
Heat Shock Proteins in Benign Prostatic Hyperplasia and Prostate Cancer



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. HSPs and Cancer Cells
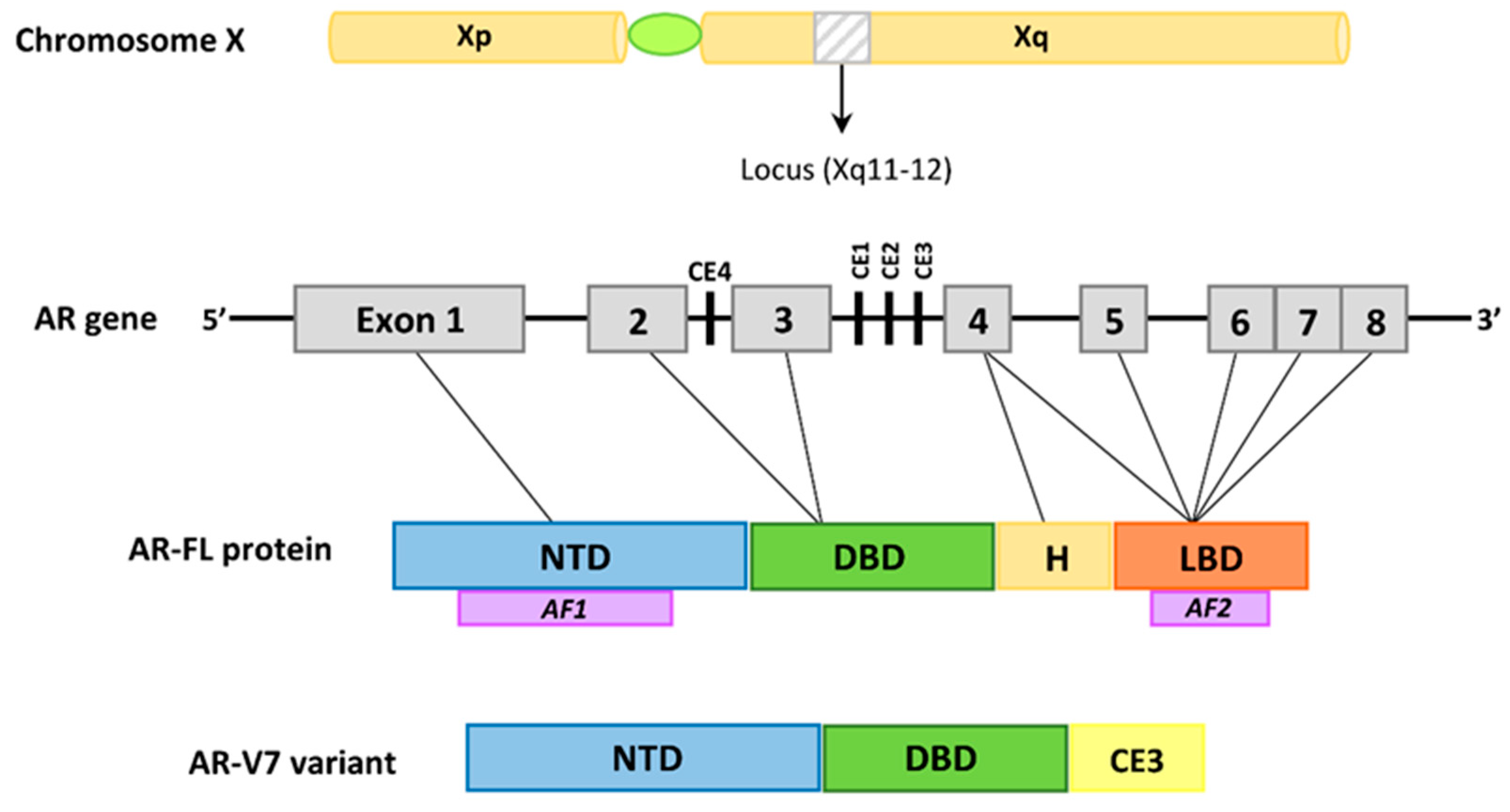
3. The Androgen Receptor in the Development of Prostate Cancer and Benign Prostatic Hyperplasia
4. The Characteristics of HSP90
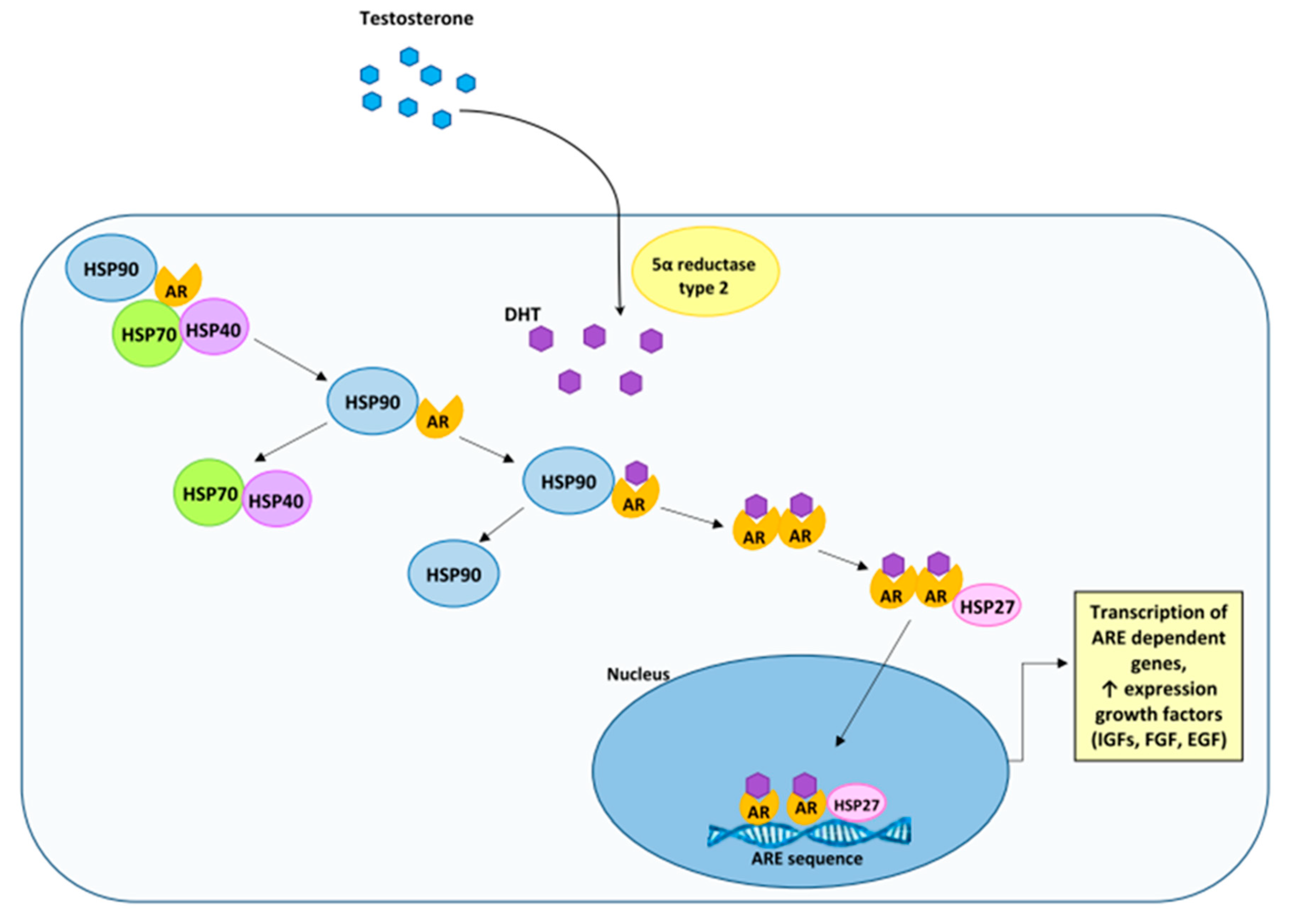
5. HSP90 and Prostate Cancer, and Benign Prostatic Hyperplasia
6. The Characteristics of HSP70 and HSP40
6.1. HSP70
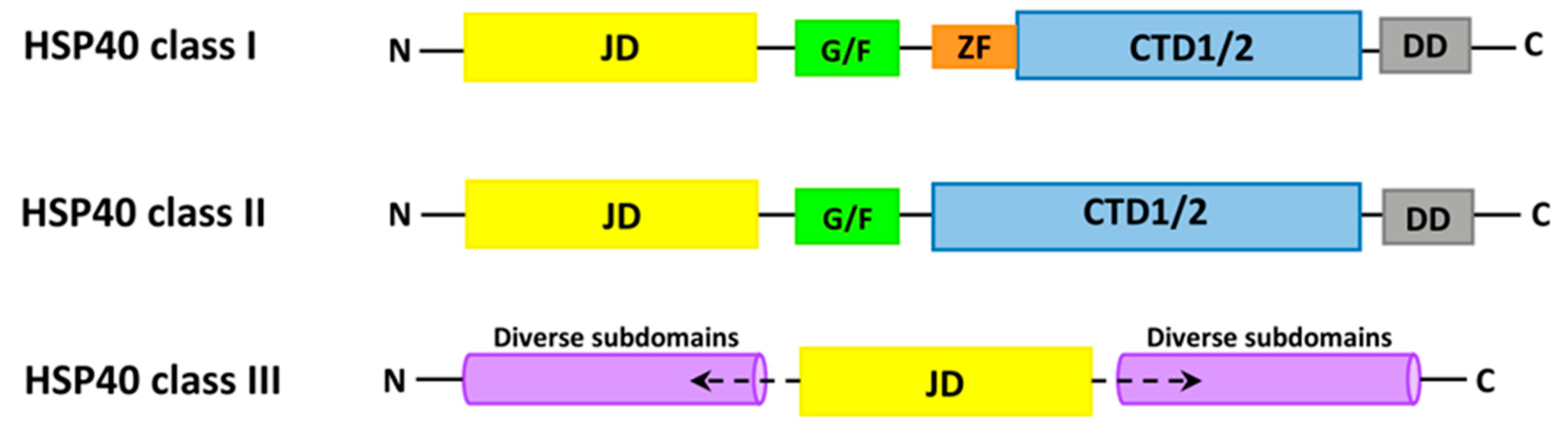
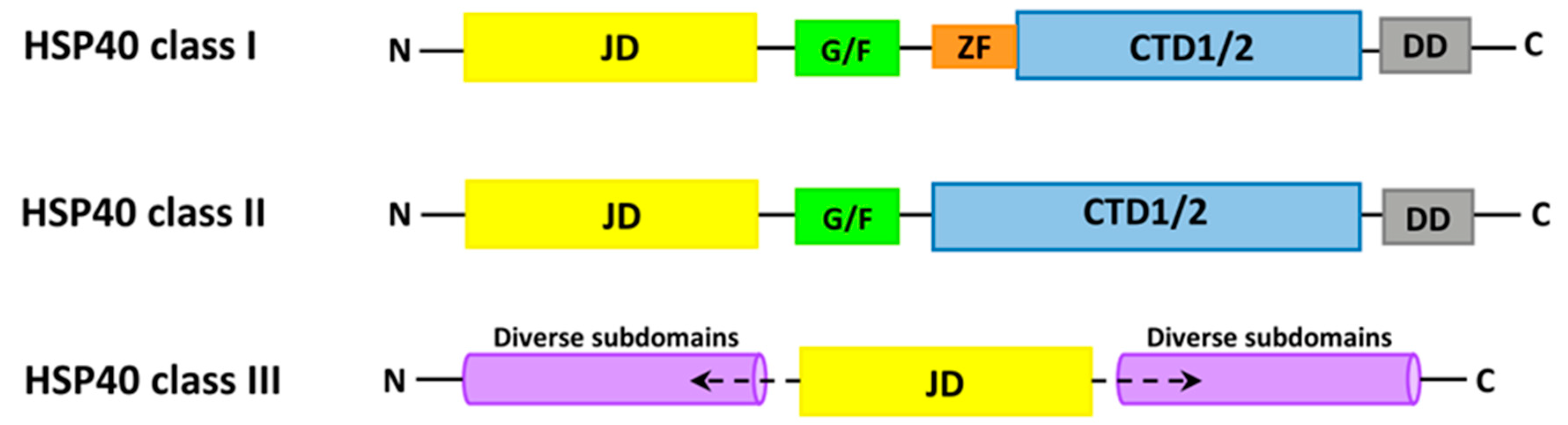
6.2. HSP40
7. HSP70 and HSP40 and Prostate Cancer
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dubrez, L.; Causse, S.; Borges Bonan, N.; Dumétier, B.; Garrido, C. Heat-shock proteins: Chaperoning DNA repair. Oncogene 2020, 39, 516–529. [Google Scholar] [CrossRef] [PubMed]
- Milani, A.; Basirnejad, M.; Bolhassani, A. Heat-shock proteins in diagnosis and treatment: An overview of different biochemical and immunological functions. Immunotherapy 2019, 11, 215–239. [Google Scholar] [CrossRef]
- Ahmad, S.; Kabir, M.; Hayat, M. Identification of heat shock protein families and J-protein types by incorporating dipeptide composition into Chou’s general PseAAC. Comput. Methods Programs Biomed. 2015, 122, 165–174. [Google Scholar] [CrossRef]
- Chatterjee, S.; Burns, T.F. Targeting Heat Shock Proteins in Cancer: A Promising Therapeutic Approach. Int. J. Mol. Sci. 2017, 18, 1978. [Google Scholar] [CrossRef] [Green Version]
- Kampinga, H.; Hageman, J.; Vos, M.; Kubota, H.; Tanguay, R.; Bruford, E.; Cheetham, M.; Chen, B.; Hightower, L. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 2009, 14, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Åkerfelt, M.; Morimoto, R.I.; Sistonen, L. Heat shock factors: Integrators of cell stress, development and lifespan. Nat. Rev. Mol. Cell Biol. 2010, 11, 545. [Google Scholar] [CrossRef]
- Gomez-Pastor, R.; Burchfiel, E.T.; Thiele, D.J. Regulation of heat shock transcription factors and their roles in physiology and disease. Nat. Rev. Mol. Cell Biol. 2017, 19, 4–19. [Google Scholar] [CrossRef]
- Zhang, B.; Fan, Y.; Cao, P.; Tan, K. Multifaceted roles of HSF1 in cell death: A state-of-the-art review. Biochim Biophys Acta—Rev Cancer 2021, 1876, 188591. [Google Scholar] [CrossRef] [PubMed]
- Alasady, M.; Mendillo, M. The Multifaceted Role of HSF1 in Tumorigenesis. Adv. Exp. Med. Biol. 2020, 1243, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Tu, K.; Fu, Q.; Schmitt, D.C.; Zhou, L.; Lu, N.; Zhao, Y. Multifaceted roles of HSF1 in cancer. Tumor. Biol. 2015, 36, 4923–4931. [Google Scholar] [CrossRef]
- Parcellier, A.; Gurbuxani, S.; Schmitt, E.; Solary, E.; Garrido, C. Heat shock proteins, cellular chaperones that modulate mitochondrial cell death pathways. Biochem. Biophys. Res. Commun. 2003, 304, 505–512. [Google Scholar] [CrossRef]
- Stankiewicz, A.R.; Livingstone, A.M.; Mohseni, N.; Mosser, D.D. Regulation of heat-induced apoptosis by Mcl-1 degradation and its inhibition by Hsp70. Cell Death Differ. 2009, 16, 638–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, M.; Jain, S.; Stühmer, T.; Andrulis, M.; Ungethüm, U.; Kuban, R.J.; Lorentz, H.; Bommert, K.; Topp, M.; Krämer, D.; et al. STAT3 and MAPK signaling maintain overexpression of heat shock proteins 90alpha and beta in multiple myeloma cells, which critically contribute to tumor-cell survival. Blood 2007, 109, 720–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.S.; Lee, J.J.; Seo, J.S. HSP70 deficiency results in activation of c-Jun N-terminal Kinase, extracellular signal-regulated kinase, and caspase-3 in hyperosmolarity-induced apoptosis. J. Biol. Chem. 2005, 280, 6634–6641. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Wang, E. Heat shock protein 90 suppresses tumor necrosis factor alpha induced apoptosis by preventing the cleavage of Bid in NIH3T3 fibroblasts. Cell Signal 2004, 16, 313–321. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Attard, G.; Murphy, L.; Clarke, N.W.; Cross, W.; Jones, R.J.; Parker, C.C.; Gillessen, S.; Cook, A.; Brawley, C.; Amos, C.L.; et al. Abiraterone acetate and prednisolone with or without enzalutamide for high-risk non-metastatic prostate cancer: A meta-analysis of primary results from two randomised controlled phase 3 trials of the STAMPEDE platform protocol. Lancet 2021. [Google Scholar] [CrossRef]
- Ghose, A.; Moschetta, M.; Pappas-Gogos, G.; Sheriff, M.; Boussios, S. Genetic Aberrations of DNA Repair Pathways in Prostate Cancer: Translation to the Clinic. Int. J. Mol. Sci. 2021, 22, 9783. [Google Scholar] [CrossRef] [PubMed]
- Nevedomskaya, E.; Baumgart, S.J.; Haendler, B. Recent Advances in Prostate Cancer Treatment and Drug Discovery. Int. J. Mol. Sci. 2018, 19, 1359. [Google Scholar] [CrossRef] [Green Version]
- Hoter, A.; Rizk, S.; Naim, H.Y. The Multiple Roles and Therapeutic Potential of Molecular Chaperones in Prostate Cancer. Cancers 2019, 11, 1194. [Google Scholar] [CrossRef] [Green Version]
- Launer, B.M.; McVary, K.T.; Ricke, W.A.; Lloyd, G.L. The rising worldwide impact of benign prostatic hyperplasia. BJU Int. 2021, 127, 722–728. [Google Scholar] [CrossRef]
- Lokeshwar, S.D.; Harper, B.T.; Webb, E.; Jordan, A.; Dykes, T.A.; Neal, D.E.; Terris, M.K.; Klaassen, Z. Epidemiology and treatment modalities for the management of benign prostatic hyperplasia. Transl. Androl. Urol. 2019, 8, 529. [Google Scholar] [CrossRef]
- Bortnick, E.; Brown, C.; Simma-Chiang, V.; Kaplan, S.A. Modern best practice in the management of benign prostatichyperplasia in the elderly. Ther. Adv. Urol. 2020, 12, 1756287220929486. [Google Scholar] [CrossRef] [PubMed]
- Ortmayr, K.; Dubuis, S.; Zampieri, M. Metabolic profiling of cancer cells reveals genome-wide crosstalk between transcriptional regulators and metabolism. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Griffin, J.L.; Shockcor, J.P. Metabolic profiles of cancer cells. Nat. Rev. Cancer 2004, 4, 551–561. [Google Scholar] [CrossRef]
- Slotta-Huspenina, J.; Becker, K.-F.; Feith, M.; Walch, A.; Langer, R. Heat Shock Protein 90 (HSP90) and Her2 in Adenocarcinomas of the Esophagus. Cancers 2014, 6, 1382–1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoppino, F.C.M.; Guerrero-Gimenez, M.E.; Castro, G.N.; Ciocca, D.R. Comprehensive transcriptomic analysis of heat shock proteins in the molecular subtypes of human breast cancer. BMC Cancer 2018, 18, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Bodzek, P.; Damasiewicz-Bodzek, A.; Janosz, I.; Witek, L.; Olejek, A. Heat shock protein 27 (hsp27) in patients with ovarian cancer. Ginekol. Pol. 2021, 92, 837–843. [Google Scholar] [CrossRef]
- Wyciszkiewicz, A.; Kalinowska-Łyszczarz, A.; Nowakowski, B.; Kaźmierczak, K.; Osztynowicz, K.; Michalak, S. Expression of small heat shock proteins in exosomes from patients with gynecologic cancers. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Ge, H.; Yan, Y.; Guo, L.; Tian, F.; Wu, D. Prognostic role of HSPs in human gastrointestinal cancer: A systematic review and meta-analysis. Onco. Targets Ther. 2018, 11, 351–359. [Google Scholar] [CrossRef] [Green Version]
- Mittal, S.; Rajala, M.S. Heat shock proteins as biomarkers of lung cancer. Cancer Biol. Ther. 2020, 21, 477–485. [Google Scholar] [CrossRef]
- Ciocca, D.R.; Fanelli, M.A.; Cuello-Carrion, F.D.; Castro, G.N. Heat shock proteins in prostate cancer: From tumorigenesis to the clinic. Int. J. Hyperth. 2010, 26, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Sliva, D. Signaling pathways responsible for cancer cell invasion as targets for cancer therapy. Curr. Cancer Drug Targets 2004, 4, 327–336. [Google Scholar] [CrossRef]
- Wu, W.K.K.; Sakamoto, K.M.; Milani, M.; Aldana-Masankgay, G.; Fan, D.; Wu, K.; Lee, C.W.; Cho, C.H.; Yu, J.; Sung, J.J.Y. Macroautophagy modulates cellular response to proteasome inhibitors in cancer therapy. Drug Resist. Updat. 2010, 13, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Mitsiades, N.; Mitsiades, C.S.; Poulaki, V.; Chauhan, D.; Fanourakis, G.; Gu, X.; Bailey, C.; Joseph, M.; Libermann, T.A.; Treon, S.P.; et al. Molecular sequelae of proteasome inhibition in human multiple myeloma cells. Proc. Natl. Acad. Sci. USA 2002, 99, 14374–14379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, W.; Bohl, C.E.; Dalton, J.T. Chemistry and Structural Biology of Androgen Receptor. Chem. Rev. 2005, 105, 3352. [Google Scholar] [CrossRef] [Green Version]
- Rył, A.; Rotter, I.; Grzywacz, A.; Małecka, I.; Skonieczna-Żydecka, K.; Grzesiak, K.; Słojewski, M.; Szylińska, A.; Sipak-Szmigiel, O.; Piasecka, M.; et al. Molecular Analysis of the SRD5A1 and SRD5A2 Genes in Patients with Benign Prostatic Hyperplasia with Regard to Metabolic Parameters and Selected Hormone Levels. Int. J. Environ. Res. Public Health 2017, 14, 1318. [Google Scholar] [CrossRef] [Green Version]
- Tan, M.E.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E. Androgen receptor: Structure, role in prostate cancer and drug discovery. Acta Pharmacol. Sin. 2014, 36, 3–23. [Google Scholar] [CrossRef] [Green Version]
- Heinlein, C.A.; Chang, C. Androgen Receptor in Prostate Cancer. Endocr. Rev. 2004, 25, 276–308. [Google Scholar] [CrossRef] [Green Version]
- Pratt, W.; Toft, D. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr. Rev. 1997, 18, 306–360. [Google Scholar] [CrossRef]
- Kazutoshi, F.; Norio, N. Role of Androgen Receptor in Prostate Cancer: A Review. World J. Mens. Health 2019, 37, 288. [Google Scholar] [CrossRef]
- Beilin, J.; Ball, E.; Favaloro, J.; Zajac, J. Effect of the androgen receptor CAG repeat polymorphism on transcriptional activity: Specificity in prostate and non-prostate cell lines. J. Mol. Endocrinol. 2000, 25, 85–96. [Google Scholar] [CrossRef] [Green Version]
- Wärnmark, A.; Treuter, E.; Wright, A.P.H.; Gustafsson, J.-A. Activation Functions 1 and 2 of Nuclear Receptors: Molecular Strategies for Transcriptional Activation. Mol. Endocrinol. 2003, 17, 1901–1909. [Google Scholar] [CrossRef] [PubMed]
- Davey, R.A.; Grossmann, M. Androgen Receptor Structure, Function and Biology: From Bench to Bedside. Clin. Biochem. Rev. 2016, 37, 3. [Google Scholar] [PubMed]
- Isaacs, J.T. Resolving the Coffey Paradox: What does the androgen receptor do in normal vs. malignant prostate epithelial cells? Am. J. Clin. Exp. Urol. 2018, 6, 55. [Google Scholar] [PubMed]
- Cunha, G.; Foster, B.; Thomson, A.; Sugimura, Y.; Tanji, N.; Tsuji, M.; Terada, N.; Finch, P.; Donjacour, A. Growth factors as mediators of androgen action during the development of the male urogenital tract. World J. Urol. 1995, 13, 264–276. [Google Scholar] [CrossRef]
- Chughtai, B.; Forde, J.C.; Thomas, D.D.M.; Laor, L.; Hossack, T.; Woo, H.H.; Te, A.E.; Kaplan, S.A. Benign prostatic hyperplasia. Nat. Rev. Dis. Prim. 2016, 2, 16031. [Google Scholar] [CrossRef] [Green Version]
- Vickman, R.E.; Franco, O.E.; Moline, D.C.; Vander Griend, D.J.; Thumbikat, P.; Hayward, S.W. The role of the androgen receptor in prostate development and benign prostatic hyperplasia: A review. Asian J. Urol. 2020, 7, 191–202. [Google Scholar] [CrossRef]
- Vander Griend, D.J.; D’Antonio, J.; Gurel, B.; Antony, L.; DeMarzo, A.M.; Isaacs, J.T. Cell-autonomous intracellular androgen receptor signaling drives the growth of human prostate cancer initiating cells. Prostate 2010, 70, 90–99. [Google Scholar] [CrossRef] [Green Version]
- El-Alfy, M.; Luu-The, V.; Huang, X.; Berger, L.; Labrie, F.; Pelletier, G. Localization of type 5 17beta-hydroxysteroid dehydrogenase, 3beta-hydroxysteroid dehydrogenase, and androgen receptor in the human prostate by in situ hybridization and immunocytochemistry. Endocrinology 1999, 140, 1481–1491. [Google Scholar] [CrossRef]
- Qiu, Y.; Leuschner, I.; Braun, P. Androgen receptor expression in clinically localized prostate cancer: Immunohistochemistry study and literature review. Asian J. Androl. 2008, 10, 855–863. [Google Scholar] [CrossRef]
- Huggins, C.; Hodges, C. Studies on prostatic cancer. I. The effect of castration, of estrogen and androgen injection on serum phosphatases in metastatic carcinoma of the prostate. CA Cancer J. Clin. 1972, 22, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Cornford, P.; Van Den Bergh, R.C.N.; Briers, E.; Van Den Broeck, T.; Cumberbatch, M.G.; De Santis, M.; Fanti, S.; Fossati, N.; Gandaglia, G.; Gillessen, S.; et al. EAU-EANM-ESTRO-ESUR-SIOG Guidelines on Prostate Cancer. Part II-2020 Update: Treatment of Relapsing and Metastatic Prostate Cancer. Eur. Urol. 2021, 79, 263–282. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, A.; Bastian, P.; Bellmunt, J.; Bolla, M.; Joniau, S.; van der Kwast, T.; Mason, M.; Matveev, V.; Wiegel, T.; Zattoni, F.; et al. EAU guidelines on prostate cancer. Part II: Treatment of advanced, relapsing, and castration-resistant prostate cancer. Eur. Urol. 2014, 65, 467–479. [Google Scholar] [CrossRef]
- Feng, Q.; He, B. Androgen Receptor Signaling in the Development of Castration-Resistant Prostate Cancer. Front. Oncol. 2019, 9, 858. [Google Scholar] [CrossRef] [Green Version]
- Lai, K.; Yamashita, S.; Huang, C.; Yeh, S.; Chang, C. Loss of stromal androgen receptor leads to suppressed prostate tumourigenesis via modulation of pro-inflammatory cytokines/chemokines. EMBO Mol. Med. 2012, 4, 791–807. [Google Scholar] [CrossRef]
- Visakorpi, T.; Hyytinen, E.; Koivisto, P.; Tanner, M.; Keinänen, R.; Palmberg, C.; Palotie, A.; Tammela, T.; Isola, J.; Kallioniemi, O. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat. Genet. 1995, 9, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Azad, A.; Volik, S.; Wyatt, A.; Haegert, A.; Le Bihan, S.; Bell, R.; Anderson, S.; McConeghy, B.; Shukin, R.; Bazov, J.; et al. Androgen Receptor Gene Aberrations in Circulating Cell-Free DNA: Biomarkers of Therapeutic Resistance in Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2015, 21, 2315–2324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, R.; Dunn, T.; Wei, S.; Isharwal, S.; Veltri, R.; Humphreys, E.; Han, M.; Partin, A.; Vessella, R.; Isaacs, W.; et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009, 69, 16–22. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Chan, S.; Brand, L.; Hwang, T.; Silverstein, K.; Dehm, S. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 2013, 73, 483–489. [Google Scholar] [CrossRef] [Green Version]
- Sharp, A.; Coleman, I.; Yuan, W.; Sprenger, C.; Dolling, D.; Rodrigues, D.N.; Russo, J.W.; Figueiredo, I.; Bertan, C.; Seed, G.; et al. Androgen receptor splice variant-7 expression emerges with castration resistance in prostate cancer. J. Clin. Invest 2019, 129, 192–208. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Dalrymple, S.L.; Coleman, I.; Zheng, S.L.; Xu, J.; Hooper, J.E.; Antonarakis, E.S.; De Marzo, A.M.; Meeker, A.K.; Nelson, P.S.; et al. Role of androgen receptor splice variant-7 (AR-V7) in prostate cancer resistance to 2nd-generation androgen receptor signaling inhibitors. Oncogene 2020, 39, 6935–6949. [Google Scholar] [CrossRef]
- Saxby, H.; Mikropoulos, C.; Boussios, S. An Update on the Prognostic and Predictive Serum Biomarkers in Metastatic Prostate Cancer. Diagnostics 2020, 10, 549. [Google Scholar] [CrossRef]
- Zhang, T.; Karsh, L.I.; Nissenblatt, M.J.; Canfield, S.E. Androgen Receptor Splice Variant, AR-V7, as a Biomarker of Resistance to Androgen Axis-Targeted Therapies in Advanced Prostate Cancer. Clin. Genitourin. Cancer 2020, 18, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryce, A.; Antonarakis, E. Androgen receptor splice variant 7 in castration-resistant prostate cancer: Clinical considerations. Int. J. Urol. 2016, 23, 646–653. [Google Scholar] [CrossRef]
- Sreedhar, A.; Kalmár, E.; Csermely, P.; Shen, Y. Hsp90 isoforms: Functions, expression and clinical importance. FEBS Lett. 2004, 562, 11–15. [Google Scholar] [CrossRef]
- Prodromou, C.; Roe, S.M.; O’Brien, R.; Ladbury, J.E.; Piper, P.W.; Pearl, L.H. Identification and Structural Characterization of the ATP/ADP-Binding Site in the Hsp90 Molecular Chaperone. Cell 1997, 90, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Meyer, P.; Prodromou, C.; Hu, B.; Vaughan, C.; Roe, S.M.; Panaretou, B.; Piper, P.W.; Pearl, L.H. Structural and Functional Analysis of the Middle Segment of Hsp90: Implications for ATP Hydrolysis and Client Protein and Cochaperone Interactions. Mol. Cell 2003, 11, 647–658. [Google Scholar] [CrossRef] [Green Version]
- Harris, S.F.; Shiau, A.K.; Agard, D.A. The Crystal Structure of the Carboxy-Terminal Dimerization Domain of htpG, the Escherichia coli Hsp90, Reveals a Potential Substrate Binding Site. Structure 2004, 12, 1087–1097. [Google Scholar] [CrossRef] [Green Version]
- Marzec, M.; Eletto, D.; Argon, Y. GRP94: An HSP90-like protein specialized for protein folding and quality control in the Endoplasmic Reticulum. Biochim. Biophys. Acta 2012, 1823, 774. [Google Scholar] [CrossRef] [Green Version]
- Dutta, R.; Inouye, M. GHKL, an emergent ATPase/kinase superfamily. Trends Biochem. Sci. 2000, 25, 24–28. [Google Scholar] [CrossRef]
- Jahn, M.; Rehn, A.; Pelz, B.; Hellenkamp, B.; Richter, K.; Rief, M.; Buchner, J.; Hugel, T. The charged linker of the molecular chaperone Hsp90 modulates domain contacts and biological function. Proc. Natl. Acad. Sci. USA 2014, 111, 17881–17886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoter, A.; El-Sabban, M.E.; Naim, H.Y. The HSP90 Family: Structure, Regulation, Function, and Implications in Health and Disease. Int. J. Mol. Sci. 2018, 19, 2560. [Google Scholar] [CrossRef] [Green Version]
- Minami, Y.; Kawasaki, H.; Suzuki, K.; Yahara, I. The calmodulin-binding domain of the mouse 90-kDa heat shock protein. J. Biol. Chem. 1993, 268, 9604–9610. [Google Scholar] [CrossRef]
- Soti, C.; Vermes, A.; Haystead, T.; Csermely, P. Comparative analysis of the ATP-binding sites of Hsp90 by nucleotide affinity cleavage: A distinct nucleotide specificity of the C-terminal ATP-binding site. Eur. J. Biochem. 2003, 270, 2421–2428. [Google Scholar] [CrossRef] [Green Version]
- Biebl, M.M.; Buchner, J. Structure, Function, and Regulation of the Hsp90 Machinery. Cold Spring Harb. Perspect. Biol. 2019, 11, a034017. [Google Scholar] [CrossRef] [Green Version]
- Voellmy, R.; Boellmann, F. Chaperone Regulation of the Heat Shock Protein Response. Adv. Exp. Med. Biol. 2007, 594, 89–99. [Google Scholar] [CrossRef]
- Dong, B.; Jaeger, A.M.; Hughes, P.F.; Loiselle, D.R.; Spencer Hauck, J.; Fu, Y.; Haystead, T.A.; Huang, J.; Thiele, D.J. Targeting therapy-resistant prostate cancer via a direct inhibitor of the human heat shock transcription factor 1. Sci. Transl. Med. 2020, 12, eabb5647. [Google Scholar] [CrossRef]
- Backe, S.J.; Sager, R.A.; Woodford, M.R.; Makedon, A.M.; Mollapour, M. Post-translational modifications of Hsp90 and translating the chaperone code. J. Biol. Chem. 2020, 295, 11099–11117. [Google Scholar] [CrossRef]
- Prodromou, C. Mechanisms of Hsp90 regulation. Biochem. J. 2016, 473, 2439. [Google Scholar] [CrossRef] [Green Version]
- Haslbeck, V.; Eckl, J.M.; Drazic, A.; Rutz, D.A.; Lorenz, O.R.; Zimmermann, K.; Kriehuber, T.; Lindemann, C.; Madl, T.; Richter, K. The activity of protein phosphatase 5 towards native clients is modulated by the middle- and C-terminal domains of Hsp90. Sci. Reports 2015, 5, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Wandinger, S.; Suhre, M.; Wegele, H.; Buchner, J. The phosphatase Ppt1 is a dedicated regulator of the molecular chaperone Hsp90. EMBO J. 2006, 25, 367–376. [Google Scholar] [CrossRef] [Green Version]
- Wolmarans, A.; Kwantes, A.; LaPointe, P. A novel method for site-specific chemical SUMOylation: SUMOylation of Hsp90 modulates co-chaperone binding in vitro. Biol. Chem. 2019, 400, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sun, M.; Myasnikov, A.G.; Elnatan, D.; Delaeter, N.; Nguyenquang, M.; Agard, D.A. Cryo-EM structures reveal a multistep mechanism of Hsp90 activation by co-chaperone Aha1. bioRxiv 2020. [Google Scholar] [CrossRef]
- Synoradzki, K.; Bieganowski, P. Middle domain of human Hsp90 isoforms differentially binds Aha1 in human cells and alters Hsp90 activity in yeast. Biochim. Biophys. Acta-Mol. Cell Res. 2015, 1853, 445–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mollapour, M.; Bourboulia, D.; Beebe, K.; Woodford, M.R.; Polier, S.; Hoang, A.; Chelluri, R.; Li, Y.; Guo, A.; Lee, M.-J.; et al. Asymmetric Hsp90 N-domain SUMOylation recruits Aha1 and ATP-competitive inhibitors. Mol. Cell 2014, 53, 317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef]
- Bhattacharya, K.; Weidenauer, L.; Luengo, T.M.; Pieters, E.C.; Echeverría, P.C.; Bernasconi, L.; Wider, D.; Sadian, Y.; Koopman, M.B.; Villemin, M.; et al. The Hsp70-Hsp90 co-chaperone Hop/Stip1 shifts the proteostatic balance from folding towards degradation. Nat. Commun. 2020, 11, 1–21. [Google Scholar] [CrossRef]
- Baindur-Hudson, S.; Edkins, A.; Blatch, G. Hsp70/Hsp90 organising protein (hop): Beyond interactions with chaperones and prion proteins. Subcell. Biochem. 2015, 78, 69–90. [Google Scholar] [CrossRef]
- Lott, A.; Oroz, J.; Zweckstetter, M. Molecular basis of the interaction of Hsp90 with its co-chaperone Hop. Protein. Sci. 2020, 29, 2422–2432. [Google Scholar] [CrossRef]
- Cliff, M.J.; Harris, R.; Barford, D.; Ladbury, J.E.; Williams, M.A. Conformational Diversity in the TPR Domain-Mediated Interaction of Protein Phosphatase 5 with Hsp90. Structure 2006, 14, 415–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zgajnar, N.R.; De Leo, S.A.; Lotufo, C.M.; Erlejman, A.G.; Piwien-Pilipuk, G.; Galigniana, M.D. Biological Actions of the Hsp90-binding Immunophilins FKBP51 and FKBP52. Biomolecules 2019, 9, 52. [Google Scholar] [CrossRef] [Green Version]
- Roe, S.; Ali, M.; Meyer, P.; Vaughan, C.; Panaretou, B.; Piper, P.; Prodromou, C.; Pearl, L. The Mechanism of Hsp90 regulation by the protein kinase-specific cochaperone p50(cdc37). Cell 2004, 116, 87–98. [Google Scholar] [CrossRef] [Green Version]
- Biebl, M.M.; Lopez, A.; Rehn, A.; Freiburger, L.; Lawatscheck, J.; Blank, B.; Sattler, M.; Buchner, J. Structural elements in the flexible tail of the co-chaperone p23 coordinate client binding and progression of the Hsp90 chaperone cycle. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.M.U.; Roe, S.M.; Vaughan, C.K.; Meyer, P.; Panaretou, B.; Piper, P.W.; Prodromou, C.; Pearl, L.H. Crystal structure of an Hsp90–nucleotide–p23/Sba1 closed chaperone complex. Nature 2006, 440, 1013–1017. [Google Scholar] [CrossRef] [Green Version]
- Wolmarans, A.; Lee, B.; Spyracopoulos, L.; LaPointe, P. The Mechanism of Hsp90 ATPase Stimulation by Aha1. Sci. Reports 2016, 6, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Paul, A.; Garcia, Y.A.; Zierer, B.; Patwardhan, C.; Gutierrez, O.; Hildenbrand, Z.; Harris, D.C.; Balsiger, H.A.; Sivils, J.C.; Johnson, J.L.; et al. The Cochaperone SGTA (Small Glutamine-rich Tetratricopeptide Repeat-containing Protein Alpha) Demonstrates Regulatory Specificity for the Androgen, Glucocorticoid, and Progesterone Receptors. J. Biol. Chem. 2014, 289, 15297. [Google Scholar] [CrossRef] [Green Version]
- Buchanan, G.; Ricciardelli, C.; Harris, J.; Prescott, J.; Yu, Z.; Jia, L.; Butler, L.; Marshall, V.; Scher, H.; Gerald, W.; et al. Control of androgen receptor signaling in prostate cancer by the cochaperone small glutamine rich tetratricopeptide repeat containing protein alpha. Cancer Res. 2007, 67, 10087–10096. [Google Scholar] [CrossRef] [Green Version]
- Hong, D.; Banerji, U.; Tavana, B.; George, G.; Aaron, J.; Kurzrock, R. Targeting the molecular chaperone heat shock protein 90 (HSP90): Lessons learned and future directions. Cancer Treat Rev. 2013, 39, 375–387. [Google Scholar] [CrossRef]
- Workman, P.; Burrows, F.; Neckers, L.; Rosen, N. Drugging the cancer chaperone HSP90: Combinatorial therapeutic exploitation of oncogene addiction and tumor stress. Ann. N. Y. Acad. Sci. 2007, 1113, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Jafari, A.; Rezaei-Tavirani, M.; Farhadihosseinabadi, B.; Taranejoo, S.; Zali, H. HSP90 and Co-chaperones: Impact on Tumor Progression and Prospects for Molecular-Targeted Cancer Therapy. Cancer Invest. 2020, 38, 310–328. [Google Scholar] [CrossRef] [PubMed]
- Trepel, J.; Mollapour, M.; Giaccone, G.; Neckers, L. Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 2010, 10, 537–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitesell, L.; Santagata, S.; Mendillo, M.L.; Lin, N.U.; Proia, D.A.; Lindquist, S. HSP90 empowers evolution of resistance to hormonal therapy in human breast cancer models. Proc. Natl. Acad. Sci. USA 2014, 111, 18297–18302. [Google Scholar] [CrossRef] [Green Version]
- Park, H.-K.; Yoon, N.G.; Lee, J.-E.; Hu, S.; Yoon, S.; Kim, S.Y.; Hong, J.-H.; Nam, D.; Chae, Y.C.; Park, J.B.; et al. Unleashing the full potential of Hsp90 inhibitors as cancer therapeutics through simultaneous inactivation of Hsp90, Grp94, and TRAP1. Exp. Mol. Med. 2020, 52, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Bakir, W.; Gaidan, H.; Al-kaabi, M. Immunohistochemical expression of interlukin10 (IL10) and heat shock protein-90 (HSP-90) in prostatic carcinoma. Indian J. Pathol. Microbiol. 2020, 63, 230. [Google Scholar] [CrossRef]
- Jansson, K.H.; Tucker, J.B.; Stahl, L.E.; Simmons, J.K.; Fuller, C.; Beshiri, M.L.; Agarwal, S.; Fang, L.; Hynes, P.G.; Alilin, A.N.; et al. High-throughput screens identify HSP90 inhibitors as potent therapeutics that target inter-related growth and survival pathways in advanced prostate cancer. Sci. Reports 2018, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Wang, X.; Gan, S.; Tang, X.; Kang, X.; Zhu, S. The Mitochondrial Chaperone TRAP1 as a Candidate Target of Oncotherapy. Front. Oncol. 2021, 10, 585047. [Google Scholar] [CrossRef]
- Leav, I.; Plescia, J.; Goel, H.L.; Li, J.; Jiang, Z.; Cohen, R.J.; Languino, L.R.; Altieri, D.C. Cytoprotective mitochondrial chaperone TRAP-1 as a novel molecular target in localized and metastatic prostate cancer. Am. J. Pathol. 2010, 176, 393–401. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Shinogle, H.E.; Galeva, N.A.; Dobrowsky, R.T.; Blagg, B.S.J. Endoplasmic Reticulum-resident Heat Shock Protein 90 (HSP90) Isoform Glucose-regulated Protein 94 (GRP94) Regulates Cell Polarity and Cancer Cell Migration by Affecting Intracellular Transport. J. Biol. Chem. 2016, 291, 8309–8323. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Wang, Y.; Xu, K.; Zhou, Z.; Gong, J.; Zhang, Y.; Gong, H.; Dai, Q.; Yang, J.; Xiong, B.; et al. Co-downregulation of GRP78 and GRP94 induces apoptosis and inhibits migration in prostate cancer cells. Open Life Sci. 2019, 14, 384–391. [Google Scholar] [CrossRef] [Green Version]
- Moon, S.J.; Jeong, B.C.; Kim, H.J.; Lim, J.E.; Kim, H.J.; Kwon, G.Y.; Jackman, J.A.; Kim, J.H. Bruceantin targets HSP90 to overcome resistance to hormone therapy in castration-resistant prostate cancer. Theranostics 2020, 11, 958–973. [Google Scholar] [CrossRef]
- Ferraldeschi, R.; Welti, J.; Powers, M.V.; Yuan, W.; Smyth, T.; Seed, G.; Riisnaes, R.; Hedayat, S.; Wang, H.; Crespo, M.; et al. Second-Generation HSP90 Inhibitor Onalespib Blocks mRNA Splicing of Androgen Receptor Variant 7 in Prostate Cancer Cells. Cancer Res. 2016, 76, 2731–2742. [Google Scholar] [CrossRef] [Green Version]
- Hata, J.; Machida, T.; Matsuoka, K.; Hoshi, S.; Akaihata, H.; Hiraki, H.; Suzuki, T.; Ogawa, S.; Kataoka, M.; Haga, N.; et al. Complement activation by autoantigen recognition in the growth process of benign prostatic hyperplasia. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-T.; Kim, Y.-J.; Park, S.-R.; Ryu, S.-Y.; Jung, J.-Y. NAD(P)H-quinone oxidoreductase 1 silencing aggravates hormone-induced prostatic hyperplasia in mice. Andrologia 2018, 50, e12906. [Google Scholar] [CrossRef]
- Mayer, M.P.; Bukau, B. Hsp70 chaperones: Cellular functions and molecular mechanism. Cell Mol. Life Sci. 2005, 62, 670. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Gierasch, L.M.; Bahar, I. Role of Hsp70 ATPase Domain Intrinsic Dynamics and Sequence Evolution in Enabling its Functional Interactions with NEFs. PLoS Comput. Biol. 2010, 6, e1000931. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Liang, C.; Zhou, L. Structural and functional analysis of the Hsp70/Hsp40 chaperone system. Protein Sci. 2020, 29, 378–390. [Google Scholar] [CrossRef]
- Mayer, M.; Gierasch, L. Recent advances in the structural and mechanistic aspects of Hsp70 molecular chaperones. J. Biol. Chem. 2019, 294, 2085–2097. [Google Scholar] [CrossRef] [Green Version]
- Kampinga, H.H.; Andreasson, C.; Barducci, A.; Cheetham, M.E.; Cyr, D.; Emanuelsson, C.; Genevaux, P.; Gestwicki, J.E.; Goloubinoff, P.; Huerta-Cepas, J.; et al. Function, evolution, and structure of J-domain proteins. Cell Stress Chaperones 2019, 24, 7. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Rossi, P.; Kalodimos, C.G. Structural basis for client recognition and activity of Hsp40 chaperones. Science 2019, 365, 1313. [Google Scholar] [CrossRef] [PubMed]
- Sterrenberg, J.N.; Blatch, G.L.; Edkins, A.L. Human DNAJ in cancer and stem cells. Cancer Lett. 2011, 312, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.B.; Shao, Y.M.; Miao, S.; Wang, L. The diversity of the DnaJ/Hsp40 family, the crucial partners for Hsp70 chaperones. Cell Mol. Life Sci. 2006, 63, 2560–2570. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Qian, X.; Sha, B. Heat shock protein 40: Structural studies and their functional implications. Protein Pept. Lett. 2009, 16, 606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ignacio, D.N.; Mason, K.D.; Hackett-Morton, E.C.; Albanese, C.; Ringer, L.; Wagner, W.D.; Wang, P.C.; Carducci, M.A.; Kachhap, S.K.; Paller, C.J.; et al. Muscadine grape skin extract inhibits prostate cancer cells by inducing cell-cycle arrest, and decreasing migration through heat shock protein 40. Heliyon 2019, 5, e01128. [Google Scholar] [CrossRef] [Green Version]
- Albakova, Z.; Armeev, G.A.; Kanevskiy, L.M.; Kovalenko, E.I.; Sapozhnikov, A.M. HSP70 Multi-Functionality in Cancer. Cells 2020, 9, 587. [Google Scholar] [CrossRef] [Green Version]
- De Los Rios, P.; Barducci, A. Hsp70 chaperones are non-equilibrium machines that achieve ultra-affinity by energy consumption. Elife 2014, 2014, e02218. [Google Scholar] [CrossRef] [Green Version]
- Eftekharzadeh, B.; Banduseela, V.C.; Chiesa, G.; Martínez-Cristóbal, P.; Rauch, J.N.; Nath, S.R.; Schwarz, D.M.C.; Shao, H.; Marin-Argany, M.; Di Sanza, C.; et al. Hsp70 and Hsp40 inhibit an inter-domain interaction necessary for transcriptional activity in the androgen receptor. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Pootrakul, L.; Datar, R.H.; Shi, S.R.; Cai, J.; Hawes, D.; Groshen, S.G.; Lee, A.S.; Cote, R.J. Expression of Stress Response Protein Grp78 Is Associated with the Development of Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2006, 12, 5987–5993. [Google Scholar] [CrossRef] [Green Version]
- Cultrara, C.N.; Kozuch, S.D.; Ramasundaram, P.; Heller, C.J.; Shah, S.; Beck, A.E.; Sabatino, D.; Zilberberg, J. GRP78 modulates cell adhesion markers in prostate Cancer and multiple myeloma cell lines. BMC Cancer 2018, 18, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; Wu, Z.; Wang, D.; Pascal, L.E.; Nelson, J.B.; Wipf, P.; Wang, Z. Hsp70 Binds to the Androgen Receptor N-terminal Domain and Modulates the Receptor Function in Prostate Cancer Cells. Mol. Cancer Ther. 2019, 18, 39–50. [Google Scholar] [CrossRef] [Green Version]
- Kita, K.; Shiota, M.; Tanaka, M.; Otsuka, A.; Matsumoto, M.; Kato, M.; Tamada, S.; Iwao, H.; Miura, K.; Nakatani, T.; et al. Heat shock protein 70 inhibitors suppress androgen receptor expression in LNCaP95 prostate cancer cells. Cancer Sci. 2017, 108, 1820–1827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moses, M.A.; Kim, Y.S.; Rivera-Marquez, G.M.; Oshima, N.; Watson, M.J.; Beebe, K.E.; Wells, C.; Lee, S.; Zuehlke, A.D.; Shao, H.; et al. Targeting the Hsp40/Hsp70 chaperone axis as a novel strategy to treat aastration-resistant prostate cancer. Cancer Res. 2018, 78, 4022–4035. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ratajczak, W.; Lubkowski, M.; Lubkowska, A. Heat Shock Proteins in Benign Prostatic Hyperplasia and Prostate Cancer. Int. J. Mol. Sci. 2022, 23, 897. https://doi.org/10.3390/ijms23020897
Ratajczak W, Lubkowski M, Lubkowska A. Heat Shock Proteins in Benign Prostatic Hyperplasia and Prostate Cancer. International Journal of Molecular Sciences. 2022; 23(2):897. https://doi.org/10.3390/ijms23020897
Chicago/Turabian StyleRatajczak, Weronika, Michał Lubkowski, and Anna Lubkowska. 2022. "Heat Shock Proteins in Benign Prostatic Hyperplasia and Prostate Cancer" International Journal of Molecular Sciences 23, no. 2: 897. https://doi.org/10.3390/ijms23020897
APA StyleRatajczak, W., Lubkowski, M., & Lubkowska, A. (2022). Heat Shock Proteins in Benign Prostatic Hyperplasia and Prostate Cancer. International Journal of Molecular Sciences, 23(2), 897. https://doi.org/10.3390/ijms23020897