
Activation of Notch3 in Renal Tubular Cells Leads to Progressive Cystic Kidney Disease

 and
and
Abstract
:1. Introduction
2. Results
2.1. Notch3 Is De Novo Expressed in Epithelial Cells in Renal Carcinoma and Polycystic Kidney Disease
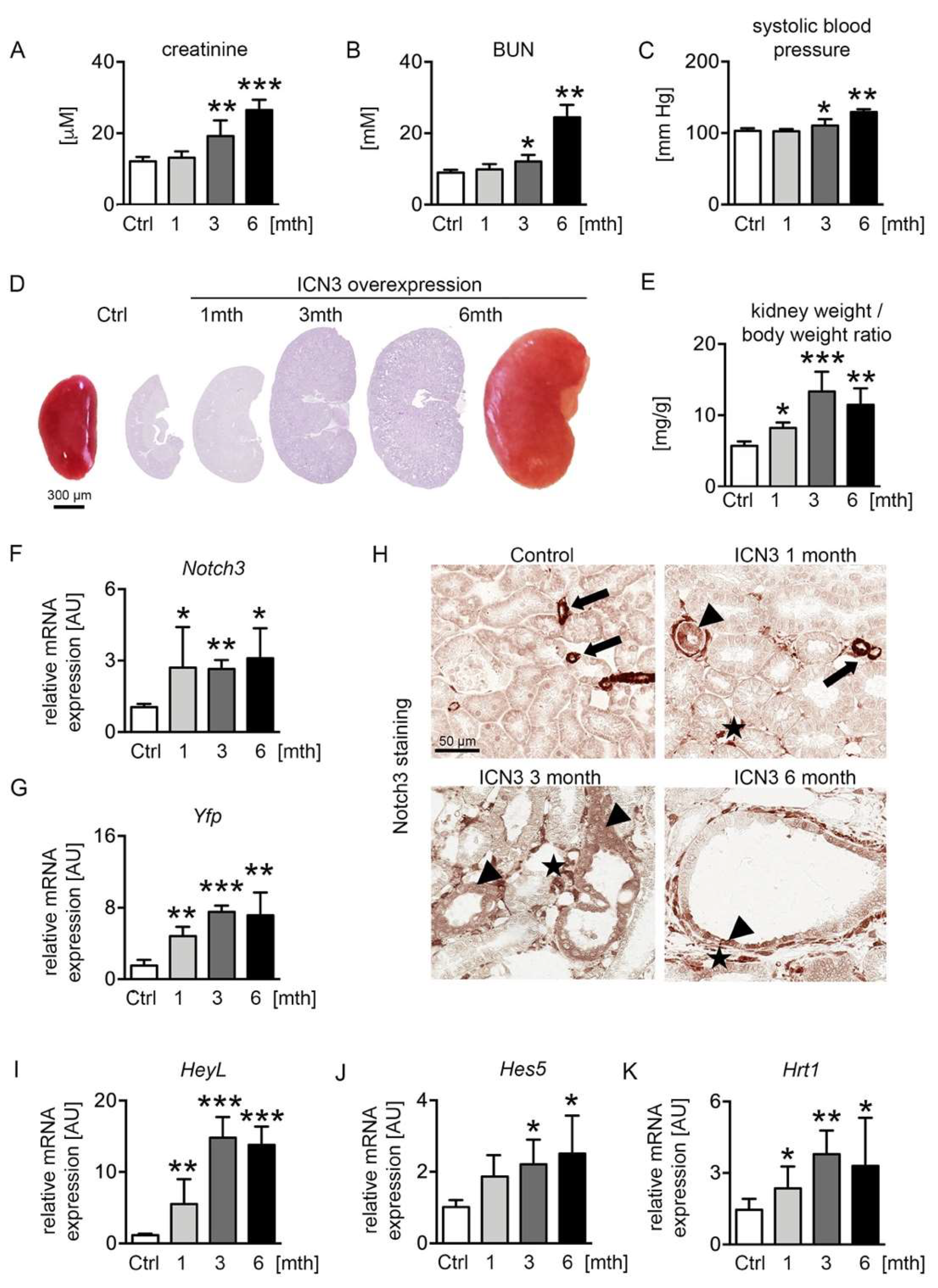
2.2. Ectopic Notch3 Signaling Activation Compromises Renal Function and Increases Kidney Size
2.3. Notch3 Activation Generates Heterogeneous Cyst Formation
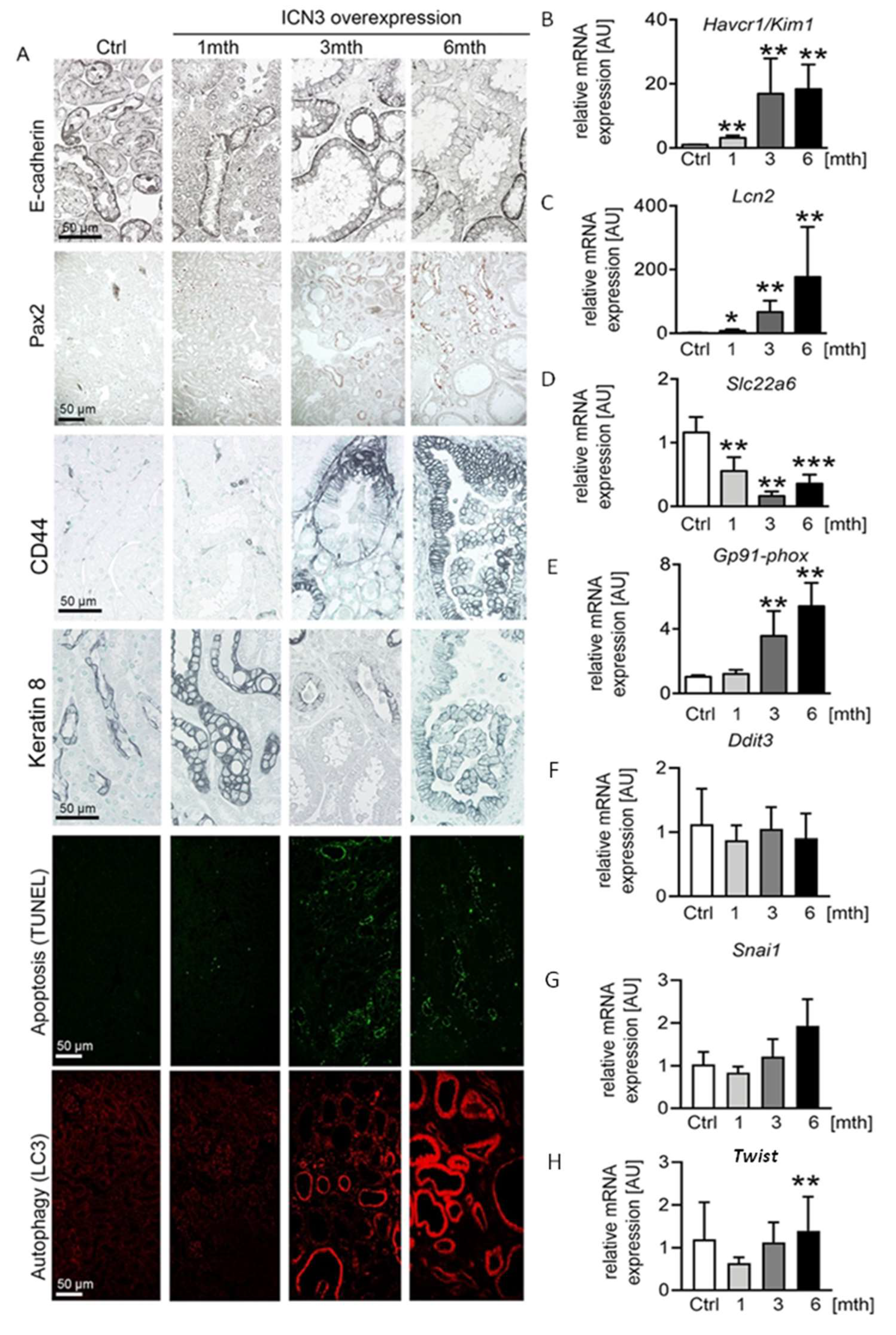
2.4. Epithelial N3ICD Induces Severe Tubular Damage and cCll Dedifferentiation
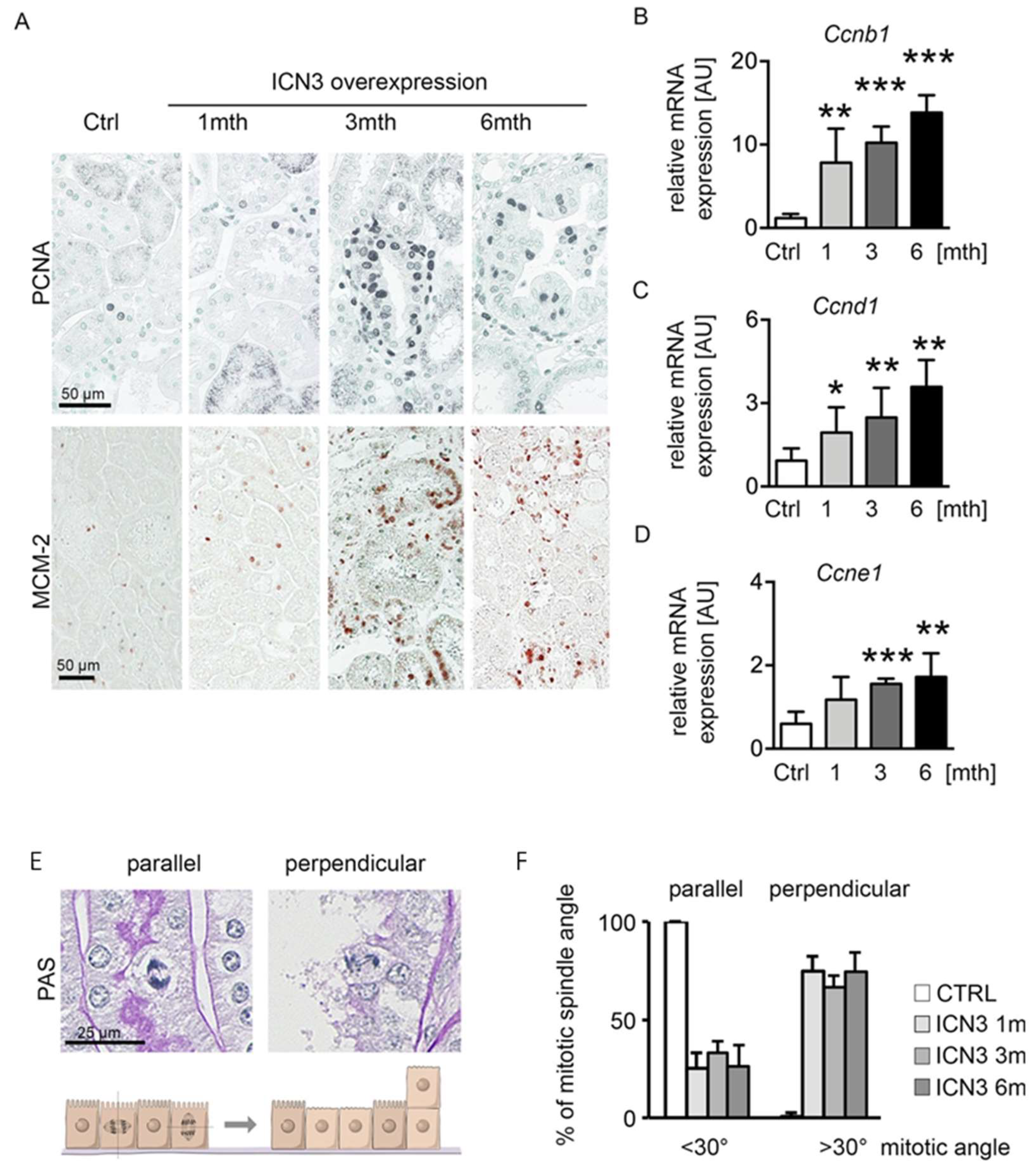
2.5. Notch3 Activation Leads to Hyperplasia and Disturbed Alignment of Cell Division
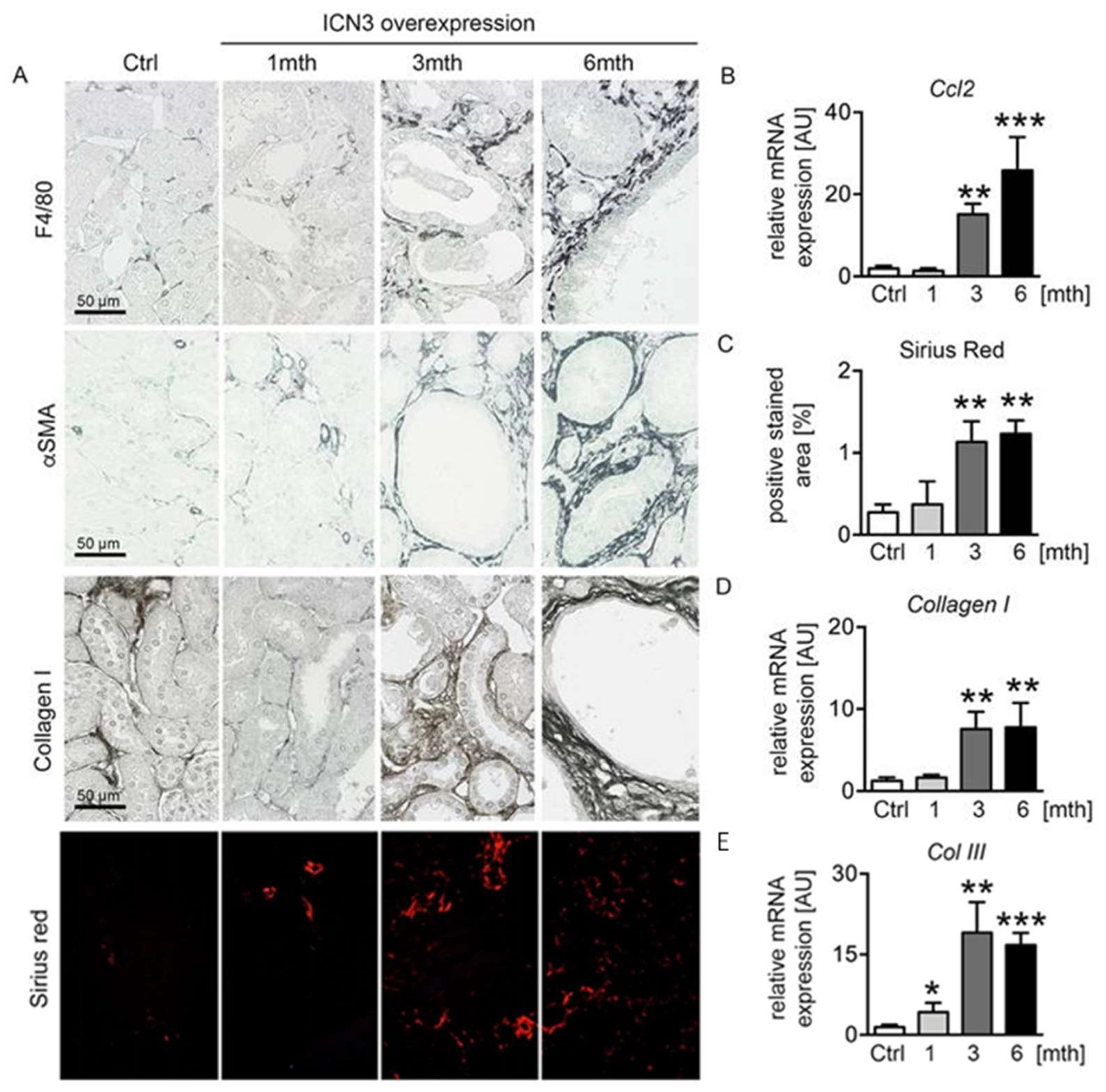
2.6. Epithelial N3ICD Resulted in Tubulointerstitial Inflammation and Fibrosis
3. Discussion
4. Materials and Methods
4.1. Pax8-rtTA-LC1-R26-N3ICSAT (N3ICD) Mice
4.2. Histological and Functional Parameters
4.3. Human Renal Tissue
4.4. Total RNA Extraction and Quantitative Real-Time PCR
4.5. Immunostaining
4.6. TUNEL Assay
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grantham, J.J.; Mulamalla, S.; Swenson-Fields, K.I. Why kidneys fail in autosomal dominant polycystic kidney disease. Nat. Rev. Nephrol. 2011, 7, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, C.; Guay-Woodford, L.M.; Harris, P.C.; Horie, S.; Peters, D.J.M.; Torres, V.E. Polycystic kidney disease. Nat. Rev. Dis Primers 2018, 4, 50. [Google Scholar] [CrossRef]
- Ghata, J.; Cowley, B.D., Jr. Polycystic Kidney Disease. Compr. Physiol. 2017, 7, 945–975. [Google Scholar] [CrossRef] [PubMed]
- Artavanis-Tsakonas, S.; Rand, M.D.; Lake, R.J. Notch signaling: Cell fate control and signal integration in development. Science 1999, 284, 770–776. [Google Scholar] [CrossRef] [Green Version]
- Iso, T.; Kedes, L.; Hamamori, Y. HES and HERP families: Multiple effectors of the Notch signaling pathway. J. Cell. Physiol. 2003, 194, 237–255. [Google Scholar] [CrossRef]
- Joutel, A.; Corpechot, C.; Ducros, A.; Vahedi, K.; Chabriat, H.; Mouton, P.; Alamowitch, S.; Domenga, V.; Cecillion, M.; Marechal, E.; et al. Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature 1996, 383, 707–710. [Google Scholar] [CrossRef] [PubMed]
- Djudjaj, S.; Chatziantoniou, C.; Raffetseder, U.; Guerrot, D.; Dussaule, J.C.; Boor, P.; Kerroch, M.; Hanssen, L.; Brandt, S.; Dittrich, A.; et al. Notch-3 receptor activation drives inflammation and fibrosis following tubulointerstitial kidney injury. J. Pathol. 2012, 228, 286–299. [Google Scholar] [CrossRef]
- El Machhour, F.; Keuylian, Z.; Kavvadas, P.; Dussaule, J.C.; Chatziantoniou, C. Activation of Notch3 in Glomeruli Promotes the Development of Rapidly Progressive Renal Disease. J. Am. Soc. Nephrol. 2015, 26, 1561–1575. [Google Scholar] [CrossRef] [Green Version]
- Kavvadas, P.; Keuylian, Z.; Prakoura, N.; Placier, S.; Dorison, A.; Chadjichristos, C.E.; Dussaule, J.C.; Chatziantoniou, C. Notch3 orchestrates epithelial and inflammatory responses to promote acute kidney injury. Kidney Int. 2018, 94, 126–138. [Google Scholar] [CrossRef]
- Sirin, Y.; Susztak, K. Notch in the kidney: Development and disease. J. Pathol. 2012, 226, 394–403. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wen, X.; Liu, Y. Tubular cell dedifferentiation and peritubular inflammation are coupled by the transcription regulator Id1 in renal fibrogenesis. Kidney Int. 2012, 81, 880–891. [Google Scholar] [CrossRef] [Green Version]
- Veerasamy, M.; Nguyen, T.Q.; Motazed, R.; Pearson, A.L.; Goldschmeding, R.; Dockrell, M.E. Differential regulation of E-cadherin and alpha-smooth muscle actin by BMP 7 in human renal proximal tubule epithelial cells and its implication in renal fibrosis. Am. J. Physiol. Ren. Physiol. 2009, 297, F1238–F1248. [Google Scholar] [CrossRef]
- Sharma, R.; Sanchez-Ferras, O.; Bouchard, M. Pax genes in renal development, disease and regeneration. Semin. Cell Dev. Biol. 2015, 44, 97–106. [Google Scholar] [CrossRef]
- Ozcan, A.; de la Roza, G.; Ro, J.Y.; Shen, S.S.; Truong, L.D. PAX2 and PAX8 expression in primary and metastatic renal tumors: A comprehensive comparison. Arch. Pathol. Lab. Med. 2012, 136, 1541–1551. [Google Scholar] [CrossRef] [Green Version]
- Shimoishi, K.; Anraku, M.; Kitamura, K.; Tasaki, Y.; Taguchi, K.; Hashimoto, M.; Fukunaga, E.; Maruyama, T.; Otagiri, M. An oral adsorbent, AST-120 protects against the progression of oxidative stress by reducing the accumulation of indoxyl sulfate in the systemic circulation in renal failure. Pharm. Res. 2007, 24, 1283–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norman, J. Fibrosis and progression of autosomal dominant polycystic kidney disease (ADPKD). Biochim. Biophys. Acta 2011, 1812, 1327–1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweeney, W.E., Jr.; Avner, E.D. Pathophysiology of childhood polycystic kidney diseases: New insights into disease-specific therapy. Pediatr. Res. 2014, 75, 148–157. [Google Scholar] [CrossRef] [Green Version]
- Idowu, J.; Home, T.; Patel, N.; Magenheimer, B.; Tran, P.V.; Maser, R.L.; Ward, C.J.; Calvet, J.P.; Wallace, D.P.; Sharma, M. Aberrant Regulation of Notch3 Signaling Pathway in Polycystic Kidney Disease. Sci. Rep. 2018, 8, 3340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bielesz, B.; Sirin, Y.; Si, H.; Niranjan, T.; Gruenwald, A.; Ahn, S.; Kato, H.; Pullman, J.; Gessler, M.; Haase, V.H.; et al. Epithelial Notch signaling regulates interstitial fibrosis development in the kidneys of mice and humans. J. Clin. Investig. 2010, 120, 4040–4054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Pathak, N.; Kramer-Zucker, A.; Drummond, I.A. Notch signaling controls the differentiation of transporting epithelia and multiciliated cells in the zebrafish pronephros. Development 2007, 134, 1111–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drummond, I.A.; Majumdar, A.; Hentschel, H.; Elger, M.; Solnica-Krezel, L.; Schier, A.F.; Neuhauss, S.C.; Stemple, D.L.; Zwartkruis, F.; Rangini, Z.; et al. Early development of the zebrafish pronephros and analysis of mutations affecting pronephric function. Development 1998, 125, 4655–4667. [Google Scholar] [CrossRef]
- Surendran, K.; Selassie, M.; Liapis, H.; Krigman, H.; Kopan, R. Reduced Notch signaling leads to renal cysts and papillary microadenomas. J. Am. Soc. Nephrol. 2010, 21, 819–832. [Google Scholar] [CrossRef] [Green Version]
- Tran, P.V.; Sharma, M.; Li, X.; Calvet, J.P. Developmental signaling: Does it bridge the gap between cilia dysfunction and renal cystogenesis? Birth Defects Res. C Embryo Today 2014, 102, 159–173. [Google Scholar] [CrossRef] [Green Version]
- Edeling, M.; Ragi, G.; Huang, S.; Pavenstadt, H.; Susztak, K. Developmental signalling pathways in renal fibrosis: The roles of Notch, Wnt and Hedgehog. Nat. Rev. Nephrol. 2016, 12, 426–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, M.; Fogarty, E.; Janga, M.; Surendran, K. Notch Signaling in Kidney Development, Maintenance, and Disease. Biomolecules 2019, 9, 692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, M.; deRiso, J.; Otterpohl, K.; Ratnayake, I.; Kota, D.; Ahrenkiel, P.; Chandrasekar, I.; Surendran, K. Endogenous Notch Signaling in Adult Kidneys Maintains Segment-Specific Epithelial Cell Types of the Distal Tubules and Collecting Ducts to Ensure Water Homeostasis. J. Am. Soc. Nephrol. 2019, 30, 110–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aparicio, L.M.; Villaamil, V.M.; Gallego, G.A.; Cainzos, I.S.; Campelo, R.G.; Rubira, L.V.; Estevez, S.V.; Mateos, L.L.; Perez, J.L.; Vazquez, M.R.; et al. Expression of Notch1 to −4 and their ligands in renal cell carcinoma: A tissue microarray study. Cancer Genom. Proteom. 2011, 8, 93–101. [Google Scholar]
- Jedroszka, D.; Orzechowska, M.; Bednarek, A.K. Predictive values of Notch signalling in renal carcinoma. Arch. Med. Sci. 2017, 13, 1249–1254. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Li, Y.; Banerjee, S.; Sarkar, F.H. Emerging role of Notch in stem cells and cancer. Cancer Lett. 2009, 279, 8–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palomero, T.; Lim, W.K.; Odom, D.T.; Sulis, M.L.; Real, P.J.; Margolin, A.; Barnes, K.C.; O’Neil, J.; Neuberg, D.; Weng, A.P.; et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc. Natl. Acad. Sci. USA 2006, 103, 18261–18266. [Google Scholar] [CrossRef] [Green Version]
- Bhagat, T.D.; Zou, Y.; Huang, S.; Park, J.; Palmer, M.B.; Hu, C.; Li, W.; Shenoy, N.; Giricz, O.; Choudhary, G.; et al. Notch Pathway Is Activated via Genetic and Epigenetic Alterations and Is a Therapeutic Target in Clear Cell Renal Cancer. J. Biol. Chem. 2017, 292, 837–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helle, F.; Jouzel, C.; Chadjichristos, C.; Placier, S.; Flamant, M.; Guerrot, D.; Francois, H.; Dussaule, J.C.; Chatziantoniou, C. Improvement of renal hemodynamics during hypertension-induced chronic renal disease: Role of EGF receptor antagonism. Am. J. Physiol. Renal. Physiol. 2009, 297, F191–F199. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequence |
|---|---|
| Notch3 | FW: CCCCAACCAGAAGTTACCCC RV: AGGAGTGTCACTTCAGCACC |
| HeyL | FW: CTGAATTGCGACGATTGGT RV: CTGAATTGCGACGATTGGT |
| Hrt1 | FW: CATGAAGAGAGCTCACCCAGA RV: GAACACAGAGCCGAACTCAA |
| Hes5 | FW: CCCAAGGAGAAAAACCGACT RV: TGCTCTATGCTGCTGTTGATG |
| Yfp | FW: CTCGTGACCACCTTCGGCT RV: TCCTGGACGTAGCCTTCG |
| Havcr1/Kim1 | FW: TCAGATTCAAGTCTTCATTTCAGG RV: CCCCCTTTACTTCCACATAAGAA |
| Lcn2 (NGAL) | FW: CCATCTATGAGCTACAAGAGAACAAT RV: TCTGATCCAGTAGCGACAGC |
| Slc12A1 (NKCC2) | FW: ATGCCTCGTATGCCAAATCT RV: CCCACATGTTGTAAATCCCATA |
| Kcnj1 (ROMK) | FW: GGTGCAAGGGACTTTCTCAC RV: TGAACATCCTTTCTGTCAGTGC |
| Aqp2 | FW: TAGCCCTGCTCTCTCCATTG RV: GAGCAGCCGGTGAAATAGAT |
| Ccnb1 | FW: TCTTCTCGAATCGGGGAAC RV: TTGGCCTTATTTTCTGCGTTA |
| Ccnd1 | FW: CATCCATGCGGAAAATCG RV: CAGGCGGCTCTTCTTCAA |
| Ccne1 | FW: ACGGGTGAGGTGCTGATG RV: GGACGCACAGGTCTAGAAGC |
| Ccl2 (MCP-1) | FW: CATCCACGTGTTGGCTCA RV: CATCCACGTGTTGGCTCA |
| Collagen I | FW: GCAGGTTCACCTACTCTGTCCT RV: CTTGCCCCATTCATTTGTCT |
| Collagen III | FW: TGGTTTCTTCTCACCCTTCTTC RV: TGCATCCCAATTCATCTACGT |
| GP91phox | FW: ACTGCGGAGAGTTTGGAAGA RV: GGTGATGACCACCTTTTGCT |
| Slc22a6 (OAT-1) | FW: CCTATGCTGTGCCCCACT RV: GGCTGACTCAATGAAGAACCA |
| Hprt | FW: GGAGCGGTAGCACCTCCT RV: CTGGTTCATCATCGCTAATCAC |
| Ddit3 (CHOP) | FW: GCGACAGAGCCAGAATAACA RV: GATGCACTTCCTTCTGGAACA |
| Snai1 (Snail homologue 1) | FW: GTCTGCACGACCTGTGGAA RV: CAGGAGAATGGCTTCTCACC |
| Twist | FW: AGCTACGCCTTCTCCGTCT RV: TCCTTCTCTGGAAACAATGACA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Djudjaj, S.; Kavvadas, P.; Prakoura, N.; Bülow, R.D.; Migeon, T.; Placier, S.; Chadjichristos, C.E.; Boor, P.; Chatziantoniou, C. Activation of Notch3 in Renal Tubular Cells Leads to Progressive Cystic Kidney Disease. Int. J. Mol. Sci. 2022, 23, 884. https://doi.org/10.3390/ijms23020884
Djudjaj S, Kavvadas P, Prakoura N, Bülow RD, Migeon T, Placier S, Chadjichristos CE, Boor P, Chatziantoniou C. Activation of Notch3 in Renal Tubular Cells Leads to Progressive Cystic Kidney Disease. International Journal of Molecular Sciences. 2022; 23(2):884. https://doi.org/10.3390/ijms23020884
Chicago/Turabian StyleDjudjaj, Sonja, Panagiotis Kavvadas, Niki Prakoura, Roman D. Bülow, Tiffany Migeon, Sandrine Placier, Christos E. Chadjichristos, Peter Boor, and Christos Chatziantoniou. 2022. "Activation of Notch3 in Renal Tubular Cells Leads to Progressive Cystic Kidney Disease" International Journal of Molecular Sciences 23, no. 2: 884. https://doi.org/10.3390/ijms23020884
APA StyleDjudjaj, S., Kavvadas, P., Prakoura, N., Bülow, R. D., Migeon, T., Placier, S., Chadjichristos, C. E., Boor, P., & Chatziantoniou, C. (2022). Activation of Notch3 in Renal Tubular Cells Leads to Progressive Cystic Kidney Disease. International Journal of Molecular Sciences, 23(2), 884. https://doi.org/10.3390/ijms23020884