
Neutrophil Cathepsin G Enhances Thrombogenicity of Mildly Injured Arteries via ADP-Mediated Platelet Sensitization



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
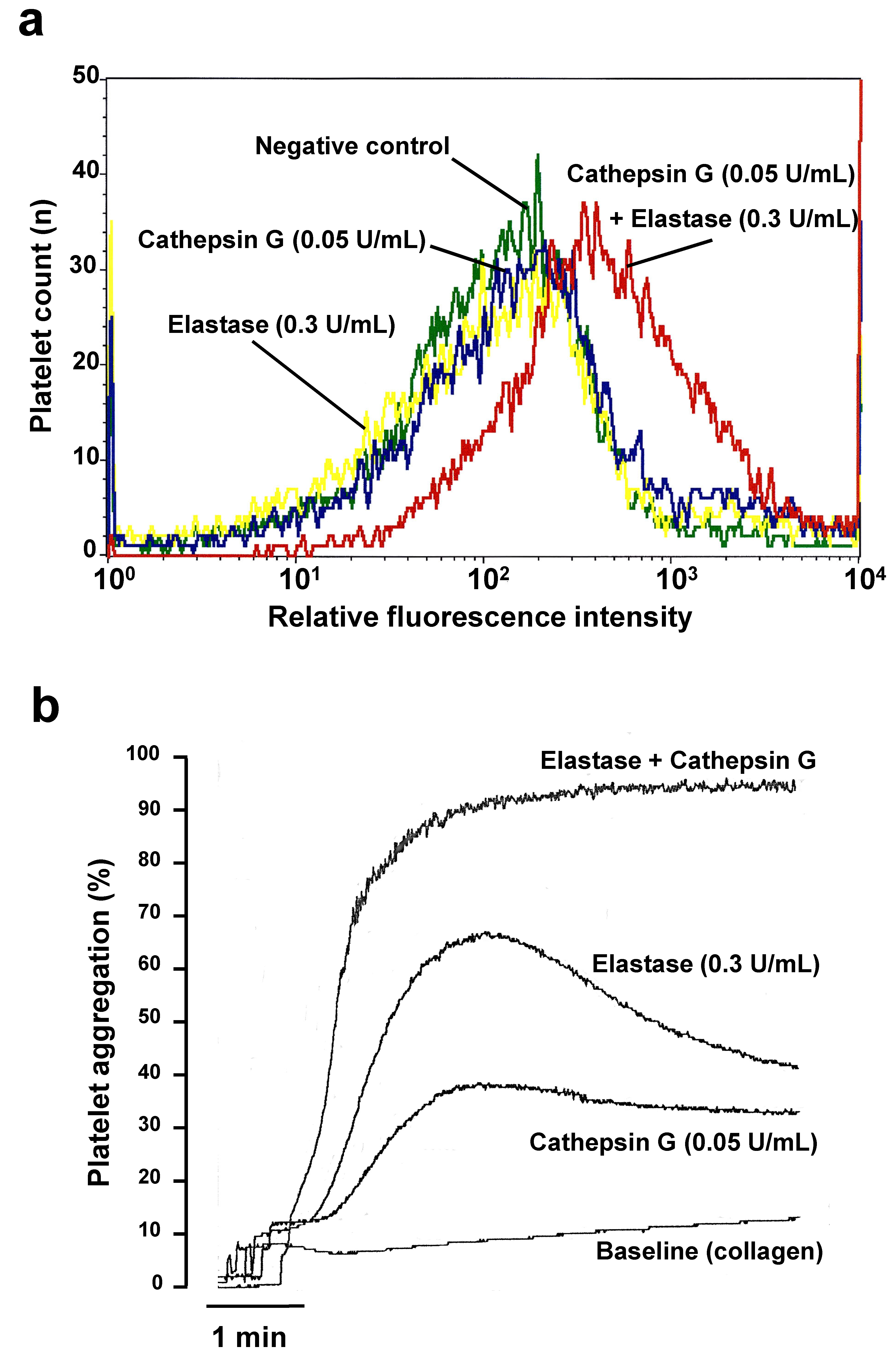
2.1. Cathepsin G and Elastase Threshold Concentration Definition
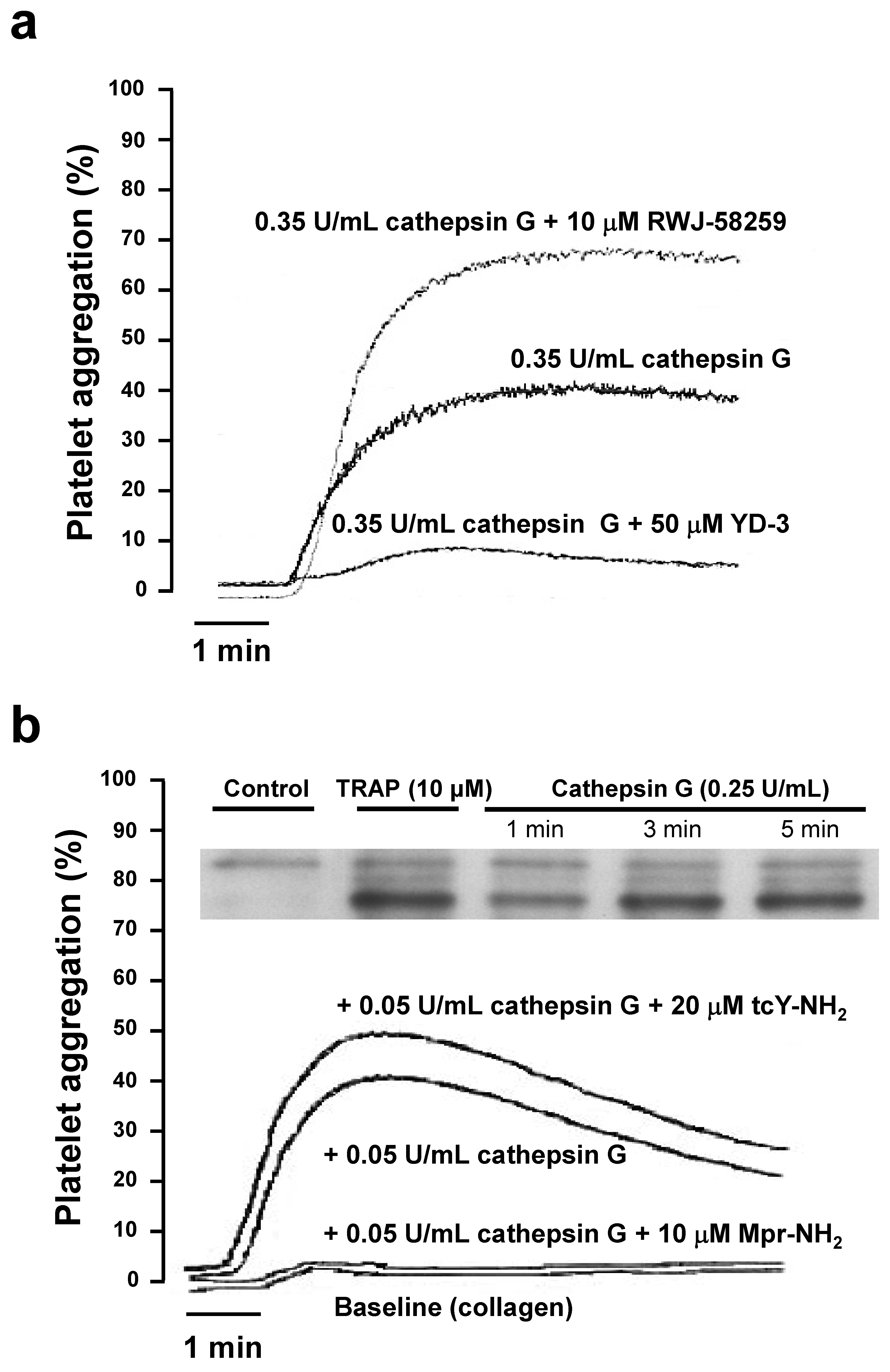
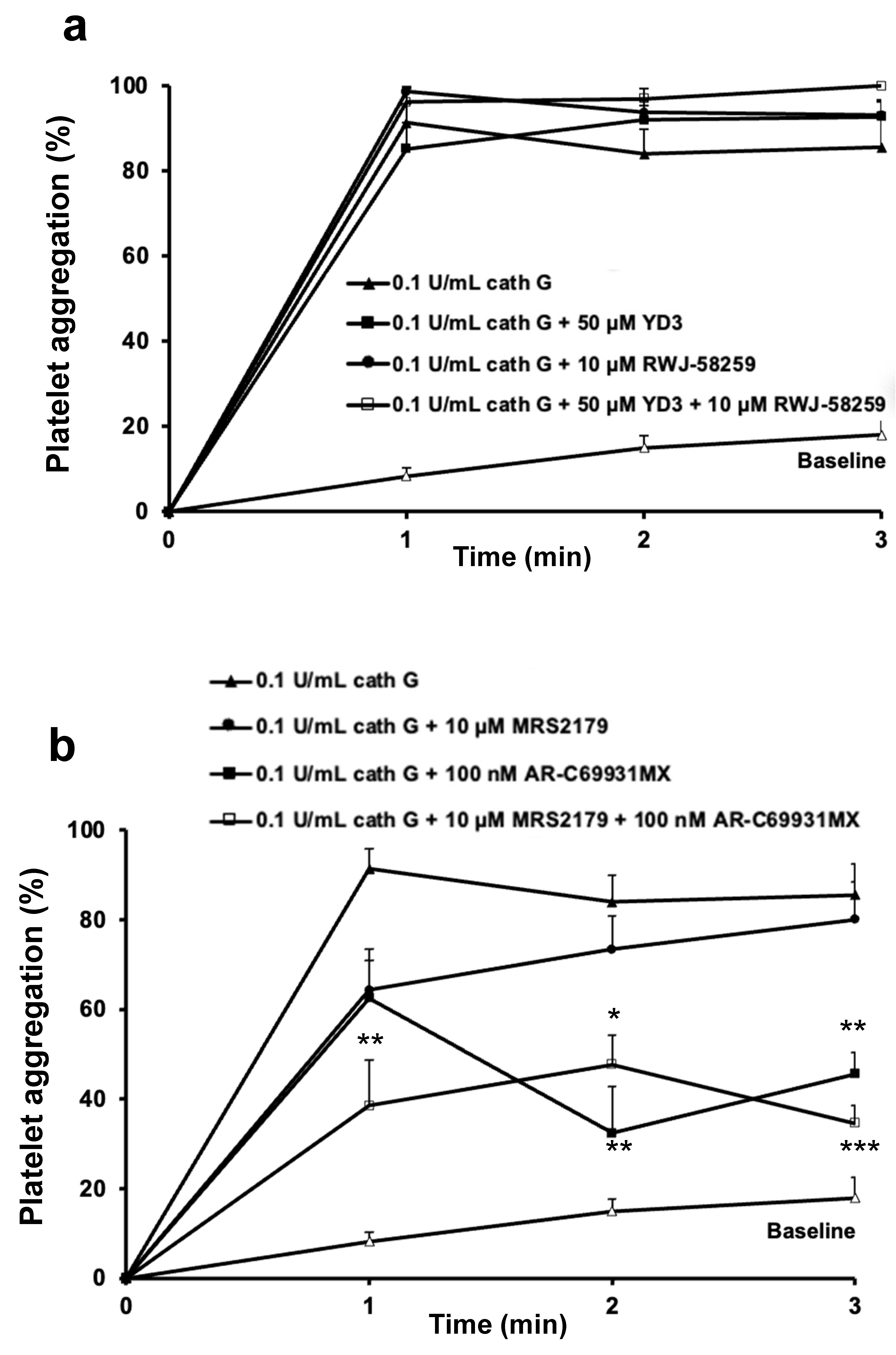
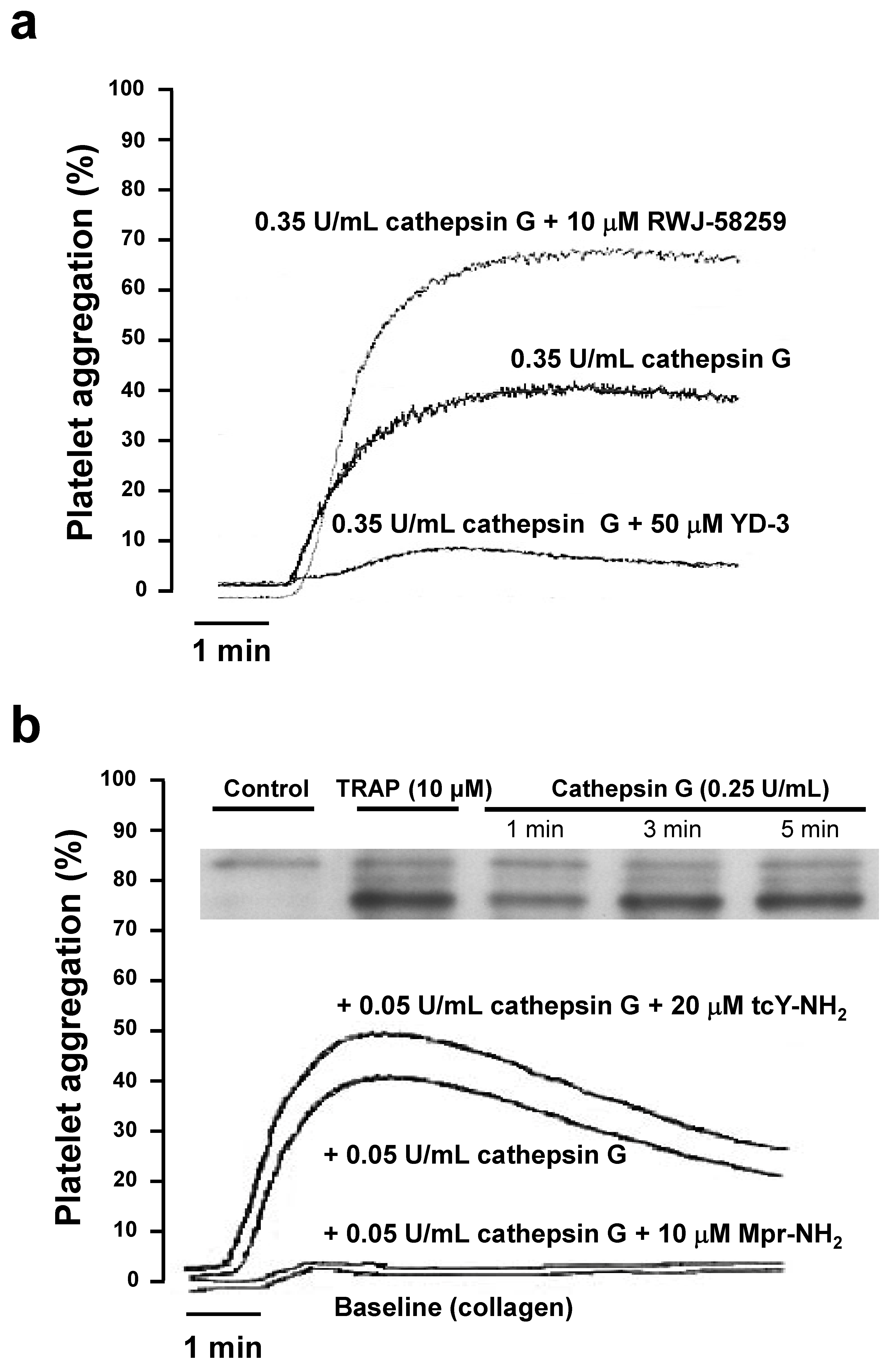
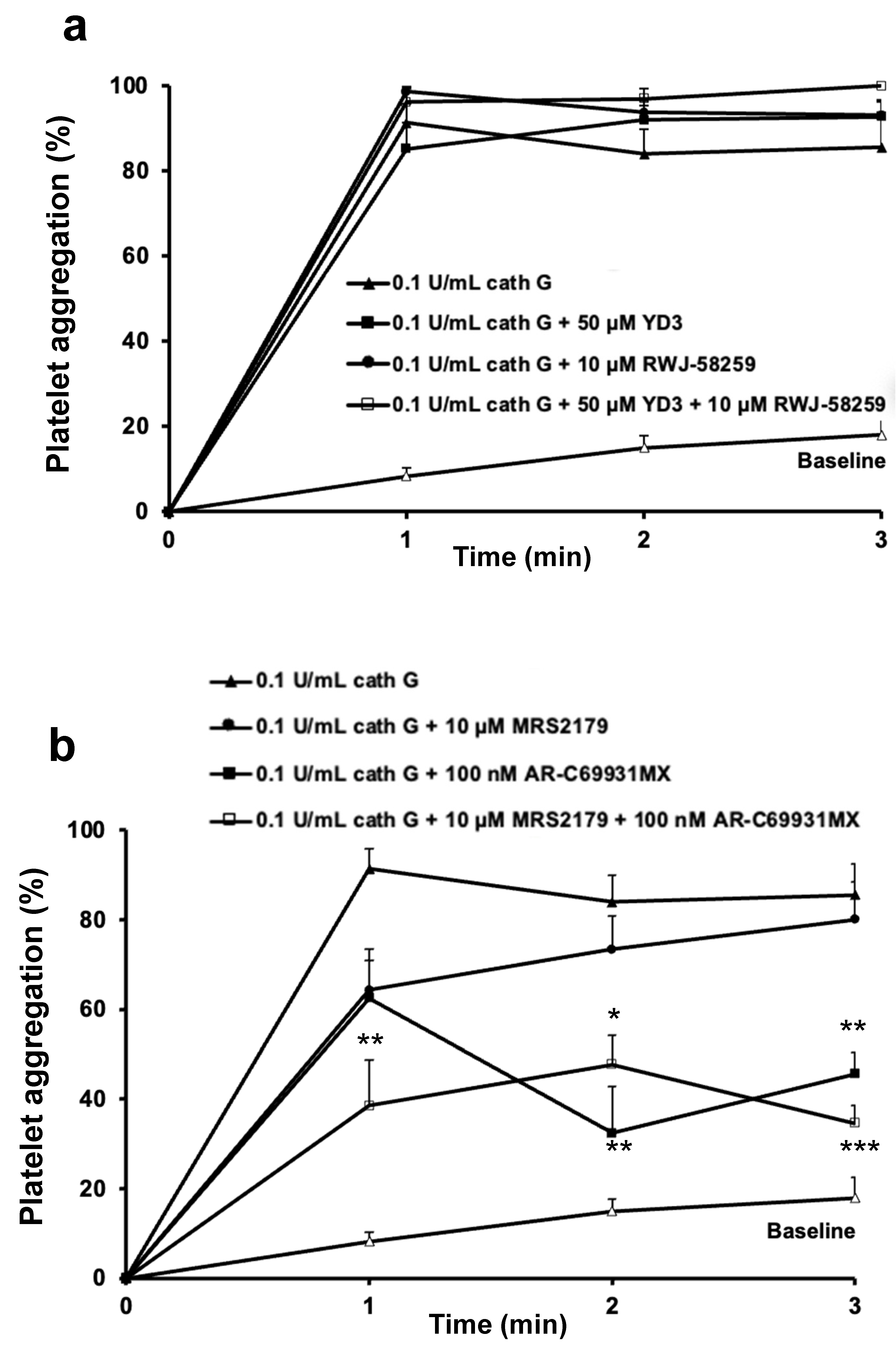
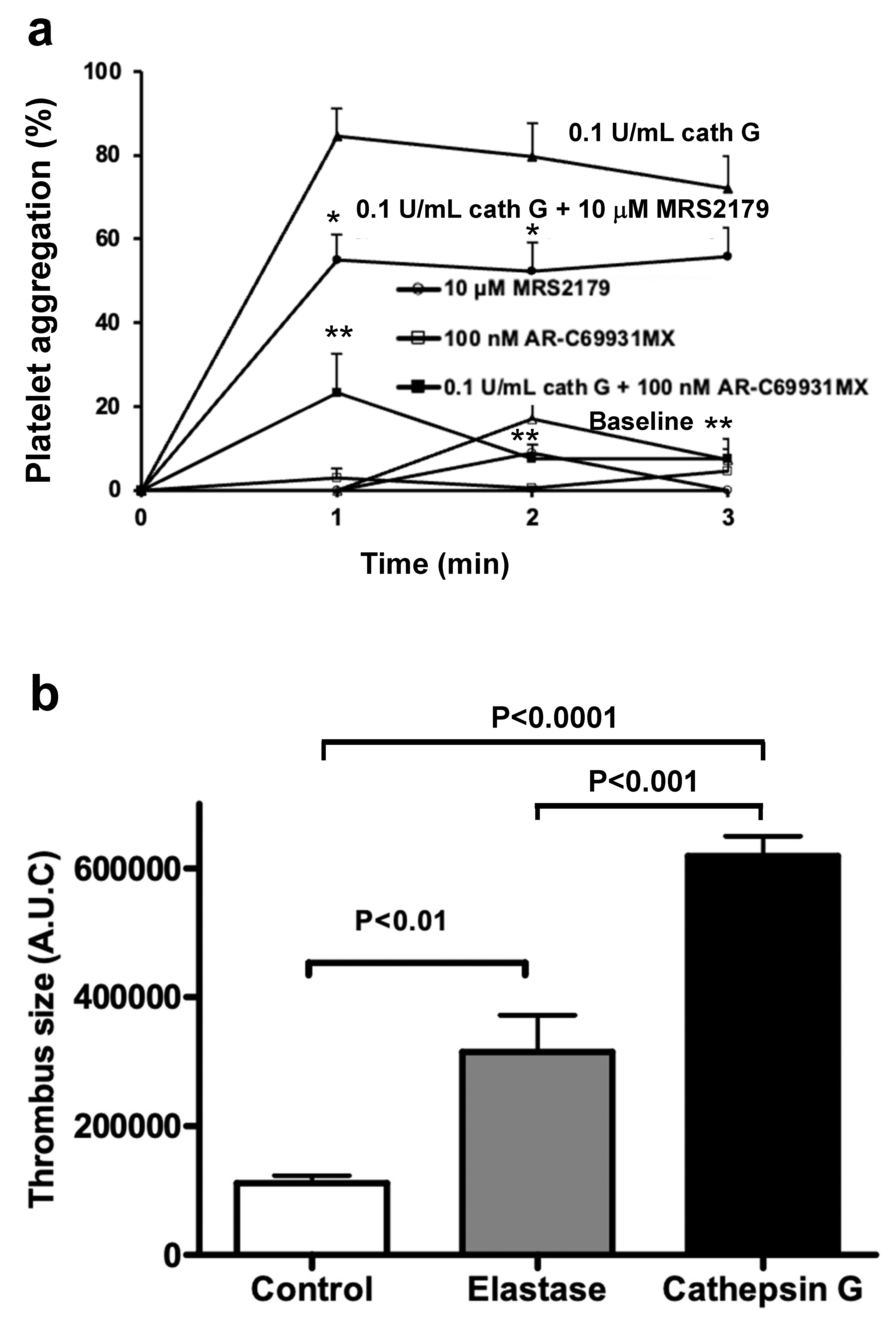
2.2. Role of PAR1 and PAR4 in Washed Human Platelet Activation by Cathepsin G
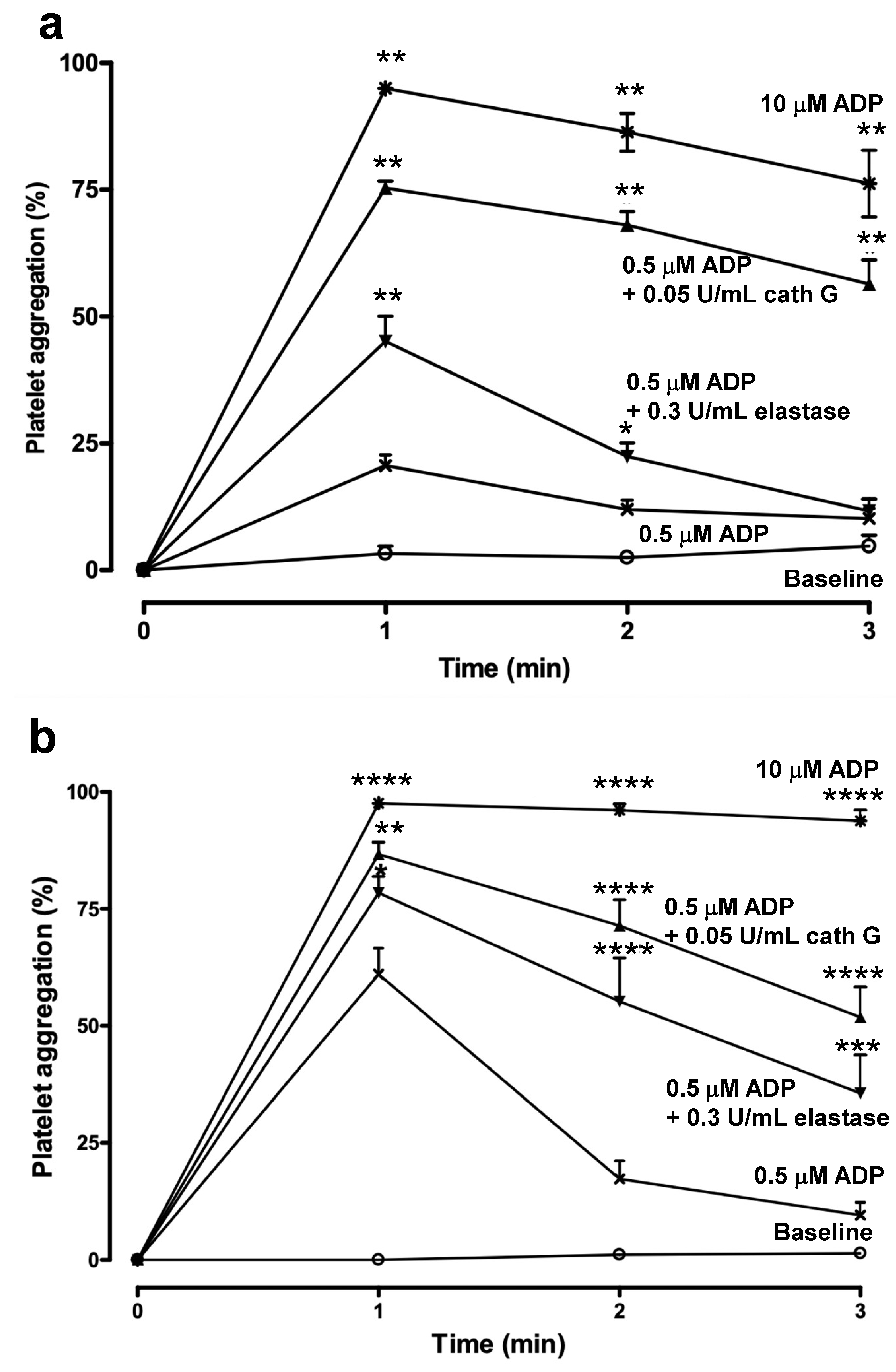
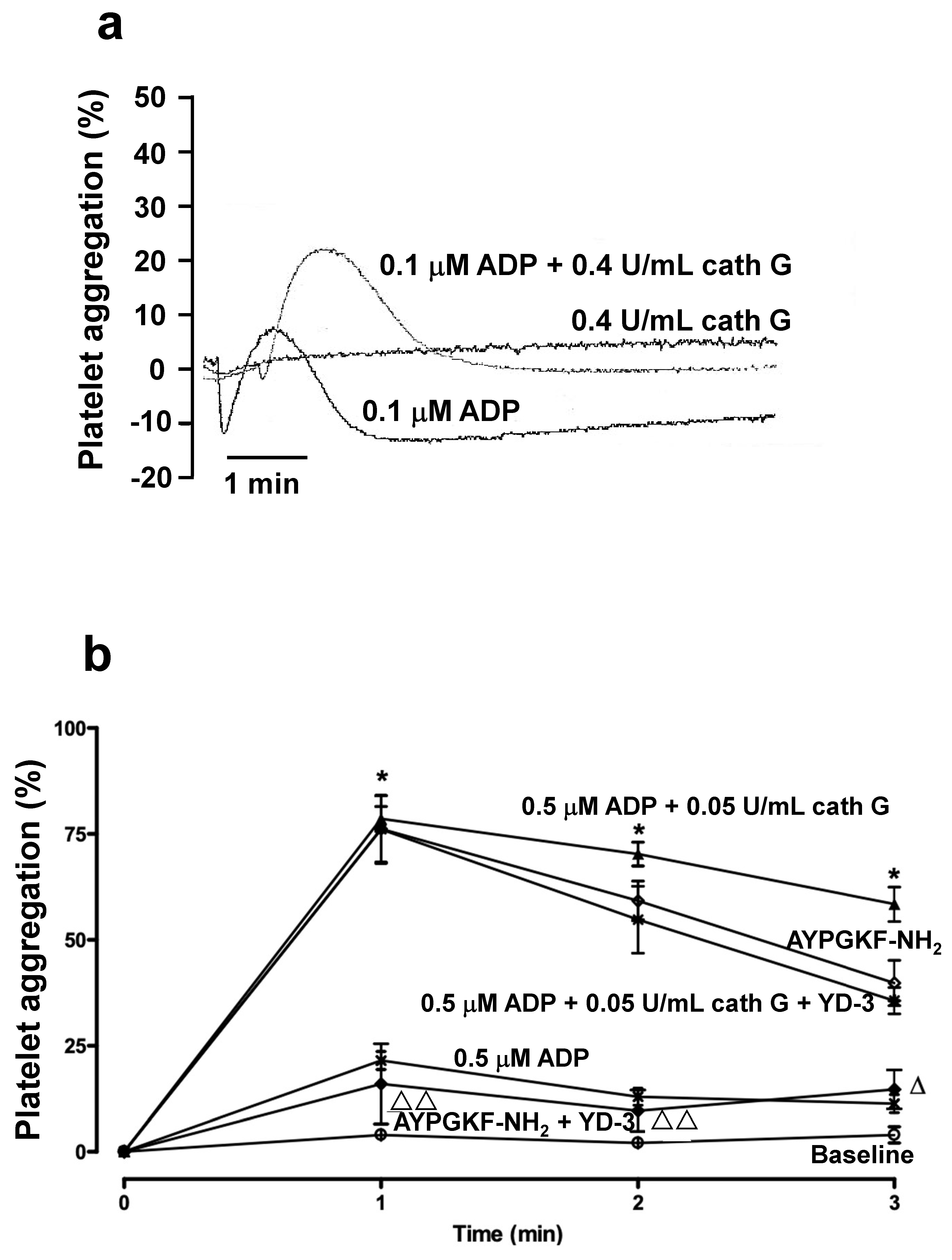
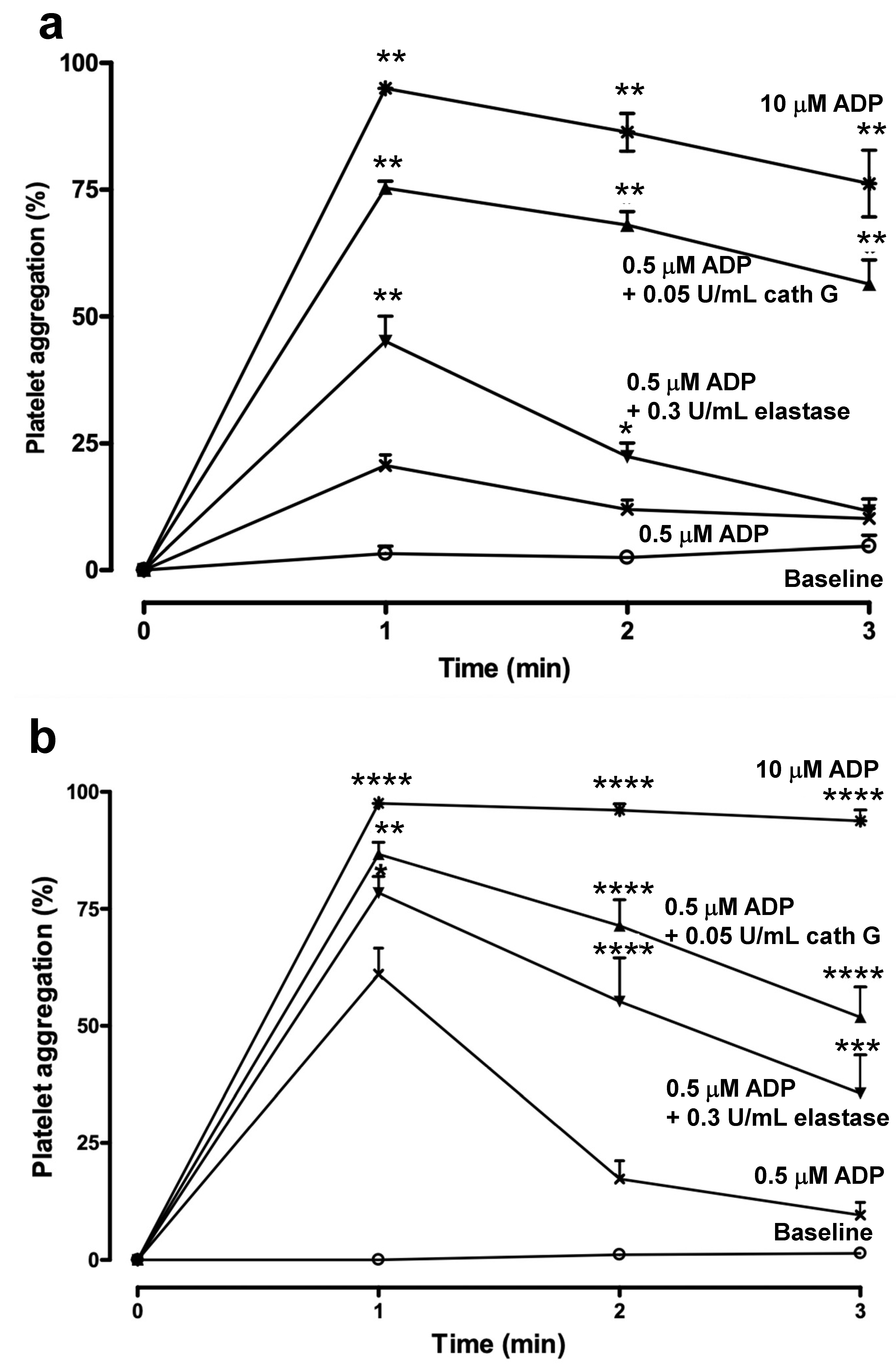
2.3. Human whole Blood Platelet Aggregation
2.4. Platelet Aggregation in Murine Platelet-Rich Plasma and in Whole Blood
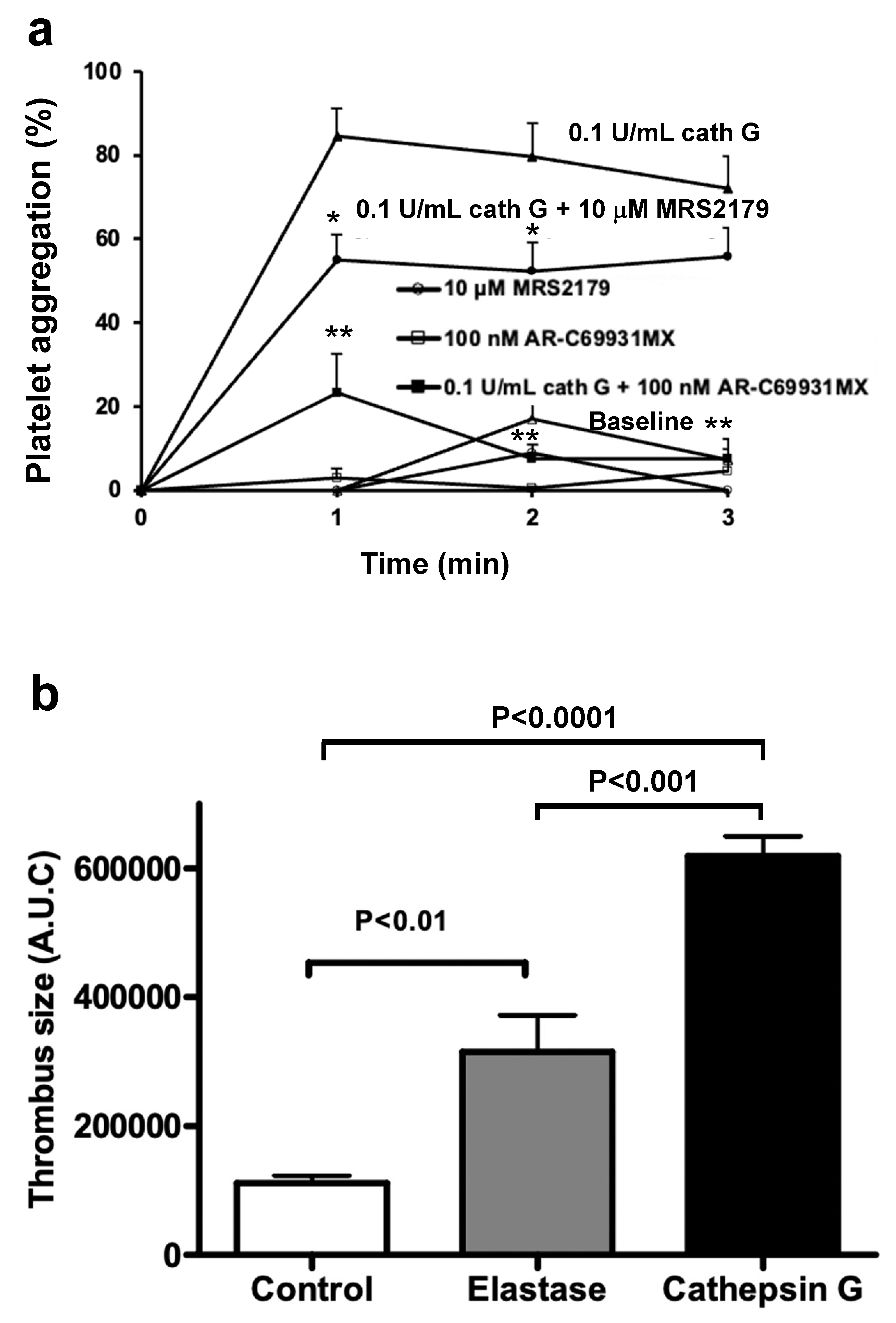
2.5. Cathepsin G in a Mouse Model of Arterial Thrombosis
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Unit Definition for Cathepsin G
4.3. Blood Collection, Plasma Isolation and Platelet Preparation
4.4. Platelet Aggregation in Murine and Human Whole Blood
4.5. Washed Human Platelet Aggregation and Mouse PRP Aggregation
4.6. Detection by Flow Cytometry of Activated Platelets Expressing P-Selectin (CD62)
4.7. Western Blot Analyses
4.8. Experimental Arterial Thrombosis Model
4.9. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brook, R.D.; Rajagopalan, S.; Pope, C.A., 3rd; Brook, J.R.; Bhatnagar, A.; Diez-Roux, A.V.; Holguin, F.; Hong, Y.; Luepker, R.V.; Mittleman, M.A.; et al. American Heart Association Council on Epidemiology and Prevention, Council on the Kidney in Cardiovascular Disease, and Council on Nutrition, Physical Activity and Metabolism. Particulate matter air pollution and cardiovascular disease: An update to the scientific statement from the American Heart Association. Circulation 2010, 121, 2331–2378. [Google Scholar]
- Franklin, B.A.; Brook, R.; Pope, C.A., 3rd. Air pollution and cardiovascular disease. Curr. Probl. Cardiol. 2015, 40, 207–238. [Google Scholar] [CrossRef]
- Newby, D.E.; Mannucci, P.M.; Tell, G.S.; Baccarelli, A.A.; Brook, R.D.; Donaldson, K.; Forastiere, F.; Franchini, M.; Franco, O.H.; Graham, I.; et al. ESC Working Group on Thrombosis, European Association for Cardiovascular Prevention and Rehabilitation; ESC Heart Failure Association. Expert position paper on air pollution and cardiovascular disease. Eur. Heart J. 2015, 36, 83–93. [Google Scholar] [CrossRef] [Green Version]
- Hennig, F.; Geisel, M.H.; Kälsch, H.; Lucht, S.; Mahabadi, A.A.; Moebus, S.; Erbel, R.; Lehmann, N.; Jöckel, K.H.; Scherag, A.; et al. Heinz Nixdorf Recall Study Investigative Group. Air Pollution and Progression of Atherosclerosis in Different Vessel Beds-Results from a Prospective Cohort Study in the Ruhr Area, Germany. Environ. Health Perspect. 2020, 128, 107003. [Google Scholar] [CrossRef]
- Jacobs, L.; Emmerechts, J.; Hoylaerts, M.F.; Mathieu, C.; Hoet, P.H.; Nemery, B.; Nawrot, T.S. Traffic air pollution and oxidized LDL. PLoS ONE 2016, 6, e16200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemmar, A.; Hoet, P.H.; Dinsdale, D.; Vermylen, J.; Hoylaerts, M.F.; Nemery, B. Diesel exhaust particles in lung acutely enhance experimental peripheral thrombosis. Circulation 2003, 107, 202–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemmar, A.; Vanbilloen, H.; Hoylaerts, M.F.; Hoet, P.H.; Verbruggen, A.; Nemery, B. Passage of intratracheally instilled ultrafine particles from the lung into the systemic circulation in hamster. Am. J. Respir. Crit. Care Med. 2001, 64, 1665–1668. [Google Scholar] [CrossRef] [PubMed]
- Nemmar, A.; Hoet, P.H.; Vanquickenborne, B.; Dinsdale, D.; Thomeer, M.; Hoylaerts, M.F.; Vanbilloen, H.; Mortelmans, L.; Nemery, B. Passage of inhaled particles into the blood circulation in humans. Circulation 2002, 105, 411–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemmar, A.; Nemery, B.; Hoet, P.H.; Vermylen, J.; Hoylaerts, M.F. Pulmonary inflammation and thrombogenicity caused by diesel particles in hamsters: Role of histamine. Am. J. Respir. Crit. Care Med. 2003, 168, 1366–1372. [Google Scholar] [CrossRef] [PubMed]
- Nemmar, A.; Hoylaerts, M.F.; Hoet, P.H.; Vermylen, J.; Nemery, B. Size effect of intratracheally instilled particles on pulmonary inflammation and vascular thrombosis. Toxicol. Appl. Pharmacol. 2003, 186, 38–45. [Google Scholar] [CrossRef]
- Burger, P.C.; Wagner, D.D. Platelet P-selectin facilitates atherosclerotic lesion development. Platelet P-selectin facilitates atherosclerotic lesion development. Blood 2003, 101, 2661–2666. [Google Scholar] [CrossRef] [Green Version]
- Theilmeier, G.; Lenaerts, T.; Remacle, C.; Collen, D.; Vermylen, J.; Hoylaerts, M.F. Circulating activated platelets assist THP-1 monocytoid/endothelial cellinteraction under shear stress. Blood 1999, 94, 2725–2734. [Google Scholar] [CrossRef]
- Nemmar, A.; Hoet, P.H.; Vandervoort, P.; Dinsdale, D.; Nemery, B.; Hoylaerts, M.F. Enhanced peripheral thrombogenicity after lung inflammation is mediated by platelet-leukocyte activation: Role of P-selectin. J. Thromb. Haemost. 2007, 5, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Steinhoff, M.; Buddenkotte, J.; Shpacovitch, V.; Rattenholl, A.; Moormann, C.; Vergnolle, N.; Luger, T.A.; Hollenberg, M.D. Proteinase-activated receptors: Transducers of proteinase-mediated signaling in inflammation and immune response. Endocr. Rev. 2005, 26, 1–43. [Google Scholar] [CrossRef] [Green Version]
- Rabhi-Sabile, S.; de Romeuf, C.; Pidard, D. On the mechanism of plasmin-induced aggregation of human platelets: Implication of secreted von Willebrand factor. Thromb. Haemost. 1998, 79, 1191–1198. [Google Scholar] [CrossRef]
- Quinton, T.M.; Kim, S.; Derian, C.K.; Jin, J.; Kunapuli, S.P. Plasmin-mediated activation of platelets occurs by cleavage of protease-activated receptor 4. J. Biol. Chem. 2004, 279, 18434–18439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asfaha, S.; Cenac, N.; Houle, S.; Altier, C.; Papez, M.D.; Nguyen, C.; Steinhoff, M.; Chapman, K.; Zamponi, G.W.; Vergnolle, N. Protease-activated receptor-4: A novel mechanism of inflammatory pain modulation. Br. J. Pharmacol. 2007, 150, 176–185. [Google Scholar] [CrossRef] [Green Version]
- Cottrell, G.S.; Amadesi, S.; Grady, E.F.; Bunnett, N.W. Trypsin IV, a novel agonist of protease-activated receptors 2 and 4. J. Biol. Chem. 2004, 279, 13532–13539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renesto, P.; Chignard, M. Neutrophil-mediated platelet activation: A key role for serine proteinases. Gen. Pharmacol. 1995, 26, 905–910. [Google Scholar] [CrossRef]
- Selak, M.A.; Chignard, M.; Smith, J.B. Cathepsin G is a strong platelet agonist released by neutrophils. Biochem. J. 1988, 251, 293–299. [Google Scholar] [CrossRef] [Green Version]
- Si-Tahar, M.; Pidard, D.; Balloy, V.; Moniatte, M.; Kieffer, N.; Van Dorsselaer, A.; Chignard, M. Human neutrophil elastase proteolytically activates the platelet integrin alphaIIbbeta3 through cleavage of the carboxyl terminus of the alphaIIb subunit heavy chain. Involvement in the potentiation of platelet aggregation. J. Biol. Chem. 1997, 272, 11636–11647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selak, M.A. Neutrophil elastase potentiates cathepsin G-induced platelet activation. Thromb. Haemost. 1992, 68, 570–576. [Google Scholar]
- Evangelista, V.; Piccardoni, P.; White, J.G.; de Gaetano, G.; Cerletti, C. Cathepsin G-dependent platelet stimulation by activated polymorphonuclear leukocytes and its inhibition by antiproteinases: Role of P-selectin-mediated cell–cell adhesion. Blood 1993, 81, 2947–2957. [Google Scholar] [CrossRef] [Green Version]
- Evangelista, V.; Rajtar, G.; de Gaetano, G.; White, J.G.; Cerletti, C. Platelet activation by fMLP-stimulated polymorphonuclear leukocytes: The activity of cathepsin G is not prevented by antiproteinases. Blood 1991, 77, 2379–2388. [Google Scholar] [CrossRef] [Green Version]
- Sambrano, G.R.; Huang, W.; Faruqi, T.; Mahrus, S.; Craik, C.; Coughlin, S.R. Cathepsin G activates protease-activated receptor-4 in human platelets. J. Biol. Chem. 2000, 275, 6819–6823. [Google Scholar] [CrossRef] [Green Version]
- Molino, M.; Blanchard, N.; Belmonte, E.; Tarver, A.P.; Abrams, C.; Hoxie, J.A.; Cerletti, C.; Brass, L.F. Proteolysis of the human platelet and endothelial cell thrombin receptor by neutrophil-derived cathepsin G. J. Biol. Chem. 1995, 270, 11168–11175. [Google Scholar] [CrossRef] [Green Version]
- Si-Tahar, M.; Renesto, P.; Falet, H.; Rendu, F.; Chignard, M. The phospholipase C/protein kinase C pathway is involved in cathepsin G-induced human platelet activation: Comparison with thrombin. Biochem. J. 1996, 313 Pt 2, 401–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faraday, N.; Schunke, K.; Saleem, S.; Fu, J.; Wang, B.; Zhang, J.; Morrell, C.; Dore, S. Cathepsin G-dependent modulation of platelet thrombus formation in vivo by blood neutrophils. PLoS ONE 2013, 8, e71447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leger, A.J.; Jacques, S.L.; Badar, J.; Kaneider, N.C.; Derian, C.K.; Andrade-Gordon, P.; Covic, L.; Kuliopulos, A. Blocking the protease-activated receptor 1–4 heterodimer in platelet-mediated thrombosis. Circulation 2006, 113, 1244–1254. [Google Scholar] [CrossRef] [Green Version]
- Franchini, M.; Veneri, D.; Lippi, G. Inflammation and hemostasis: A bidirectional interaction. Clin. Lab. 2007, 53, 63–67. [Google Scholar]
- Molino, M.; Lallo, M.D.; de Gaetano, G.; Cerletti, C. Intracellular Ca2+ rise in human platelets induced by polymorphonuclear-leucocyte-derived cathepsin G. Biochem. J. 1992, 288 Pt 3, 741–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrin, J.; Lecompte, T.; Tournier, A.; Morlon, L.; Marchand-Arvier, M.; Vigneron, C. In vitro effects of human neutrophil cathepsin G on thrombin generation: Both acceleration and decreased potential. Thromb. Haemost. 2010, 104, 514–522. [Google Scholar]
- Björk, P.; Axelsson, L.; Ohlsson, K. Release of dog polymorphonuclear leukocyte cathepsin G, normally and in endotoxin and pancreatitic shock. Isolation and partial characterization of dog polymorphonuclear leukocyte cathepsin G. Biol. Chem. Hoppe Seyler 1991, 372, 419–426. [Google Scholar] [CrossRef]
- Nakanishi-Matsui, M.; Zheng, Y.W.; Sulciner, D.J.; Weiss, E.J.; Ludeman, M.J.; Coughlin, S.R. PAR3 is a cofactor for PAR4 activation by thrombin. Nature 2000, 404, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Anand, A.; Feffer, S.E. Hematocrit and bleeding time: An update. South. Med. J. 1994, 87, 299–301. [Google Scholar] [CrossRef]
- Valles, J.; Santos, M.T.; Aznar, J.; Marcus, A.J.; Martinez-Sales, V.; Portoles, M.; Broekman, M.J.; Safier, L.B. Erythrocytes metabolically enhance collagen-induced platelet responsiveness via increased thromboxane production, adenosine diphosphate release, and recruitment. Blood 1991, 78, 154–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bykowska, K.; Duk, M.; Kusnierz-Alejska, G.; Kopec, M.; Lisowska, E. Degradation of human erythrocyte surface components by human neutrophil elastase and cathepsin G: Preferential digestion of glycophorins. Br. J. Haematol. 1993, 84, 736–742. [Google Scholar] [CrossRef]
- Bykowska, K.; Duk, M.; Kusnierz-Alejska, G.; Sikorska, A.; Letowska, M.; Mendek-Czajkowska, E.; Lopaciuk, S.; Kopec, M.; Lisowska, E. Degradation of glycophorin A of human erythrocytes in patients with myelo-or lymphoproliferative disorders: Possible role of neutrophil proteases. Br. J. Haematol. 1997, 96, 514–520. [Google Scholar] [CrossRef]
- Born, G.V.; Wehmeier, A. Inhibition of platelet thrombus formation by chlorpromazine acting to diminish haemolysis. Nature 1979, 282, 212–213. [Google Scholar] [CrossRef]
- Helms, C.C.; Marvel, M.; Zhao, W.; Stahle, M.; Vest, R.; Kato, G.J.; Lee, J.S.; Christ, G.; Gladwin, M.T.; Hantgan, R.R.; et al. Mechanisms of hemolysis-associated platelet activation. J. Thromb. Haemost. 2013, 11, 2148–2154. [Google Scholar] [CrossRef] [Green Version]
- Damiano, B.P.; Derian, C.K.; Maryanoff, B.E.; Zhang, H.C.; Gordon, P.A. RWJ-58259: A selective antagonist of protease activated receptor-1. Cardiovasc. Drug Rev. 2003, 21, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Hwang, T.L.; Liao, C.H.; Kuo, S.C.; Lee, F.Y.; Lee, C.Y.; Teng, C.M. Selective inhibition of protease-activated receptor 4-dependent platelet activation by YD-3. Thromb. Haemost. 2002, 87, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Stockmans, F.; Stassen, J.M.; Vermylen, J.; Hoylaerts, M.F.; Nystrom, A. A technique to investigate mural thrombus formation in small arteries and veins: I. Comparative morphometric and histological analysis. Ann. Plast. Surg. 1997, 38, 56–62. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nemmar, A.; Hoylaerts, M.F. Neutrophil Cathepsin G Enhances Thrombogenicity of Mildly Injured Arteries via ADP-Mediated Platelet Sensitization. Int. J. Mol. Sci. 2022, 23, 744. https://doi.org/10.3390/ijms23020744
Nemmar A, Hoylaerts MF. Neutrophil Cathepsin G Enhances Thrombogenicity of Mildly Injured Arteries via ADP-Mediated Platelet Sensitization. International Journal of Molecular Sciences. 2022; 23(2):744. https://doi.org/10.3390/ijms23020744
Chicago/Turabian StyleNemmar, Abderrahim, and Marc F. Hoylaerts. 2022. "Neutrophil Cathepsin G Enhances Thrombogenicity of Mildly Injured Arteries via ADP-Mediated Platelet Sensitization" International Journal of Molecular Sciences 23, no. 2: 744. https://doi.org/10.3390/ijms23020744
APA StyleNemmar, A., & Hoylaerts, M. F. (2022). Neutrophil Cathepsin G Enhances Thrombogenicity of Mildly Injured Arteries via ADP-Mediated Platelet Sensitization. International Journal of Molecular Sciences, 23(2), 744. https://doi.org/10.3390/ijms23020744