology, 2nd ed.; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Boulay, J.-L.; O’shea, J.J.; Paul, W.E. Molecular phylogeny within type I cytokines and their cognate receptors. Immunity 2003, 19, 159–163. [Google Scholar] [CrossRef]
- Bazan, J. Haemopoietic receptors and helical cytokines. Immunol. Today 1990, 11, 350–354. [Google Scholar] [CrossRef]
- Kishimoto, T.; Taga, T.; Akirat, S. Cytokine Signal Transduction Review. Cell 1994, 76, 253–262. [Google Scholar] [CrossRef]
- Leonard, W.J.; Lin, J.-X. Cytokine receptor signaling pathways. J. Allergy Clin. Immunol. 2000, 105, 877–888. [Google Scholar] [CrossRef]
- Metcalfe, R.D.; Putoczki, T.L.; Griffin, M.D.W. Structural Understanding of Interleukin 6 Family Cytokine Signaling and Targeted Therapies: Focus on Interleukin 11. Front. Immunol. 2020, 11, 1424. [Google Scholar] [CrossRef]
- De Vos, A.M.; Ultsch, M.; Kossiakoff, A. A Human Growth Hormone and Extracellular Domain of Its Receptor: Crystal Structure of the Complex. Science 1992, 255, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, P.; Nair, A.; Patidar, A.; Dandapat, J.; Sarkar, A.; Saha, B. A primer on cytokines. Cytokine 2021, 145, 155458. [Google Scholar] [CrossRef]
- Lokau, J.; Garbers, C. Biological functions and therapeutic opportunities of soluble cytokine receptors. Cytokine Growth Factor Rev. 2020, 55, 94–108. [Google Scholar] [CrossRef]
- Murakami, M.; Kamimura, D.; Hirano, T. Pleiotropy and Specificity: Insights from the Interleukin 6 Family of Cyto-kines. Immunity 2019, 50, 812–831. [Google Scholar] [CrossRef] [PubMed]
- Giraldez, M.D.; Carneros, D.; Garbers, C.; Rose-John, S.; Bustos, M. New Insights into IL-6 Family Cytokines in Metabolism, Hepatology and Gastroenterology. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 787–803. [Google Scholar] [CrossRef]
- Heinrich, P.C.; Behrmann, I.; Müller-Newen, G.; Schaper, F.; Graeve, L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem. J. 1998, 334, 297–314. [Google Scholar] [CrossRef]
- Heinrich, P.C.; Behrmann, I.; Haan, S.; Hermanns, H.M.; Müller-Newen, G.; Schaper, F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 2003, 374, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Taga, T.; Hibi, M.; Hirata, Y.; Yamasaki, K.; Yasukawa, K.; Matsuda, T.; Hirano, T.; Kishimoto, T. Interleukin-6 triggers the association of its receptor with a possible signal transducer, gp130. Cell 1989, 58, 573–581. [Google Scholar] [CrossRef]
- Jones, S.A.; Rose-John, S. The role of soluble receptors in cytokine biology: The agonistic properties of the sIL-6R/IL-6 complex. Biochim. Biophys. Acta 2002, 1592, 251–263. [Google Scholar] [CrossRef]
- Jones, S.A.; Scheller, J.; Rose-John, S. Therapeutic strategies for the clinical blockade of IL-6/gp130 signaling. J. Clin. Investig. 2011, 121, 3375–3383. [Google Scholar] [CrossRef]
- Morris, R.; Kershaw, N.J.; Babon, J.J. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 2018, 27, 1984–2009. [Google Scholar] [CrossRef]
- Liongue, C.; Taznin, T.; Ward, A.C. Signaling via the CytoR/JAK/STAT/SOCS pathway: Emergence during evolution. Mol. Immunol. 2016, 71, 166–175. [Google Scholar] [CrossRef]
- O’Sullivan, L.A.; Liongue, C.; Lewis, R.S.; Stephenson, S.E.M.; Ward, A.C. Cytokine Receptor Signaling through the Jak-Stat-Socs Pathway in Disease. Mol. Immunol. 2007, 44, 2497–2506. [Google Scholar] [CrossRef]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef]
- Senaldi, G.; Varnum, B.C.; Sarmiento, U.; Starnes, C.; Lile, J.; Scully, S.; Guo, J.; Elliott, G.; McNinch, J.; Shaklee, C.L.; et al. Novel neurotrophin-1/B cell-stimulating factor-3: A cytokine of the IL-6 family. Proc. Natl. Acad. Sci. USA 1999, 96, 11458–11463. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, W.; Yourey, P.; Gohari, S.; Zukauskas, D.; Zhang, J.; Ruben, S.; Alderson, R. Computational EST Database Analysis Identifies a Novel Member of the Neuropoietic Cytokine Family. Biochem. Biophys. Res. Commun. 1999, 262, 132–138. [Google Scholar] [CrossRef]
- Elson, G.C.; Graber, P.; Losberger, C.; Herren, S.; Gretener, D.; Menoud, L.N.; Wells, T.; Kosco-Vilbois, M.H.; Gauchat, J.F. Cytokine-like factor-1, a novel soluble protein, shares homology with members of the cytokine type I receptor family. J. Immunol. 1998, 161, 1371–1379. [Google Scholar] [PubMed]
- Alexander, W.; Rakar, S.; Robb, L.; Farley, A.; Willson, T.; Zhang, J.-G.; Hartley, L.; Kikuchi, Y.; Kojima, T.; Nomura, H.; et al. Suckling defect in mice lacking the soluble haemopoietin receptor NR6. Curr. Biol. 1999, 9, P605–P608. [Google Scholar] [CrossRef][Green Version]
- Elson, G.C.A.; Lelievre, E.; Guillet, C.; Chevalier, S.; Plun-Favreau, H.; Froger, J.; Suard, I.; De Coignac, A.B.; Delneste, Y.; Bonnefoy, J.-Y.; et al. CLF associates with CLC to form a functional heteromeric ligand for the CNTF receptor complex. Nat. Neurosci. 2000, 3, 867–872. [Google Scholar] [CrossRef]
- Plun-Favreau, H.; Elson, G.; Chabbert, M.; Froger, J.; Delapeyrière, O.; Lelievre, E.; Guillet, C.; Hermann, J.; Gauchat, J.; Gascan, H.; et al. The ciliary neurotrophic factor receptor alpha component induces the secretion of and is required for functional responses to cardiotrophin-like cytokine. EMBO J. 2001, 20, 1692–1703. [Google Scholar] [CrossRef]
- Herholz, J.; Crisponi, L.; Mallick, B.N.; Rutsch, F. Successful treatment of cold-induced sweating in Crisponi syndrome and its possible mechanism of action. Dev. Med. Child Neurol. 2010, 52, 494–497. [Google Scholar] [CrossRef] [PubMed]
- Perret, D.; Guillet, C.; Elson, G.; Froger, J.; Plun-Favreau, H.; Rousseau, F.; Chabbert, M.; Gauchat, J.-F.; Gascan, H. Two Different Contact Sites Are Recruited by Cardiotrophin-like Cytokine (CLC) to Generate the CLC/CLF and CLC/sCNTFRα Composite Cytokines. J. Biol. Chem. 2004, 279, 43961–43970. [Google Scholar] [CrossRef] [PubMed]
- Herholz, J.; Meloni, A.; Marongiu, M.; Chiappe, F.; Deiana, M.; Herrero, C.R.; Zampino, G.; Hamamy, H.; Zalloum, Y.; Waaler, P.E.; et al. Differential secretion of the mutated protein is a major component affecting phenotypic severity in CRLF1-associated disorders. Eur. J. Hum. Genet. 2011, 19, 525–533. [Google Scholar] [CrossRef][Green Version]
- Larsen, J.V.; Kristensen, A.M.; Pallesen, L.T.; Bauer, J.; Vægter, C.B.; Nielsen, M.S.; Madsen, P.S.; Petersen, C.M. Cytokine-Like Factor 1, an Essential Facilitator of Cardiotrophin-Like Cytokine:Ciliary Neurotrophic Factor Receptor α Signaling and sorLA-Mediated Turnover. Mol. Cell. Biol. 2016, 36, 1272–1286. [Google Scholar] [CrossRef]
- Larsen, J.V.; Hansen, M.; Møller, B.; Madsen, P.S.; Scheller, J.; Nielsen, M.S.; Petersen, C.M. Sortilin Facilitates Signaling of Ciliary Neurotrophic Factor and Related Helical Type 1 Cytokines Targeting the gp130/Leukemia Inhibitory Factor Receptor β Heterodimer. Mol. Cell. Biol. 2010, 30, 4175–4187. [Google Scholar] [CrossRef]
- Talbot, H.; Saada, S.; Naves, T.; Gallet, P.-F.; Fauchais, A.-L.; Jauberteau, M.-O. Regulatory Roles of Sortilin and SorLA in Immune-Related Processes. Front. Pharmacol. 2019, 9, 1507. [Google Scholar] [CrossRef]
- Crabe, S.; Guay-Giroux, A.; Tormo, A.J.; Duluc, D.; Lissilaa, R.; Guilhot, F.; Mavoungou-Bigouagou, U.; Lefouili, F.; Cognet, I.; Ferlin, W.; et al. The IL-27 p28 Subunit Binds Cytokine-Like Factor 1 to Form a Cytokine Regulating NK and T Cell Activities Requiring IL-6R for Signaling. J. Immunol. 2009, 183, 7692–7702. [Google Scholar] [CrossRef]
- Tormo, A.J.; Meliani, Y.; Beaupré, L.A.; Sharma, M.; Fritz, J.H.; Elson, G.; Crabé, S.; Gauchat, J.-F. The Composite Cytokine p28/Cytokine-Like Factor 1 Sustains B Cell Proliferation and Promotes Plasma Cell Differentiation. J. Immunol. 2013, 191, 1657–1665. [Google Scholar] [CrossRef] [PubMed]
- Crisponi, L.; Crisponi, G.; Meloni, A.; Toliat, M.R.; Nürnberg, G.; Usala, G.; Uda, M.; Masala, M.; Höhne, W.; Becker, C.; et al. Crisponi Syndrome Is Caused by Mutations in the CRLF1 Gene and Is Allelic to Cold-Induced Sweating Syndrome Type 1. Am. J. Hum. Genet. 2007, 80, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Dagoneau, N.; Bellais, S.; Blanchet, P.; Sarda, P.; Al-Gazali, L.; Di Rocco, M.; Huber, C.; Djouadi, F.; Goff, C.L.E.; Munnich, A.; et al. Mutations in Cytokine Receptor-Like Factor 1 (CRLF1) Account for Both Crisponi and Cold-Induced Sweating Syndromes. Am. J. Hum. Genet. 2007, 80, 966–970. [Google Scholar] [CrossRef] [PubMed]
- Buers, I.; Persico, I.; Schöning, L.; Nitschke, Y.; Di Rocco, M.; Loi, A.; Sahi, P.K.; Utine, G.E.; Bayraktar-Tanyeri, B.; Zampino, G.; et al. Crisponi/cold-induced sweating syndrome: Differential diagnosis, pathogenesis and treatment concepts. Clin. Genet. 2020, 97, 209–221. [Google Scholar] [CrossRef]
- Rousseau, F.; Gauchat, J.-F.; McLeod, J.G.; Chevalier, S.; Guillet, C.; Guilhot, F.; Cognet, I.; Froger, J.; Hahn, A.F.; Knappskog, P.M.; et al. Inactivation of cardiotrophin-like cytokine, a second ligand for ciliary neurotrophic factor receptor, leads to cold-induced sweating syndrome in a patient. Proc. Natl. Acad. Sci. USA 2006, 103, 10068–10073. [Google Scholar] [CrossRef]
- Hahn, A.; Waaler, P.; Kvistad, P.; Bamforth, J.; Miles, J.; McLeod, J.; Knappskog, P.; Boman, H. Cold-induced sweating syndrome: CISS1 and CISS2: Manifestations from infancy to adulthood. Four new cases. J. Neurol. Sci. 2010, 293, 68–75. [Google Scholar] [CrossRef]
- Zou, X.; Bolon, B.; Pretorius, J.K.; Kurahara, C.; McCabe, J.; Christiansen, K.A.; Sun, N.; Duryea, D.; Foreman, O.; Senaldi, G.; et al. Neonatal Death in Mice Lacking Cardiotrophin-like Cytokine is Associated with Multifocal Neuronal Hypoplasia. Vet. Pathol. 2008, 46, 514–519. [Google Scholar] [CrossRef]
- Forger, N.G.; Prevette, D.; Delapeyrière, O.; De Bovis, B.; Wang, S.; Bartlett, P.; Oppenheim, R.W. Cardiotrophin-Like Cytokine/Cytokine-Like Factor 1 is an Essential Trophic Factor for Lumbar and Facial MotoneuronsIn Vivo. J. Neurosci. 2003, 23, 8854–8858. [Google Scholar] [CrossRef]
- Busch, A.; Žarković, M.; Lowe, C.; Jankofsky, M.; Ganschow, R.; Buers, I.; Kurth, I.; Reutter, H.; Rutsch, F.; Hübner, C.A. Mutations in CRLF1 cause familial achalasia. Clin. Genet. 2017, 92, 104–108. [Google Scholar] [CrossRef]
- Uemuraab, A.; Takizawaac, T.; Ochiaia, W.; Yanagisawaa, M.; Nakashimaa, K.; Taga, T. Cardiotrophin-like Cytokine Induces Astrocyte Differentiation of Fetal Neuroepithelial Cells via Activation of STAT3. Cytokine 2002, 18, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kraves, S.; Weitz, C.J. A role for cardiotrophin-like cytokine in the circadian control of mammalian locomotor activity. Nat. Neurosci. 2006, 9, 212–219. [Google Scholar] [CrossRef]
- McKemy, D.D. The Molecular and Cellular Basis of Cold Sensation. ACS Chem. Neurosci. 2013, 4, 238–247. [Google Scholar] [CrossRef]
- Boulant, J.A. Hypothalamic mechanisms in thermoregulation. Fed. Proc. 1981, 40, 2843–2850. [Google Scholar]
- Melone, M.A.B.; Pellegrino, M.J.; Nolano, M.; Habecker, B.; Johansson, S.; Nathanson, N.M.; Knappskog, P.M.; Hahn, A.F.; Boman, H. Unusual Stüve-Wiedemann syndrome with complete maternal chromosome 5 isodisomy. Ann. Clin. Transl. Neurol. 2014, 1, 926–932. [Google Scholar] [CrossRef]
- Di Leo, R.; Nolano, M.; Boman, H.; Pierangeli, G.; Provitera, V.; Knappskog, P.M.; Cortelli, P.; Vita, G.; Rodolico, C. Central and peripheral autonomic failure in cold-induced sweating syndrome type 1. Neurology 2010, 75, 1567–1569. [Google Scholar] [CrossRef]
- Finch, L.; Haeusler, G.; Thoenen, H. A comparison of the effects of chemical sympathectomy by 6-hydroxydopamine in newborn and adult rats. Br. J. Pharmacol. 1973, 47, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Pantic, I.; Cumic, J.; Skodric, S.R.; Dugalic, S.; Brodski, C. Oxidopamine and oxidative stress: Recent advances in experimental physiology and pharmacology. Chem. Interact. 2021, 336, 109380. [Google Scholar] [CrossRef]
- Yodlowski, M.; Fredieu, J.; Landis, S. Neonatal 6-hydroxydopamine treatment eliminates cholinergic sympathetic innervation and induces sensory sprouting in rat sweat glands. J. Neurosci. 1984, 4, 1535–1548. [Google Scholar] [CrossRef] [PubMed]
- Asmus, S.E.; Parsons, S.; Landis, S.C. Developmental Changes in the Transmitter Properties of Sympathetic Neurons That Innervate the Periosteum. J. Neurosci. 2000, 20, 1495–1504. [Google Scholar] [CrossRef]
- Asmus, S.E.; Tian, H.; Landis, S.C. Induction of Cholinergic Function in Cultured Sympathetic Neurons by Periosteal Cells: Cellular Mechanisms. Dev. Biol. 2001, 235, 1–11. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Landis, S.C.; Keefe, D. Evidence for neurotransmitter plasticity in vivo: Developmental changes in properties of cholinergic sympathetic neurons. Dev. Biol. 1983, 98, 349–372. [Google Scholar] [CrossRef]
- Stevens, L.M.; Landis, S.C. Development and properties of the secretory response in rat sweat glands: Relationship to the induction of cholinergic function in sweat gland innervation. Dev. Biol. 1987, 123, 179–190. [Google Scholar] [CrossRef]
- Landis, S.C.; Siegel, R.E.; Schwab, M. Evidence for neurotransmitter plasticity in vivo: II. Immunocytochemical studies of rat sweat gland innervation during development. Dev. Biol. 1988, 126, 129–140. [Google Scholar] [CrossRef]
- Leblanc, G.; Landis, S. Development of choline acetyltransferase (CAT) in the sympathetic innervation of rat sweat glands. J. Neurosci. 1986, 6, 260–265. [Google Scholar] [CrossRef]
- Stanke, M.; Duong, C.V.; Pape, M.; Geissen, M.; Burbach, G.; Deller, T.; Gascan, H.; Parlato, R.; Schütz, G.; Rohrer, H. Target-dependent specification of the neurotransmitter phenotype:cholinergic differentiation of sympathetic neurons is mediated in vivo by gp130 signaling. Development 2006, 133, 141–150. [Google Scholar] [CrossRef]
- Habecker, B.A.; Symes, A.J.; Stahl, N.; Francis, N.J.; Economides, A.; Fink, J.S.; Yancopoulos, G.D.; Landis, S.C. A Sweat Gland-derived Differentiation Activity Acts through Known Cytokine Signaling Pathways. J. Biol. Chem. 1997, 272, 30421–30428. [Google Scholar] [CrossRef]
- Elsaeidi, F.; Bemben, M.A.; Zhao, X.-F.; Goldman, D. Jak/Stat Signaling Stimulates Zebrafish Optic Nerve Regeneration and Overcomes the Inhibitory Actions of Socs3 and Sfpq. J. Neurosci. 2014, 34, 2632–2644. [Google Scholar] [CrossRef]
- Barrette, B.; Calvo, E.; Vallières, N.; Lacroix, S. Transcriptional profiling of the injured sciatic nerve of mice carrying the Wld(S) mutant gene: Identification of genes involved in neuroprotection, neuroinflammation, and nerve regeneration. Brain Behav. Immun. 2010, 24, 1254–1267. [Google Scholar] [CrossRef]
- Tirmenstein, M.A.; Hu, C.X.; Scicchitano, M.S.; Narayanan, P.K.; McFarland, D.C.; Thomas, H.C.; Schwartz, L.W. Effects of 6-hydroxydopamine on mitochondrial function and glutathione status in SH-SY5Y human neuroblastoma cells. Toxicol. Vitro 2005, 19, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Looyenga, B.D.; Resau, J.; MacKeigan, J. Cytokine Receptor-Like Factor 1 (CRLF1) Protects against 6-Hydroxydopamine Toxicity Independent of the gp130/JAK Signaling Pathway. PLoS ONE 2013, 8, e66548. [Google Scholar] [CrossRef] [PubMed]
- Kaur, N.; Lu, B.; Ward, S.; Halvorsen, S. Inducers of oxidative stress block ciliary neurotrophic factor activation of Jak/STAT signaling in neurons. J. Neurochem. 2005, 92, 1521–1530. [Google Scholar] [CrossRef]
- Aljabari, S.; Howard, E.; Bell, T.; Vasylyeva, T.L. Cold Induced Sweating Syndrome with Urinary System Anomaly Association. Case Rep. Pediatr. 2013, 2013, 173890. [Google Scholar] [CrossRef] [PubMed]
- Schwab, K.; Patterson, L.T.; Aronow, B.J.; Luckas, R.; Liang, H.-C.; Potter, S.S. A catalogue of gene expression in the developing kidney. Kidney Int. 2003, 64, 1588–1604. [Google Scholar] [CrossRef] [PubMed]
- GTEx Expression Data. Available online: https://www.gtexportal.org/home/ (accessed on 22 December 2021).
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ (accessed on 22 December 2021).
- Karlsson, M.; Zhang, C.; Méar, L.; Zhong, W.; Digre, A.; Katona, B.; Sjöstedt, E.; Butler, L.; Odeberg, J.; Dusart, P.; et al. A single–cell type transcriptomics map of human tissues. Sci. Adv. 2021, 7, abh2169. [Google Scholar] [CrossRef]
- Cebrian, C.; Borodo, K.; Charles, N.; Herzlinger, D.A. Morphometric index of the developing murine kidney. Dev. Dyn. 2004, 231, 601–608. [Google Scholar] [CrossRef]
- Short, K.M.; Smyth, I.M. Branching morphogenesis as a driver of renal development. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 2020, 303, 2578–2587. [Google Scholar] [CrossRef]
- Schmidt-Ott, K.M.; Yang, J.; Chen, X.; Wang, H.; Paragas, N.; Mori, K.; Li, J.-Y.; Lu, B.; Costantini, F.; Schiffer, M.; et al. Novel Regulators of Kidney Development from the Tips of the Ureteric Bud. J. Am. Soc. Nephrol. 2005, 16, 1993–2002. [Google Scholar] [CrossRef]
- Caruana, G.; Cullen-McEwen, L.; Nelson, A.L.; Kostoulias, X.; Woods, K.; Gardiner, B.; Davis, M.J.; Taylor, D.F.; Teasdale, R.; Grimmond, S.; et al. Spatial gene expression in the T-stage mouse metanephros. Gene Expr. Patterns 2006, 6, 807–825. [Google Scholar] [CrossRef] [PubMed]
- Kurtzeborn, K.; Cebrian, C.; Kuure, S. Regulation of Renal Differentiation by Trophic Factors. Front. Physiol. 2018, 9, 1588. [Google Scholar] [CrossRef] [PubMed]
- Rutledge, E.A.; Benazet, J.-D.; McMahon, A.P. Cellular heterogeneity in the ureteric progenitor niche and distinct profiles of branching morphogenesis in organ development. Development 2017, 144, 3177–3188. [Google Scholar] [CrossRef]
- Costantini, F. GDNF/Ret signaling and renal branching morphogenesis. Organogenesis 2010, 6, 252–262. [Google Scholar] [CrossRef]
- Attanasio, M.; Uhlenhaut, N.H.; Sousa, V.H.; O’Toole, J.F.; Otto, E.; Anlag, K.; Klugmann, C.; Treier, A.-C.; Helou, J.; Sayer, J.A.; et al. Loss of GLIS2 causes nephronophthisis in humans and mice by increased apoptosis and fibrosis. Nat. Genet. 2007, 39, 1018–1024. [Google Scholar] [CrossRef] [PubMed]
- Savin, V.J.; Sharma, M.; Zhou, J.; Gennochi, D.; Fields, T.; Sharma, R.; McCarthy, E.T.; Srivastava, T.; Domen, J.; Tormo, A.; et al. Renal and Hematological Effects of CLCF-1, a B-Cell-Stimulating Cytokine of the IL-6 Family. J. Immunol. Res. 2015, 2015, 714964. [Google Scholar] [CrossRef]
- Savin, V.J.; Sharma, M.; Zhou, J.; Genochi, D.; Sharma, R.; Srivastava, T.; Ilahe, A.; Budhiraja, P.; Gupta, A.; McCarthy, E.T. Multiple Targets for Novel Therapy of FSGS Associated with Circulating Permeability Factor. BioMed Res. Int. 2017, 2017, 6232616. [Google Scholar] [CrossRef]
- Königshausen, E.; Sellin, L. Circulating Permeability Factors in Primary Focal Segmental Glomerulosclerosis: A Review of Proposed Candidates. BioMed Res. Int. 2016, 2016, 3765608. [Google Scholar] [CrossRef]
- Sharma, M.; Zhou, J.; Gauchat, J.-F.; Sharma, R.; McCarthy, E.T.; Srivastava, T.; Savin, V.J. Janus kinase 2/signal transducer and activator of transcription 3 inhibitors attenuate the effect of cardiotrophin-like cytokine factor 1 and human focal segmental glomerulosclerosis serum on glomerular filtration barrier. Transl. Res. 2015, 166, 384–398. [Google Scholar] [CrossRef] [PubMed]
- Müller-Deile, J.; Sarau, G.; Kotb, A.M.; Jaremenko, C.; Rolle-Kampczyk, U.E.; Daniel, C.; Kalkhof, S.; Christiansen, S.H.; Schiffer, M. Novel diagnostic and therapeutic techniques reveal changed metabolic profiles in recurrent focal segmental glomerulosclerosis. Sci. Rep. 2021, 11, 4577. [Google Scholar] [CrossRef] [PubMed]
- Brosius, F.C. New insights into the mechanisms of fibrosis and sclerosis in diabetic nephropathy. Rev. Endocr. Metab. Disord. 2008, 9, 245–254. [Google Scholar] [CrossRef]
- Clancy, B.M.; Johnson, J.D.; Lambert, A.-J.; Rezvankhah, S.; Wong, A.; Resmini, C.; Feldman, J.L.; Leppanen, S.; Pittman, D.D. A gene expression profile for endochondral bone formation: Oligonucleotide microarrays establish novel connections between known genes and BMP-2-induced bone formation in mouse quadriceps. Bone 2003, 33, 46–63. [Google Scholar] [CrossRef]
- Walker, E.C.; Poulton, I.J.; McGregor, N.E.; Ho, P.W.M.; Allan, E.H.; Quach, J.M.; Martin, T.J.; Sims, N.A. Sustained RANKL response to parathyroid hormone in oncostatin M receptor-deficient osteoblasts converts anabolic treatment to a catabolic effect in vivo. J. Bone Miner. Res. 2012, 27, 902–912. [Google Scholar] [CrossRef]
- McGregor, N.E.; Poulton, I.J.; Walker, E.C.; Pompolo, S.; Quinn, J.M.W.; Martin, T.J.; Sims, N.A. Ciliary Neurotrophic Factor Inhibits Bone Formation and Plays a Sex-Specific Role in Bone Growth and Remodeling. Calcif. Tissue Int. 2010, 86, 261–270. [Google Scholar] [CrossRef]
- Sims, N.A. Influences of the IL-6 cytokine family on bone structure and function. Cytokine 2021, 146, 155655. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Turner, C.H.; Yokota, H. Joint loading-driven bone formation and signaling pathways predicted from genome-wide expression profiles. Bone 2009, 44, 989–998. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Frost, H.M. Bone’s mechanostat: A 2003 update. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 2003, 275A, 1081–1101. [Google Scholar] [CrossRef]
- Heino, T.; Hentunen, T. Differentiation of Osteoblasts and Osteocytes from Mesenchymal Stem Cells. Curr. Stem Cell Res. Ther. 2008, 3, 131–145. [Google Scholar] [CrossRef]
- Kangari, P.; Talaei-Khozani, T.; Razeghian-Jahromi, I.; Razmkhah, M. Mesenchymal stem cells: Amazing remedies for bone and cartilage defects. Stem Cell Res. Ther. 2020, 11, 492. [Google Scholar] [CrossRef]
- Ash, P.; Loutit, J.F.; Townsend, K.M.S. Osteoclasts Derive from Hematopoietic Stem Cells According to Marker, Giant Lysosomes of Beige Mice. Clin. Orthop. Relat. Res. 1981, 155, 249–258. [Google Scholar] [CrossRef]
- Breeland, G.; Sinkler, M.A.; Menezes, R.G. Embryology, Bone Ossification; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Sims, N.A. The JAK1/STAT3/SOCS3 Axis in Bone Development, Physiology, and Pathology. Exp. MoLecular Med. 2020, 52, 1185–1197. [Google Scholar] [CrossRef] [PubMed]
- Ware, C.B.; Horowitz, M.C.; Renshaw, B.R.; Hunt, J.S.; Liggitt, D.; Koblar, S.A.; Gliniak, B.C.; McKenna, H.J.; Papayannopoulou, T.; Thoma, B.; et al. Targeted disruption of the low-affinity leukemia inhibitory factor receptor gene causes placental, skeletal, neural and metabolic defects and results in perinatal death. Development 1995, 121, 1283–1299. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, K.; Gao, Y.-H.; Yokose, S.; Kaji, Y.; Nakamura, T.; Suda, T.; Yoshida, K.; Taga, T.; Kishimoto, T.; Kataoka, H.; et al. Osteoclasts Are Present in gp130-Deficient Mice. Endocrinology 1997, 138, 4959–4965. [Google Scholar] [CrossRef][Green Version]
- Itoh, S.; Udagawa, N.; Takahashi, N.; Yoshitake, F.; Narita, H.; Ebisu, S.; Ishihara, K. A critical role for interleukin-6 family-mediated Stat3 activation in osteoblast differentiation and bone formation. Bone 2006, 39, 505–512. [Google Scholar] [CrossRef]
- DeChiara, T.M.; Vejsada, R.; Poueymirou, W.T.; Acheson, A.; Suri, C.; Conover, J.; Friedman, B.; McClain, J.; Pan, L.; Stahl, N.; et al. Mice lacking the CNTF receptor, unlike mice lacking CNTF, exhibit profound motor neuron deficits at birth. Cell 1995, 83, 313–322. [Google Scholar] [CrossRef]
- Bailey, J.R.; Tapscott, D.C. Osteopetrosis; StatPearls Publishing LLC: Treasure Island, FL, USA, 2021. [Google Scholar]
- Ge, J.-R.; Xie, L.-H.; Chen, J.; Li, S.-Q.; Xu, H.-J.; Lai, Y.-L.; Qiu, L.-L.; Ni, C.-B. Liuwei Dihuang Pill Treats Postmenopausal Osteoporosis with Shen (Kidney) Yin Deficiency via Janus Kinase/Signal Transducer and Activator of Transcription Signal Pathway by Up-regulating Cardiotrophin-Like Cytokine Factor 1 Expression. Chin. J. Integr. Med. 2016, 24, 415–422. [Google Scholar] [CrossRef]
- Chen, X.; Li, J.; Ye, Y.; Huang, J.; Xie, L.; Chen, J.; Li, S.; Chen, S.; Ge, J. Association of cardiotrophin-like cytokine factor 1 levels in peripheral blood mononuclear cells with bone mineral density and osteoporosis in postmenopausal women. BMC Musculoskelet. Disord. 2021, 22, 62. [Google Scholar] [CrossRef] [PubMed]
- Yokota, S.; Matsumae, G.; Shimizu, T.; Hasegawa, T.; Ebata, T.; Takahashi, D.; Heguo, C.; Tian, Y.; Alhasan, H.; Takahata, M.; et al. Cardiotrophin Like Cytokine Factor 1 (CLCF1) alleviates bone loss in osteoporosis mouse models by suppressing osteoclast differentiation through activating interferon signaling and repressing the nuclear factor-κB signaling pathway. Bone 2021, 153, 116140. [Google Scholar] [CrossRef]
- Boyce, B.F.; Xing, L. Biology of RANK, RANKL, and osteoprotegerin. Arthritis Res. Ther. 2007, 9, S1. [Google Scholar] [CrossRef]
- Nahlé, S.; Pasquin, S.; Laplante, V.; Rousseau, F.; Sharma, M.; Gauchat, J.-F. Cardiotrophin-like cytokine (CLCF1) modulates mesenchymal stem cell osteoblastic differentiation. J. Biol. Chem. 2019, 294, 11952–11959. [Google Scholar] [CrossRef]
- Stefanovic, L.; Stefanovic, B. Role of cytokine receptor-like factor 1 in hepatic stellate cells and fibrosis. World J. Hepatol. 2012, 4, 356–364. [Google Scholar] [CrossRef]
- Ealba, E.L.; Jheon, A.H.; Hall, J.; Curantz, C.; Butcher, K.D.; Schneider, R.A. Neural crest-mediated bone resorption is a determinant of species-specific jaw length. Dev. Biol. 2015, 408, 151–163. [Google Scholar] [CrossRef]
- Bataille, C.; Mauprivez, C.; Haÿ, E.; Baroukh, B.; Brun, A.; Chaussain, C.; Marie, P.J.; Saffar, J.-L.; Cherruau, M. Different sympathetic pathways control the metabolism of distinct bone envelopes. Bone 2012, 50, 1162–1172. [Google Scholar] [CrossRef]
- Krishnan, Y.; Grodzinsky, A.J. Cartilage diseases. Matrix Biol. 2018, 71–72, 51–69. [Google Scholar] [CrossRef] [PubMed]
- Molnar, V.; Matišić, V.; Kodvanj, I.; Bjelica, R.; Jeleč, Ž.; Hudetz, D.; Rod, E.; Čukelj, F.; Vrdoljak, T.; Vidović, D.; et al. Cytokines and Chemokines Involved in Osteoarthritis Pathogenesis. Int. J. Mol. Sci. 2021, 22, 9208. [Google Scholar] [CrossRef] [PubMed]
- Tsuritani, K.; Takeda, J.; Sakagami, J.; Ishii, A.; Eriksson, T.; Hara, T.; Ishibashi, H.; Koshihara, Y.; Yamada, K.; Yoneda, Y. Cytokine Receptor-Like Factor 1 is Highly Expressed in Damaged Human Knee Osteoarthritic Cartilage and Involved in Osteoarthritis Downstream of TGF-β. Calcif. Tissue Int. 2010, 86, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.S.; Kim, H.A. Chondrocyte Apoptosis in the Pathogenesis of Osteoarthritis. Int. J. MoLecular Sci. 2015, 16, 26035–26054. [Google Scholar] [CrossRef]
- Barreto, G.; Manninen, M.; Eklund, K.K. Osteoarthritis and Toll-Like Receptors: When Innate Immunity Meets Chondrocyte Apoptosis. Biology 2020, 9, 65. [Google Scholar] [CrossRef]
- Xu, H.; Ding, C.; Guo, C.; Xiang, S.; Wang, Y.; Luo, B.; Xiang, H. Suppression of CRLF1 promotes the chondrogenic differentiation of bone marrow-derived mesenchymal stem and protects cartilage tissue from damage in osteoarthritis via Activation of MiR-320. Mol. Med. 2021, 27, 116. [Google Scholar] [CrossRef] [PubMed]
- Hollander, W.D.; Pulyakhina, I.; Boer, C.; Bomer, N.; Van Der Breggen, R.; Arindrarto, W.; De Almeida, R.C.; Lakenberg, N.; Sentner, T.; Laros, J.F.J.; et al. Annotating Transcriptional Effects of Genetic Variants in Disease-Relevant Tissue: Transcriptome-Wide Allelic Imbalance in Osteoarthritic Cartilage. Arthritis Rheumatol. 2019, 71, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Ramos, Y.F.M.; Hollander, W.D.; Bovee, J.; Bomer, N.; Van Der Breggen, R.; Lakenberg, N.; Keurentjes, J.C.; Goeman, J.; Slagboom, P.; Nelissen, R.; et al. Genes Involved in the Osteoarthritis Process Identified through Genome Wide Expression Analysis in Articular Cartilage; the RAAK Study. PLoS ONE 2014, 9, e103056. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Yang, B.; Guo, H.; Kang, F. MicroRNAs regulate osteogenesis and chondrogenesis. Biochem. Biophys. Res. Commun. 2012, 418, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Laxman, N.; Mallmin, H.; Nilsson, O.; Kindmark, A. miR-203 and miR-320 Regulate Bone Morphogenetic Protein-2-Induced Osteoblast Differentiation by Targeting Distal-Less Homeobox 5 (Dlx5). Genes 2016, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Ao, X.; Li, P.; Lian, Z.; Jiang, T.; Zhang, Z.; Wang, L. CRLF1 Is a Key Regulator in the Ligamentum Flavum Hypertrophy. Front. Cell Dev. Biol. 2020, 8, 858. [Google Scholar] [CrossRef] [PubMed]
- Kass, D.J.; Yu, G.; Loh, K.S.; Savir, A.; Borczuk, A.; Kahloon, R.; Juan-Guardela, B.; Deiuliis, G.; Tedrow, J.; Choi, J.; et al. Cytokine-Like Factor 1 Gene Expression Is Enriched in Idiopathic Pulmonary Fibrosis and Drives the Accumulation of CD4+ T Cells in Murine Lungs: Evidence for an Antifibrotic Role in Bleomycin Injury. Am. J. Pathol. 2012, 180, 1963–1978. [Google Scholar] [CrossRef] [PubMed]
- Kreus, M.; Lehtonen, S.; Skarp, S.; Kaarteenaho, R. Extracellular matrix proteins produced by stromal cells in idiopathic pulmonary fibrosis and lung adenocarcinoma. PLoS ONE 2021, 16, e0250109. [Google Scholar] [CrossRef]
- Haemopedia. Available online: https://www.haemosphere.org (accessed on 22 December 2021).
- Choi, J.; Baldwin, T.M.; Wong, M.; Bolden, J.E.; Fairfax, K.A.; Lucas, E.C.; Cole, R.; Biben, C.; Morgan, C.; Ramsay, K.A.; et al. Haemopedia RNA-seq: A database of gene expression during haematopoiesis in mice and humans. Nucleic Acids Res. 2019, 47, D780–D785. [Google Scholar] [CrossRef]
- Belyavsky, A.; Petinati, N.; Drize, N. Hematopoiesis during Ontogenesis, Adult Life, and Aging. Int. J. Mol. Sci. 2021, 22, 9231. [Google Scholar] [CrossRef] [PubMed]
- Senaldi, G.; Stolina, M.; Guo, J.; Faggioni, R.; McCabe, S.; Kaufman, S.A.; Van, G.; Xu, W.; Fletcher, F.A.; Boone, T.; et al. Regulatory Effects of Novel Neurotrophin-1/B Cell-Stimulating Factor-3 (Cardiotrophin-Like Cytokine) on B Cell Function. J. Immunol. 2002, 168, 5690–5698. [Google Scholar] [CrossRef]
- Pasquin, S.; Tormo, A.; Moreau, J.; Laplante, V.; Sharma, M.; Gauchat, J.-F.; Rafei, M. Cardiotrophin-Like Cytokine Factor 1 Exhibits a Myeloid-Biased Hematopoietic-Stimulating Function. Front. Immunol. 2019, 10, 2133. [Google Scholar] [CrossRef]
- Pasquin, S.; Laplante, V.; Kouadri, S.; Milasan, A.; Mayer, G.; Tormo, A.J.; Savin, V.; Sharma, M.; Martel, C.; Gauchat, J.-F. Cardiotrophin-like Cytokine Increases Macrophage–Foam Cell Transition. J. Immunol. 2018, 201, 2462–2471. [Google Scholar] [CrossRef]
- Pflanz, S.; Timans, J.C.; Cheung, J.; Rosales, R.; Kanzler, H.; Gilbert, J.; Hibbert, L.; Churakova, T.; Travis, M.; Vaisberg, E.; et al. IL-27, a Heterodimeric Cytokine Composed of EBI3 and p28 Protein, Induces Proliferation of Naive CD4+ T Cells. Immunity 2002, 16, 779–790. [Google Scholar] [CrossRef]
- Hasle, H.; Niemeyer, C.M.; Chessells, J.M.; Baumann, I.; Bennett, J.M.; Kerndrup, G.; Head, D.R. A pediatric approach to the WHO classification of myelodysplastic and myeloproliferative diseases. Leukemia 2003, 17, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Calkoen, F.; Vervat, C.; van Pel, M.; de Haas, V.; Vijfhuizen, L.; Eising, E.; Kroes, W.; Hoen, P.T.; Heuvel-Eibrink, M.V.D.; Egeler, R.; et al. Despite differential gene expression profiles pediatric MDS derived mesenchymal stromal cells display functionality in vitro. Stem Cell Res. 2015, 14, 198–210. [Google Scholar] [CrossRef]
- Kim, J.W.; Marquez, C.; Kostyrko, K.; Koehne, A.L.; Marini, K.; Simpson, D.R.; Lee, A.G.; Leung, S.G.; Sayles, L.C.; Shrager, J.; et al. Antitumor activity of an engineered decoy receptor targeting CLCF1–CNTFR signaling in lung adenocarcinoma. Nat. Med. 2019, 25, 1783–1795. [Google Scholar] [CrossRef] [PubMed]
- Vicent, S.; Sayles, L.C.; Vaka, D.; Khatri, P.; Gevaert, O.; Chen, R.; Zheng, Y.; Gillespie, A.K.; Clarke, N.; Xu, Y.; et al. Cross-Species Functional Analysis of Cancer-Associated Fibroblasts Identifies a Critical Role for CLCF1 and IL-6 in Non–Small Cell Lung Cancer In Vivo. Cancer Res. 2012, 72, 5744–5756. [Google Scholar] [CrossRef]
- Kim, J.W.; Marquez, C.; Sperberg, R.A.P.; Wu, J.; Bae, W.G.; Huang, P.-S.; Sweet-Cordero, E.A.; Cochran, J.R. Engineering a potent receptor superagonist or antagonist from a novel IL-6 family cytokine ligand. Proc. Natl. Acad. Sci. USA 2020, 117, 14110–14118. [Google Scholar] [CrossRef]
- Song, M.; He, J.; Pan, Q.; Yang, J.; Zhao, J.; Zhang, Y.; Huang, Y.; Tang, Y.; Wang, Q.; He, J.; et al. Cancer-Associated Fibroblast-Mediated Cellular Crosstalk Supports Hepatocellular Carcinoma Progression. Hepatology 2021, 73, 1717–1735. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.-T.; Zhong, Q.; Chen, R.-H.; Han, P.; Li, S.-B.; Zhang, H.; Yuan, L.; Xia, T.-L.; Zeng, M.-S.; Huang, X.-M. CRLF1 promotes malignant phenotypes of papillary thyroid carcinoma by activating the MAPK/ERK and PI3K/AKT pathways. Cell Death Dis. 2018, 9, 371. [Google Scholar] [CrossRef]
- Li, Y.; Xun, J.; Wang, B.; Ma, Y.; Zhang, L.; Yang, L.; Gao, R.; Guan, J.; Liu, T.; Gao, H.; et al. miR-3065-3p promotes stemness and metastasis by targeting CRLF1 in colorectal cancer. J. Transl. Med. 2021, 19, 429. [Google Scholar] [CrossRef]
- Jones, S.A.; Jenkins, B.J. Recent insights into targeting the IL-6 cytokine family in inflammatory diseases and cancer. Nat. Rev. Immunol. 2018, 18, 773–789. [Google Scholar] [CrossRef]
- Xin, P.; Xu, X.; Deng, C.; Liu, S.; Wang, Y.; Zhou, X.; Ma, H.; Wei, D.; Sun, S. The role of JAK/STAT signaling pathway and its inhibitors in diseases. Int. Immunopharmacol. 2020, 80, 106210. [Google Scholar] [CrossRef] [PubMed]
- Cutler, A.; Brombacher, F. Cytokine Therapy. Ann. N. Y. Acad. Sci. 2005, 1056, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Baldo, B.A. Side Effects of Cytokines Approved for Therapy. Drug Saf. 2014, 37, 921–943. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crisponi, L.; Buers, I.; Rutsch, F. CRLF1 and CLCF1 in Development, Health and Disease. Int. J. Mol. Sci. 2022, 23, 992. https://doi.org/10.3390/ijms23020992
Crisponi L, Buers I, Rutsch F. CRLF1 and CLCF1 in Development, Health and Disease. International Journal of Molecular Sciences. 2022; 23(2):992. https://doi.org/10.3390/ijms23020992
Chicago/Turabian StyleCrisponi, Laura, Insa Buers, and Frank Rutsch. 2022. "CRLF1 and CLCF1 in Development, Health and Disease" International Journal of Molecular Sciences 23, no. 2: 992. https://doi.org/10.3390/ijms23020992
APA StyleCrisponi, L., Buers, I., & Rutsch, F. (2022). CRLF1 and CLCF1 in Development, Health and Disease. International Journal of Molecular Sciences, 23(2), 992. https://doi.org/10.3390/ijms23020992