
Hypoxia Inhibits Subretinal Inflammation Resolution Thrombospondin-1 Dependently


 , ,
, ,  and
and 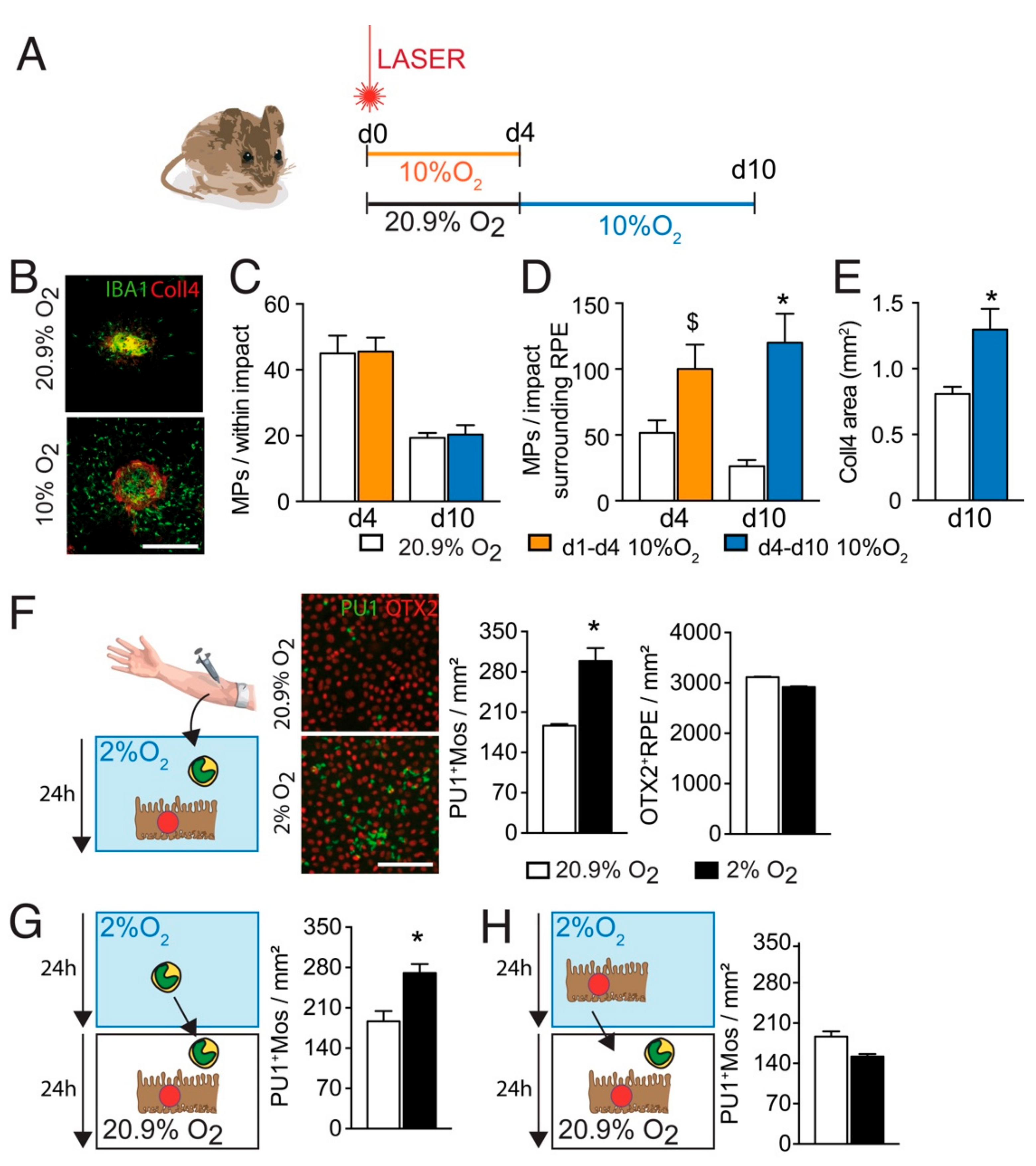{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
2.1. Laser-Injury Model, Intravitreal Injections, and Hypoxia
2.2. Monocyte RPE Co-Cultures
2.3. Gene Expression Analysis
2.4. Immunohistochemistry, CNV and MPs Quantification
2.5. Statistical Analyses
3. Results
3.1. Hypoxia Increases the Infiltration of Subretinal MPs after Laser-Injury
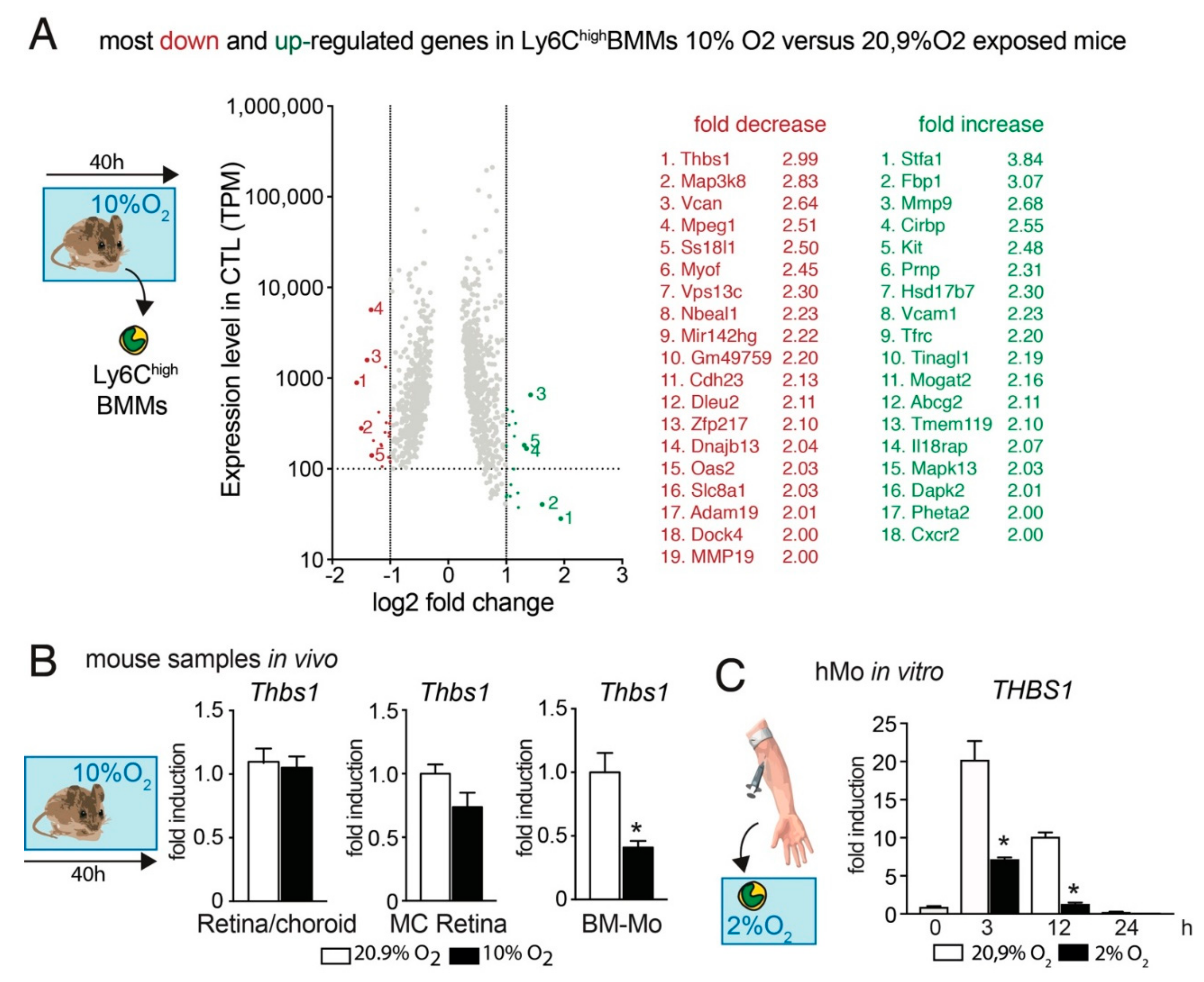
3.2. Hypoxia Decreases Thbs1-Expression in Mos
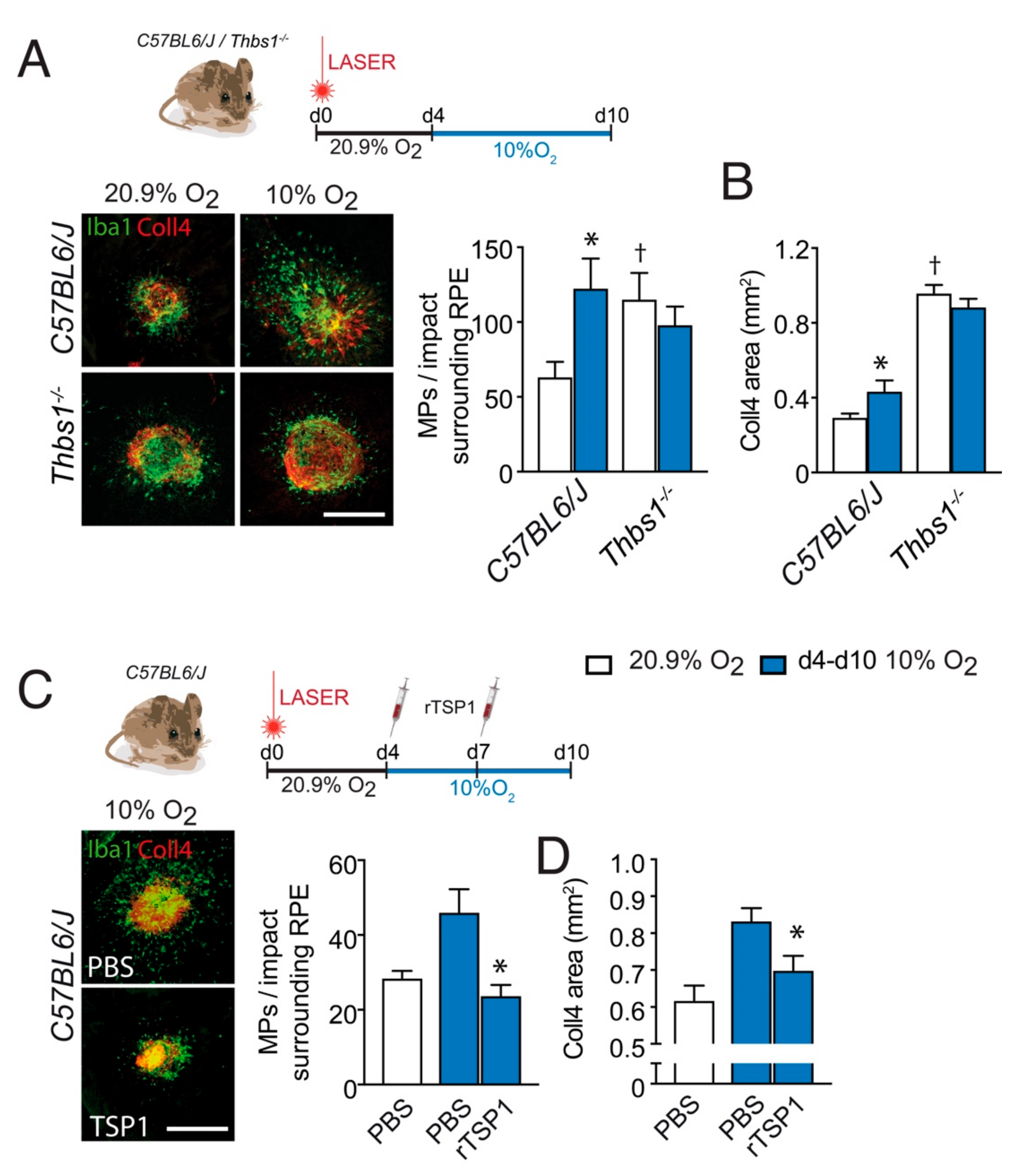
3.3. TSP-1 Is Necessary for Hypoxia-Induced Inhibition of Inflammation Resolution after Laser-Injury
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Sarks, S.H. Ageing and degeneration in the macular region: A clinico-pathological study. Br. J. Ophthalmol. 1976, 60, 324–341. [Google Scholar] [CrossRef]
- Guillonneau, X.; Eandi, C.M.; Paques, M.; Sahel, J.A.; Sapieha, P.; Sennlaub, F. On phagocytes and macular degeneration. Prog. Retin Eye Res. 2017, 61, 98–128. [Google Scholar] [CrossRef]
- Sennlaub, F.; Auvynet, C.; Calippe, B.; Lavalette, S.; Poupel, L.; Hu, S.J.; Dominguez, E.; Camelo, S.; Levy, O.; Guyon, E.; et al. CCR2(+) monocytes infiltrate atrophic lesions in age-related macular disease and mediate photoreceptor degeneration in experimental subretinal inflammation in Cx3cr1 deficient mice. EMBO Mo.l Med. 2013, 5, 1775–1793. [Google Scholar] [CrossRef]
- Calippe, B.; Augustin, S.; Beguier, F.; Charles-Messance, H.; Poupel, L.; Conart, J.B.; Hu, S.J.; Lavalette, S.; Fauvet, A.; Rayes, J.; et al. Complement Factor H Inhibits CD47-Mediated Resolution of Inflammation. Immunity 2017, 46, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Beguier, F.; Housset, M.; Roubeix, C.; Augustin, S.; Zagar, Y.; Nous, C.; Mathis, T.; Eandi, C.; Benchaboune, M.; Drame-Maigne, A.; et al. The 10q26 Risk Haplotype of Age-Related Macular Degeneration Aggravates Subretinal Inflammation by Impairing Monocyte Elimination. Immunity 2020, 53, 429–441.e8. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.F.; Turpie, B.; Masli, S. Thrombospondin-1-mediated regulation of microglia activation after retinal injury. Investig. Ophthalmol. Vis. Sci. 2009, 50, 5472–5478. [Google Scholar] [CrossRef][Green Version]
- Wang, S.; Sorenson, C.M.; Sheibani, N. Lack of thrombospondin 1 and exacerbation of choroidal neovascularization. Arch. Ophthalmol. 2012, 130, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Blasiak, J.; Petrovski, G.; Vereb, Z.; Facsko, A.; Kaarniranta, K. Oxidative stress, hypoxia, and autophagy in the neovascular processes of age-related macular degeneration. Biomed Res. Int. 2014, 2014, 768026. [Google Scholar] [CrossRef]
- Stefansson, E.; Geirsdottir, A.; Sigurdsson, H. Metabolic physiology in age related macular degeneration. Prog Retin Eye Res 2011, 30, 72–80. [Google Scholar] [CrossRef]
- Chakravarthy, U.; Wong, T.Y.; Fletcher, A.; Piault, E.; Evans, C.; Zlateva, G.; Buggage, R.; Pleil, A.; Mitchell, P. Clinical risk factors for age-related macular degeneration: A systematic review and meta-analysis. BMC Ophthalmol. 2010, 10, 31. [Google Scholar] [CrossRef]
- Klein, R.; Klein, B.E.; Tomany, S.C.; Cruickshanks, K.J. The association of cardiovascular disease with the long-term incidence of age-related maculopathy: The Beaver Dam Eye Study. Ophthalmology 2003, 110, 1273–1280. [Google Scholar] [CrossRef]
- Feigl, B. Age-related maculopathy-linking aetiology and pathophysiological changes to the ischaemia hypothesis. Prog. Retin Eye Res. 2009, 28, 63–86. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef]
- Lavalette, S.; Raoul, W.; Houssier, M.; Camelo, S.; Levy, O.; Calippe, B.; Jonet, L.; Behar-Cohen, F.; Chemtob, S.; Guillonneau, X.; et al. Interleukin-1beta inhibition prevents choroidal neovascularization and does not exacerbate photoreceptor degeneration. Am. J. Pathol. 2011, 178, 2416–2423. [Google Scholar] [CrossRef]
- Levy, O.; Calippe, B.; Lavalette, S.; Hu, S.J.; Raoul, W.; Dominguez, E.; Housset, M.; Paques, M.; Sahel, J.A.; Bemelmans, A.P.; et al. Apolipoprotein E promotes subretinal mononuclear phagocyte survival and chronic inflammation in age-related macular degeneration. EMBO Mol. Med. 2015, 7, 211–226. [Google Scholar] [CrossRef] [PubMed]
- Mathis, T.; Housset, M.; Eandi, C.; Beguier, F.; Touhami, S.; Reichman, S.; Augustin, S.; Gondouin, P.; Sahel, J.A.; Kodjikian, L.; et al. Activated monocytes resist elimination by retinal pigment epithelium and downregulate their OTX2 expression via TNF-alpha. Aging Cell 2017, 16, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Touhami, S.; Beguier, F.; Augustin, S.; Charles-Messance, H.; Vignaud, L.; Nandrot, E.F.; Reichman, S.; Forster, V.; Mathis, T.; Sahel, J.A.; et al. Chronic exposure to tumor necrosis factor alpha induces retinal pigment epithelium cell dedifferentiation. J. Neuroinflammation 2018, 15, 85. [Google Scholar] [CrossRef]
- Charles-Messance, H.; Blot, G.; Couturier, A.; Vignaud, L.; Touhami, S.; Beguier, F.; Siqueiros, L.; Forster, V.; Barmo, N.; Augustin, S.; et al. IL-1beta induces rod degeneration through the disruption of retinal glutamate homeostasis. J. Neuroinflammation 2020, 17, 1. [Google Scholar] [CrossRef] [PubMed]
- Levy, O.; Lavalette, S.; Hu, S.J.; Housset, M.; Raoul, W.; Eandi, C.; Sahel, J.A.; Sullivan, P.M.; Guillonneau, X.; Sennlaub, F. APOE-isoforms control pathogenic subretinal inflammation in age related macular degeneration. J. Neurosci. 2015, 35, 13568–13576. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Dyer, N.P.; Shahrezaei, V.; Hebenstreit, D. LiBiNorm: An htseq-count analogue with improved normalisation of Smart-seq2 data and library preparation diagnostics. PeerJ 2019, 7, e6222. [Google Scholar] [CrossRef] [PubMed]
- Caicedo, A.; Espinosa-Heidmann, D.G.; Pina, Y.; Hernandez, E.P.; Cousins, S.W. Blood-derived macrophages infiltrate the retina and activate Muller glial cells under experimental choroidal neovascularization. Exp. Eye Res. 2005, 81, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, E.; Anand, A.; Ambati, B.K.; van Rooijen, N.; Ambati, J. Macrophage depletion inhibits experimental choroidal neovascularization. Investig. Ophthalmol. Vis. Sci. 2003, 44, 3578–3585. [Google Scholar] [CrossRef]
- Tsutsumi, C.; Sonoda, K.H.; Egashira, K.; Qiao, H.; Hisatomi, T.; Nakao, S.; Ishibashi, M.; Charo, I.F.; Sakamoto, T.; Murata, T.; et al. The critical role of ocular-infiltrating macrophages in the development of choroidal neovascularization. J. Leukoc. Biol. 2003, 74, 25–32. [Google Scholar] [CrossRef]
- Luhmann, U.F.; Robbie, S.; Munro, P.M.; Barker, S.E.; Duran, Y.; Luong, V.; Fitzke, F.W.; Bainbridge, J.; Ali, R.R.; Maclaren, R. The drusen-like phenotype in aging Ccl2 knockout mice is caused by an accelerated accumulation of swollen autofluorescent subretinal macrophages. Investig. Ophthalmol. Vis. Sci. 2009, 50, 5934–5943. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Copland, D.A.; Horie, S.; Wu, W.K.; Chen, M.; Xu, Y.; Paul Morgan, B.; Mack, M.; Xu, H.; Nicholson, L.B.; et al. Myeloid cells expressing VEGF and arginase-1 following uptake of damaged retinal pigment epithelium suggests potential mechanism that drives the onset of choroidal angiogenesis in mice. PLoS ONE 2013, 8, e72935. [Google Scholar] [CrossRef]
- Robbie, S.J.; Georgiadis, A.; Barker, S.E.; Duran, Y.; Smith, A.J.; Ali, R.R.; Luhmann, U.F.; Bainbridge, J.W. Enhanced Ccl2-Ccr2 signaling drives more severe choroidal neovascularization with aging. Neurobiol. Aging 2016, 40, 110–119. [Google Scholar] [CrossRef]
- Sakurai, E.; Taguchi, H.; Anand, A.; Ambati, B.K.; Gragoudas, E.S.; Miller, J.W.; Adamis, A.P.; Ambati, J. Targeted disruption of the CD18 or ICAM-1 gene inhibits choroidal neovascularization. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2743–2749. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.Y.; Shimoda, L.A.; Iyer, N.V.; Huso, D.L.; Sun, X.; McWilliams, R.; Beaty, T.; Sham, J.S.; Wiener, C.M.; Sylvester, J.T.; et al. Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1alpha. J. Clin. Investig. 1999, 103, 691–696. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, C.; Muthana, M.; Lewis, C.E. Hypoxia regulates macrophage functions in inflammation. J. Immunol. 2005, 175, 6257–6263. [Google Scholar] [CrossRef]
- Lewis, J.S.; Lee, J.A.; Underwood, J.C.; Harris, A.L.; Lewis, C.E. Macrophage responses to hypoxia: Relevance to disease mechanisms. J. Leukoc. Biol. 1999, 66, 889–900. [Google Scholar] [CrossRef]
- Rahat, M.A.; Bitterman, H.; Lahat, N. Molecular mechanisms regulating macrophage response to hypoxia. Front/ Immunol/ 2011, 2, 45. [Google Scholar] [CrossRef]
- Ausserer, W.A.; Bourrat-Floeck, B.; Green, C.J.; Laderoute, K.R.; Sutherland, R.M. Regulation of c-jun expression during hypoxic and low-glucose stress. Mol. Cell. Biol. 1994, 14, 5032–5042. [Google Scholar] [PubMed]
- Mettouchi, A.; Cabon, F.; Montreau, N.; Vernier, P.; Mercier, G.; Blangy, D.; Tricoire, H.; Vigier, P.; Binetruy, B. SPARC and thrombospondin genes are repressed by the c-jun oncogene in rat embryo fibroblasts. EMBO J. 1994, 13, 5668–5678. [Google Scholar] [CrossRef]
- Keenan, T.D.; Goldacre, R.; Goldacre, M.J. Associations between obstructive sleep apnoea, primary open angle glaucoma and age-related macular degeneration: Record linkage study. Br. J. Ophthalmol. 2017, 101, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Jurado-Gamez, B.; Gomez-Chaparro, J.L.; Munoz-Calero, M.; Serna Sanz, A.; Munoz-Cabrera, L.; Lopez-Barea, J.; Gozal, D. Serum proteomic changes in adults with obstructive sleep apnoea. J. Sleep Res. 2012, 21, 139–146. [Google Scholar] [CrossRef]
- Pack, A.I.; Gislason, T. Obstructive sleep apnea and cardiovascular disease: A perspective and future directions. Prog. Cardiovasc. Dis. 2009, 51, 434–451. [Google Scholar] [CrossRef]
- Daulatzai, M.A. Evidence of neurodegeneration in obstructive sleep apnea: Relationship between obstructive sleep apnea and cognitive dysfunction in the elderly. J. Neurosci. Res. 2015, 93, 1778–1794. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Touhami, S.; Béguier, F.; Yang, T.; Augustin, S.; Roubeix, C.; Blond, F.; Conart, J.B.; Sahel, J.A.; Bodaghi, B.; Delarasse, C.; et al. Hypoxia Inhibits Subretinal Inflammation Resolution Thrombospondin-1 Dependently. Int. J. Mol. Sci. 2022, 23, 681. https://doi.org/10.3390/ijms23020681
Touhami S, Béguier F, Yang T, Augustin S, Roubeix C, Blond F, Conart JB, Sahel JA, Bodaghi B, Delarasse C, et al. Hypoxia Inhibits Subretinal Inflammation Resolution Thrombospondin-1 Dependently. International Journal of Molecular Sciences. 2022; 23(2):681. https://doi.org/10.3390/ijms23020681
Chicago/Turabian StyleTouhami, Sara, Fanny Béguier, Tianxiang Yang, Sébastien Augustin, Christophe Roubeix, Frederic Blond, Jean Baptiste Conart, José Alain Sahel, Bahram Bodaghi, Cécile Delarasse, and et al. 2022. "Hypoxia Inhibits Subretinal Inflammation Resolution Thrombospondin-1 Dependently" International Journal of Molecular Sciences 23, no. 2: 681. https://doi.org/10.3390/ijms23020681
APA StyleTouhami, S., Béguier, F., Yang, T., Augustin, S., Roubeix, C., Blond, F., Conart, J. B., Sahel, J. A., Bodaghi, B., Delarasse, C., Guillonneau, X., & Sennlaub, F. (2022). Hypoxia Inhibits Subretinal Inflammation Resolution Thrombospondin-1 Dependently. International Journal of Molecular Sciences, 23(2), 681. https://doi.org/10.3390/ijms23020681