
Signatures of Co-Deregulated Genes and Their Transcriptional Regulators in Lung Cancer



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
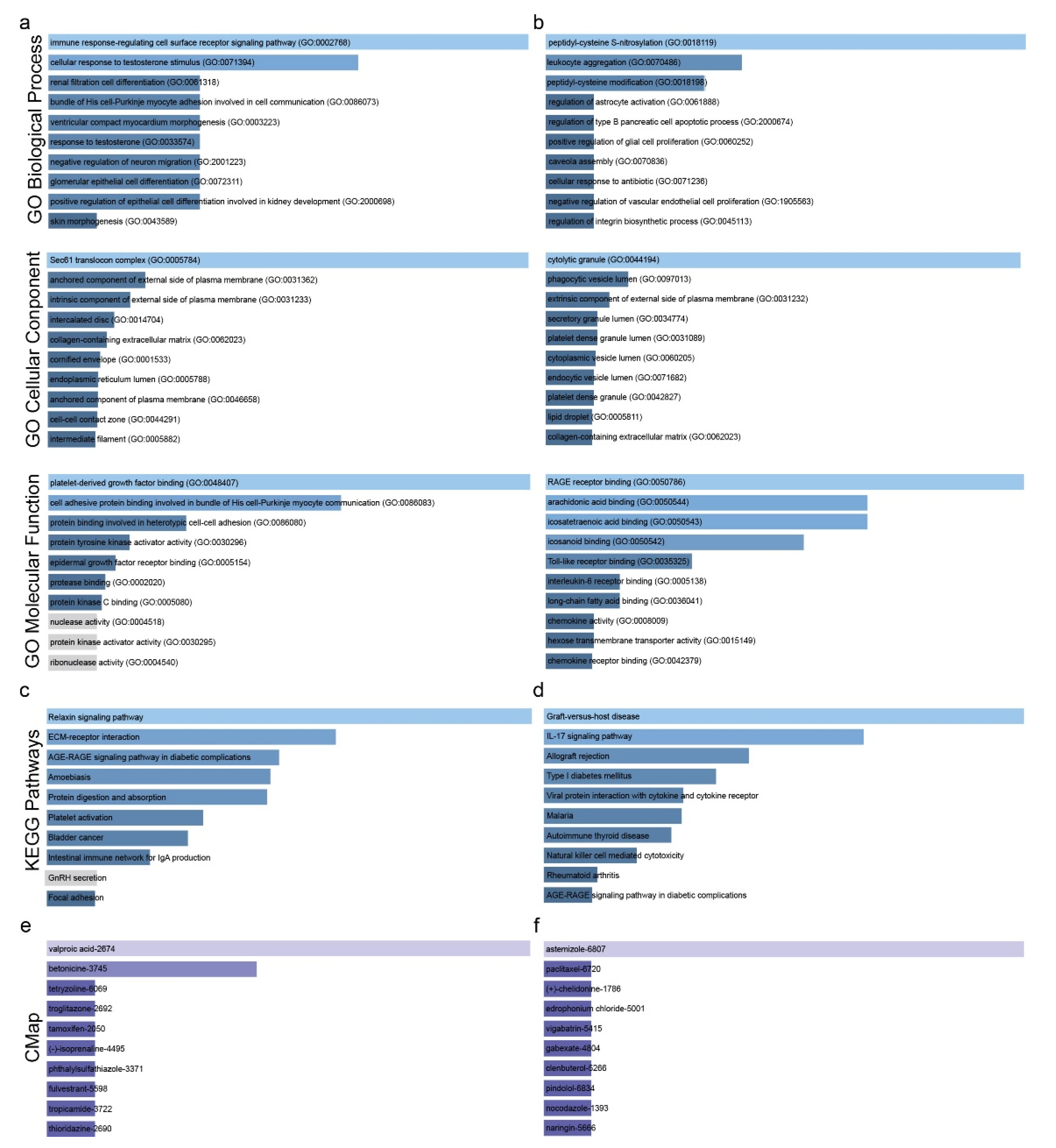
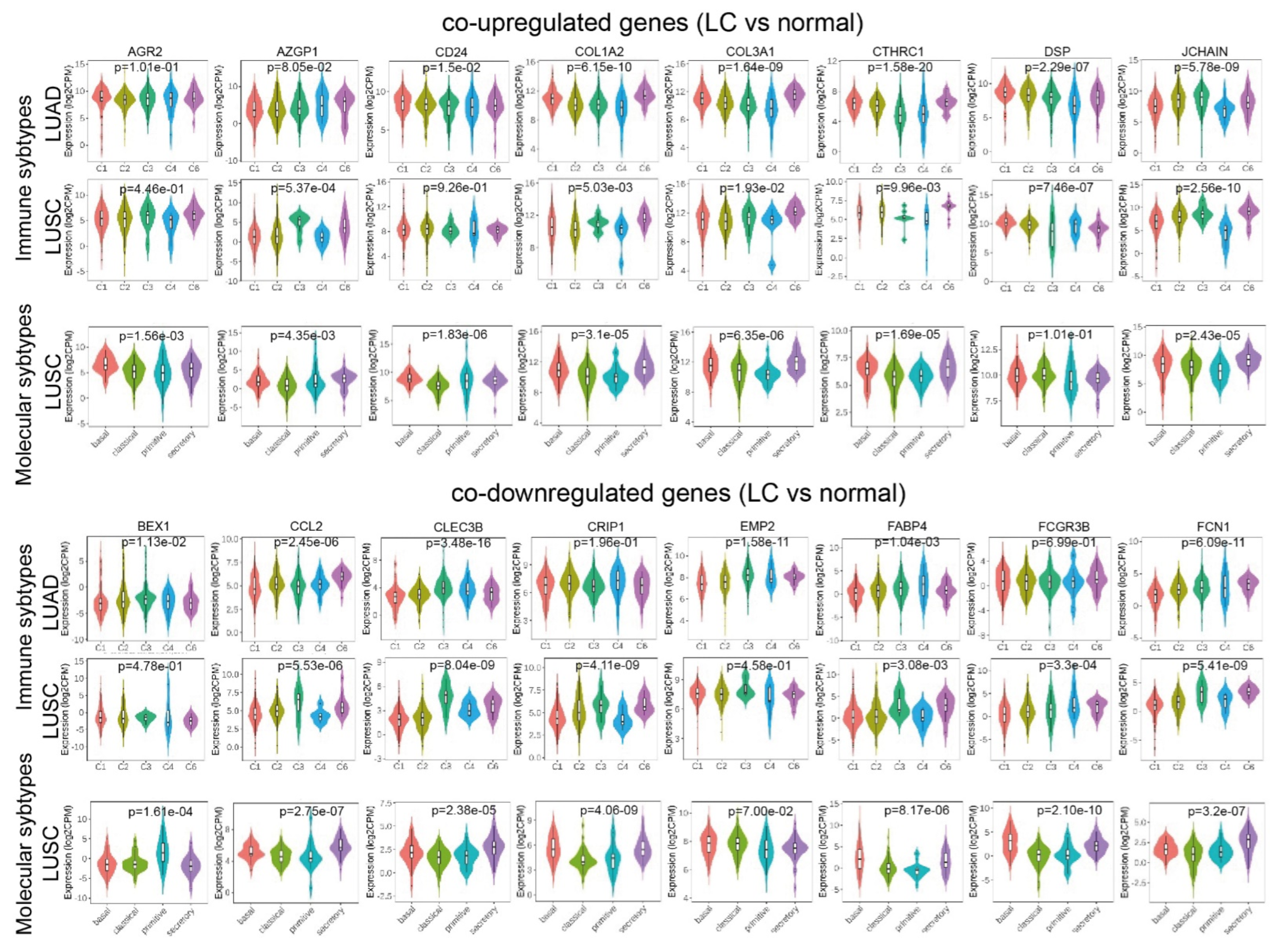
2.1. Co-Deregulated Genes in Lung Cancers
2.2. Drug Repurposing against Lung Cancer
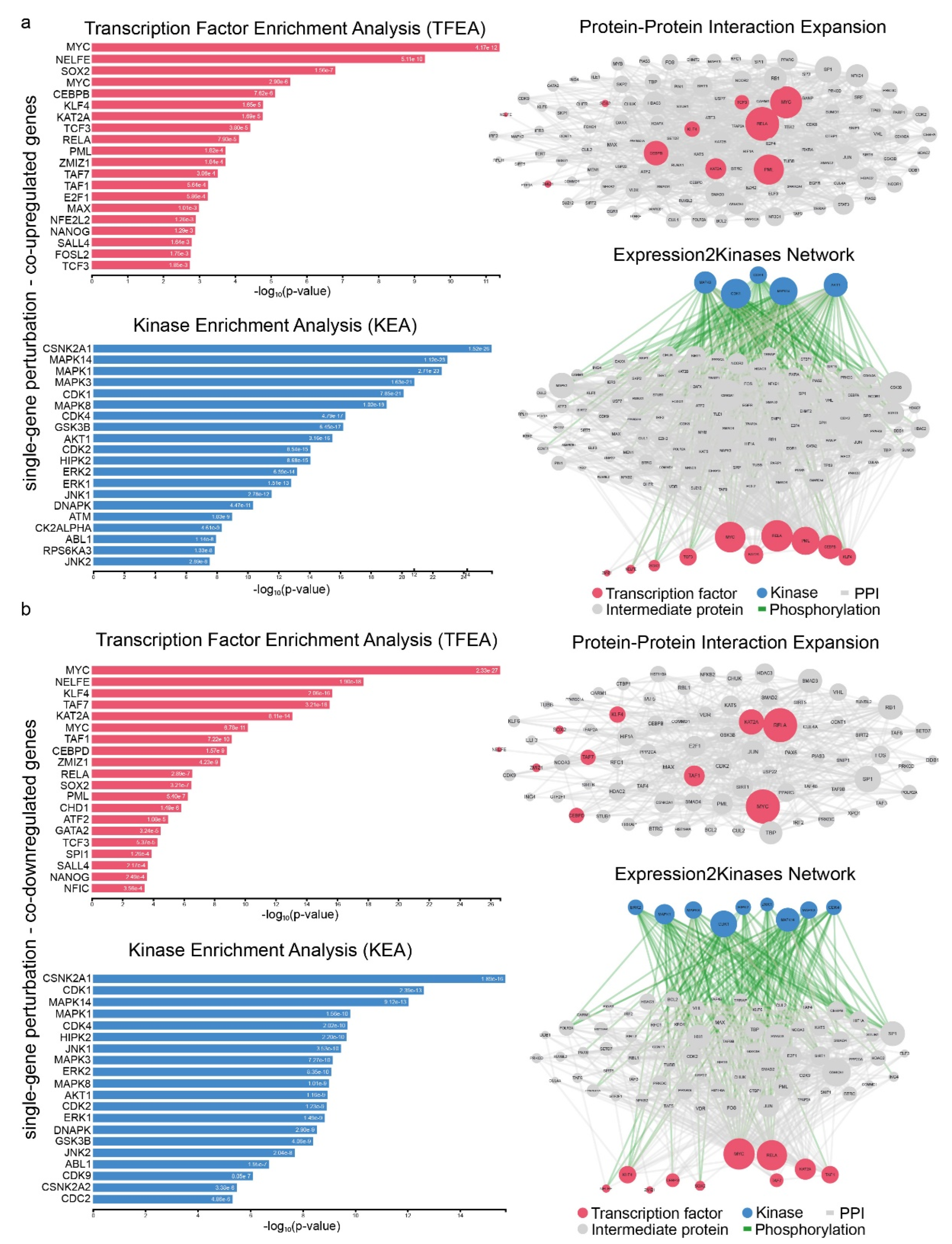
2.3. Signatures of Co-Deregulated Genes in Single-Gene Perturbation Experiments
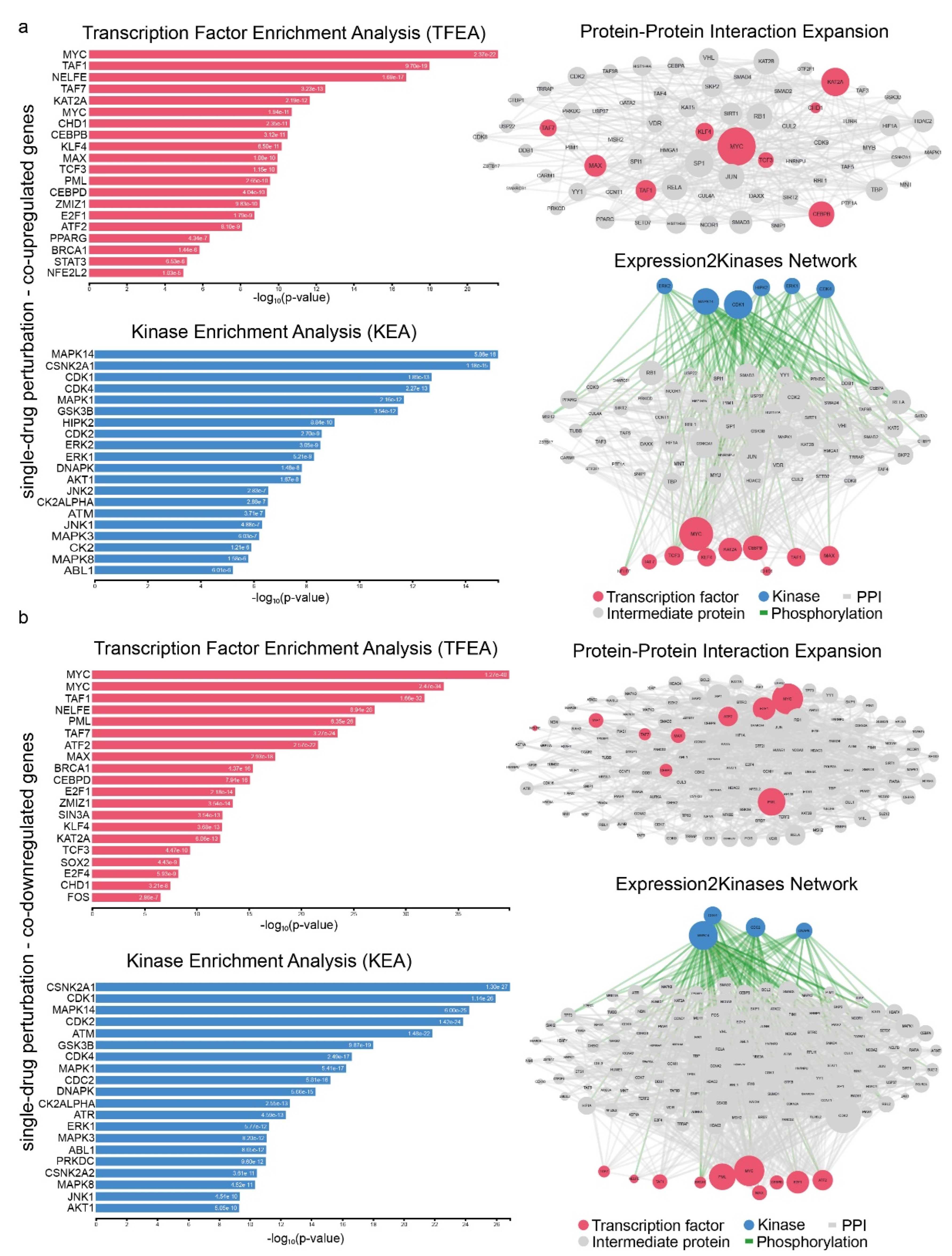
2.4. Signatures of Co-Deregulated Genes in Single-Drug Perturbation Experiments
3. Discussion
4. Materials and Methods
4.1. GEO Data Extraction and Filtering
4.2. Differentially Expressed Genes and Co-DEGs
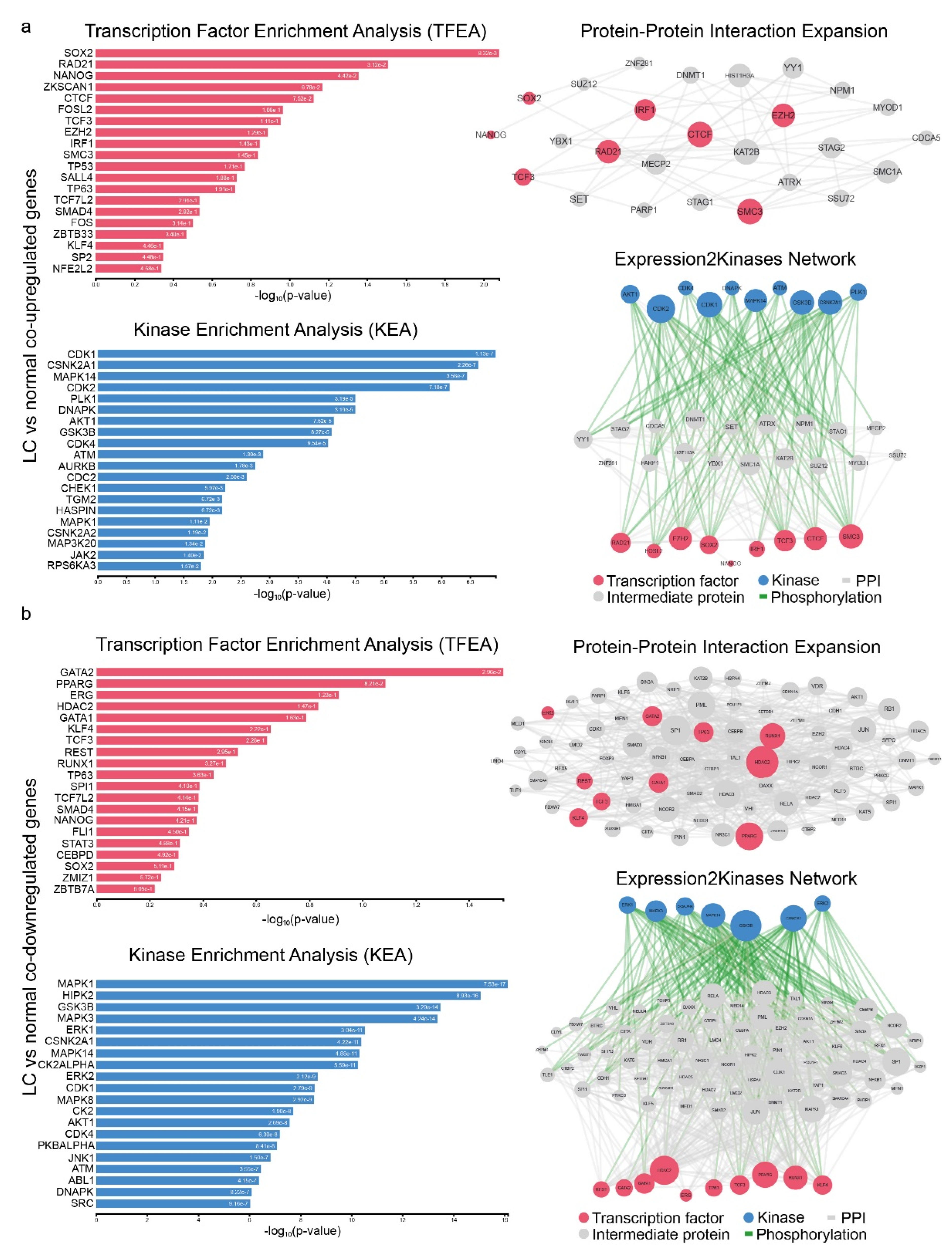
4.3. Upstream Regulators of co-DEGs and Protein–Protein Interaction Networks
4.4. GO and KEGG Enrichment Analysis
4.5. Connectivity Map Analysis
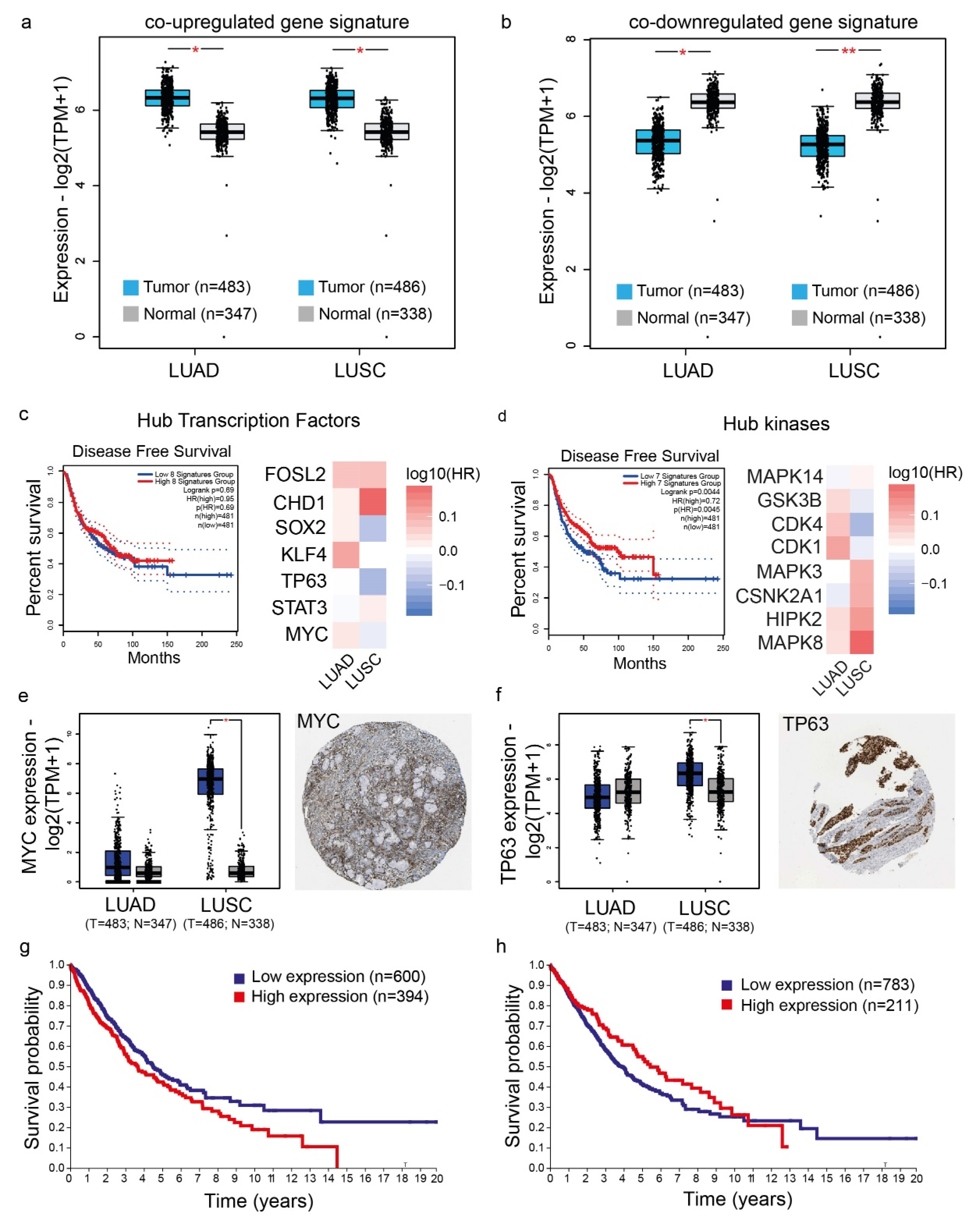
4.6. Validation of the Co-Deregulated Gene Signatures and Hub Genes in the TCGA and the Human Protein Atlas
4.7. Patient Survival Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Liu, W.-J.; Du, Y.; Wen, R.; Yang, M.; Xu, J. Drug resistance to targeted therapeutic strategies in non-small cell lung cancer. Pharmacol. Ther. 2019, 206, 107438. [Google Scholar] [CrossRef] [PubMed]
- Mastrogamvraki, N.; Zaravinos, A. Signatures of co-deregulated genes and their transcriptional regulators in colorectal cancer. NPJ Syst. Biol. Appl. 2020, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.; Peng, W.; Chen, Y.; Du, X.; Xia, J.; Shen, B.; Zhou, G. Four differentially expressed genes can predict prognosis and microenvironment immune infiltration in lung cancer: A study based on data from the GEO. BMC Cancer 2022, 22, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zengin, T.; Önal-Süzek, T. Analysis of genomic and transcriptomic variations as prognostic signature for lung adenocarcinoma. BMC Bioinform. 2020, 21 (Suppl. S14), 368. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Kong, W.; Huang, Z.; Xie, Z. Identification of key genes related to lung squamous cell carcinoma using bioinformatics analysis. Int. J. Mol. Sci. 2020, 21, 2994. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, X.; Liu, L.; Li, J.; Hu, Q.; Sun, R. Expression and Prognostic Significance of m6A-Related Genes in Lung Adenocarcinoma. Med Sci. Monit. 2020, 26, e919644. [Google Scholar] [CrossRef]
- Long, T.; Liu, Z.; Zhou, X.; Yu, S.; Tian, H.; Bao, Y. Identification of differentially expressed genes and enriched pathways in lung cancer using bioinformatics analysis. Mol. Med. Rep. 2019, 19, 2029–2040. [Google Scholar] [CrossRef]
- Li, R.; Liu, Y.; Wang, T.; Tang, J.; Xie, L.; Yao, Z.; Li, K.; Liao, Y.; Zhou, L.; Geng, Z.; et al. The characteristics of lung cancer in Xuanwei County: A review of differentially expressed genes and noncoding RNAs on cell proliferation and migration. Biomed. Pharmacother. 2019, 119, 109312. [Google Scholar] [CrossRef]
- Yu, H.; Pang, Z.; Li, G.; Gu, T. Bioinformatics analysis of differentially expressed miRNAs in non-small cell lung cancer. J. Clin. Lab. Anal. 2021, 35, e23588. [Google Scholar] [CrossRef]
- Tian, S. Identification of monotonically differentially expressed genes for non-small cell lung cancer. BMC Bioinform. 2019, 20, 177. [Google Scholar] [CrossRef] [PubMed]
- Clark, N.R.; Hu, K.S.; Feldmann, A.S.; Kou, Y.; Chen, E.Y.; Duan, Q.; Ma’Ayan, A. The characteristic direction: A geometrical approach to identify differentially expressed genes. BMC Bioinform. 2014, 15, 79. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.-H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef] [PubMed]
- Shults, N.V.; Seeherman, S.; Sariipek, N.E.; Rybka, V.; Marcocci, L.; Gychka, S.G.; Ibrahim, Y.F.; Suzuki, Y.J. Tau Protein in Lung Smooth Muscle Cells. J. Respir. 2020, 1, 30–39. [Google Scholar] [CrossRef]
- Papin, S.; Paganetti, P. Emerging Evidences for an Implication of the Neurodegeneration-Associated Protein TAU in Cancer. Brain Sci. 2020, 10, 862. [Google Scholar] [CrossRef] [PubMed]
- Gavrilovic, I.; Posner, J.B. Brain metastases: Epidemiology and pathophysiology. J. Neuro-Oncol. 2005, 75, 5–14. [Google Scholar] [CrossRef]
- Wang, G.; Xu, J.; Qi, Y.; Xiu, J.; Li, R.; Han, M. Distribution of Brain Metastasis from Lung Cancer. Cancer Manag. Res. 2019, 11, 9331–9338. [Google Scholar] [CrossRef]
- Pappot, H.; Brünner, N. The plasminogen activation system and its role in lung cancer. A review. Lung Cancer 1995, 12, 1–12. [Google Scholar] [CrossRef]
- Vallabhapurapu, S.D.; Blanco, V.M.; Sulaiman, M.K.; Vallabhapurapu, S.L.; Chu, Z.; Franco, R.S.; Qi, X. Variation in human cancer cell external phosphatidylserine is regulated by flippase activity and intracellular calcium. Oncotarget 2015, 6, 34375–34388. [Google Scholar] [CrossRef]
- Birge, R.B.; Boeltz, S.; Kumar, S.; Carlson, J.; Wanderley, J.; Calianese, D.; Barcinski, M.; Brekken, R.A.; Huang, X.; Hutchins, J.T.; et al. Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ. 2016, 23, 962–978. [Google Scholar] [CrossRef] [PubMed]
- Soon Nan, K.; Karuppanan, K.; Kumar, S.; Alam, S. Identification of common key genes and pathways between COVID-19 and lung cancer by using protein-protein interaction network analysis. bioRxiv 2021. [Google Scholar] [CrossRef]
- Kang, J.H.; Lee, S.-H.; Hong, D.; Lee, J.-S.; Ahn, H.-S.; Ahn, J.-H.; Seong, T.W.; Lee, C.-H.; Jang, H.; Hong, K.M.; et al. Aldehyde dehydrogenase is used by cancer cells for energy metabolism. Exp. Mol. Med. 2016, 48, e272. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Liu, Q.; Li, G.; Dong, F.; Qiu, M.; Lv, X.; Zhang, S.; Guo, Z. Identification of ubiquinol cytochrome c reductase hinge (UQCRH) as a potential diagnostic biomarker for lung adenocarcinoma. Open Biol. 2016, 6, 150256. [Google Scholar] [CrossRef] [PubMed]
- Cassandri, M.; Smirnov, A.; Novelli, F.; Pitolli, C.; Agostini, M.; Malewicz, M.; Melino, G.; Raschellà, G. Zinc-finger proteins in health and disease. Cell Death Discov. 2017, 3, 17071. [Google Scholar] [CrossRef]
- Yu, Y.; Tian, X. Analysis of genes associated with prognosis of lung adenocarcinoma based on GEO and TCGA databases. Medicine 2020, 99, e20183. [Google Scholar] [CrossRef] [PubMed]
- Fallowfield, J.A.; Hayden, A.L.; Snowdon, V.K.; Aucott, R.L.; Stutchfield, B.M.; Mole, D.J.; Pellicoro, A.; Gordon-Walker, T.T.; Henke, A.; Schrader, J.; et al. Relaxin modulates human and rat hepatic myofibroblast function and ameliorates portal hypertension in vivo. Hepatology 2014, 59, 1492–1504. [Google Scholar] [CrossRef]
- Wyatt, T.; Sisson, J.; Forgèt, M.; Bennett, R.; Hamel, F.; Spurzem, J. Relaxin Stimulates Bronchial Epithelial Cell PKA Activation, Migration, and Ciliary Beating. Exp. Biol. Med. 2002, 227, 1047–1053. [Google Scholar] [CrossRef]
- Rizvi, S.; Gores, G.J. The Two Faces of Relaxin in Cancer: Antitumor or Protumor? Hepatology 2020, 71, 1117–1119. [Google Scholar] [CrossRef]
- Thanasupawat, T.; Glogowska, A.; Nivedita-Krishnan, S.; Wilson, B.; Klonisch, T.; Hombach-Klonisch, S. Emerging roles for the relaxin/RXFP1 system in cancer therapy. Mol. Cell. Endocrinol. 2019, 487, 85–93. [Google Scholar] [CrossRef]
- Samuel, C.S.; Lekgabe, E.D.; Mookerjee, I. The Effects of Relaxin on Extracellular Matrix Remodeling in Health and Fibrotic Disease. Adv. Exp. Med. Biol. 2007, 612, 88–103. [Google Scholar] [CrossRef] [PubMed]
- Ben-Lulu, S.; Ziv, T.; Weisman-Shomer, P.; Benhar, M. Nitrosothiol-trapping-based proteomic analysis of s-nitrosylation in human lung carcinoma cells. PLoS ONE 2017, 12, e0169862. [Google Scholar] [CrossRef] [PubMed]
- Meikle, C.K.; Meisler, A.J.; Bird, C.M.; Jeffries, J.A.; Azeem, N.; Garg, P.; Crawford, E.L.; Kelly, C.A.; Gao, T.Z.; Wuescher, L.M.; et al. Platelet-T cell aggregates in lung cancer patients: Implications for thrombosis. PLoS ONE 2020, 15, e0236966. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, J. From trends to transformation: Where cardio-oncology is to make a difference. Eur. Hear. J. 2019, 40, 3898–3900. [Google Scholar] [CrossRef] [PubMed]
- Kay, A.M.; Simpson, C.L.; Stewart, J.A. The Role of AGE/RAGE Signaling in Diabetes-Mediated Vascular Calcification. J. Diabetes Res. 2016, 2016, 1–8. [Google Scholar] [CrossRef]
- Krysan, K.; Reckamp, K.L.; Dalwadi, H.; Sharma, S.; Rozengurt, E.; Dohadwala, M.; Dubinett, S.M. Prostaglandin E2 Activates Mitogen-Activated Protein Kinase/Erk Pathway Signaling and Cell Proliferation in Non–Small Cell Lung Cancer Cells in an Epidermal Growth Factor Receptor–Independent Manner. Cancer Res. 2005, 65, 6275–6281. [Google Scholar] [CrossRef]
- Gorjao, R.; dos Santos, C.M.M.; Serdan, T.D.A.; Diniz, V.L.S.; Alba-Loureiro, T.C.; Cury-Boaventura, M.F.; Hatanaka, E.; Levada-Pires, A.C.; Sato, F.T.; Pithon-Curi, T.C.; et al. New insights on the regulation of cancer cachexia by N-3 polyunsaturated fatty acids. Pharmacol. Ther. 2018, 196, 117–134. [Google Scholar] [CrossRef]
- Yang, P.; Cartwright, C.; Chan, D.; Ding, J.; Felix, E.; Pan, Y.; Pang, J.; Rhea, P.; Block, K.; Fischer, S.M.; et al. Anticancer activity of fish oils against human lung cancer is associated with changes in formation of PGE2 and PGE3 and alteration of Akt phosphorylation. Mol. Carcinog. 2013, 53, 566–577. [Google Scholar] [CrossRef]
- Poczobutt, J.M.; Gijon, M.; Amin, J.; Hanson, D.; Li, H.; Walker, D.; Weiser-Evans, M.; Lu, X.; Murphy, R.C.; Nemenoff, R.A. Eicosanoid profiling in an orthotopic model of lung cancer progression by mass spectrometry demonstrates selective production of leukotrienes by inflammatory cells of the microenvironment. PLoS ONE 2013, 8, e79633. [Google Scholar] [CrossRef]
- Gomes, R.N.; da Costa, S.F.; Colquhoun, A. Eicosanoids and cancer. Clinics 2018, 73, e530s. [Google Scholar] [CrossRef]
- Oczypok, E.A.; Perkins, T.N.; Oury, T.D. All the “RAGE” in lung disease: The receptor for advanced glycation endproducts (RAGE) is a major mediator of pulmonary inflammatory responses. Paediatr. Respir. Rev. 2017, 23, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.-F.; Luo, X.-D.; Ning, J.; Xu, D.; Fan, Z.-P.; Sun, J.; Zhang, Y.; Xu, B.; Wei, Y.-Q. Association between acute graft versus host disease and lung injury after allogeneic haematopoietic stem cell transplantation. Hematology 2009, 14, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Yang, Q.; Lin, L.; Xu, C.; Zheng, C.; Chen, X.; Han, Y.; Li, M.; Cao, W.; Cao, K.; et al. Interleukin-17 enhances immunosuppression by mesenchymal stem cells. Cell Death Differ. 2014, 21, 1758–1768. [Google Scholar] [CrossRef]
- Wu, F.; Xu, J.; Huang, Q.; Han, J.; Duan, L.; Fan, J.; Lv, Z.; Guo, M.; Hu, G.; Chen, L.; et al. The Role of Interleukin-17 in Lung Cancer. Mediat. Inflamm. 2016, 2016, 8494079. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Gou, H.; Yao, J.; Yi, K.; Jin, Z.; Matsuoka, M.; Zhao, T. The noncanonical role of EZH2 in cancer. Cancer Sci. 2021, 112, 1376–1382. [Google Scholar] [CrossRef] [PubMed]
- Gardner, E.E.; Lok, B.H.; Schneeberger, V.E.; Desmeules, P.; Miles, L.A.; Arnold, P.K.; Ni, A.; Khodos, I.; de Stanchina, E.; Nguyen, T.; et al. Chemosensitive Relapse in Small Cell Lung Cancer Proceeds through an EZH2-SLFN11 Axis. Cancer Cell 2017, 31, 286–299. [Google Scholar] [CrossRef]
- Kim, S.; Yu, N.-K.; Kaang, B.-K. CTCF as a multifunctional protein in genome regulation and gene expression. Exp. Mol. Med. 2015, 47, e166. [Google Scholar] [CrossRef]
- Rhodes, D.R.; Yu, J.; Shanker, K.; Deshpande, N.; Varambally, R.; Ghosh, D.; Barrette, T.; Pandey, A.; Chinnaiyan, A.M. Large-scale meta-analysis of cancer microarray data identifies common transcriptional profiles of neo-plastic transformation and progression. Proc. Natl. Acad. Sci. USA 2004, 101, 9309–9314. [Google Scholar] [CrossRef]
- Atienza, J.M.; Roth, R.B.; Rosette, C.; Smylie, K.J.; Kammerer, S.; Rehbock, J.; Ekblom, J.; Denissenko, M.F. Suppression of RAD21 gene expression decreases cell growth and enhances cytotoxicity of etoposide and bleomycin in human breast cancer cells. Mol. Cancer Ther. 2005, 4, 361–368. [Google Scholar] [CrossRef]
- Ramsey, J.; Butnor, K.; Peng, Z.; LeClair, T.; van der Velden, J.; Stein, G.; Lian, J.; Kinsey, C.M. Loss of RUNX1 is associated with aggressive lung adenocarcinomas. J. Cell. Physiol. 2017, 233, 3487–3497. [Google Scholar] [CrossRef]
- Kim, Y.; Bin Lee, B.; Kim, D.; Um, S.; Cho, E.Y.; Han, J.; Shim, Y.M.; Kim, D.-H. Clinicopathological Significance of RUNX1 in Non-Small Cell Lung Cancer. J. Clin. Med. 2020, 9, 1694. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, M.; Tan, T.Z.; Sulaiman, N.B.S.; Lamar, J.M.; Bansal, P.; Cui, J.; Qiao, Y.; Ito, Y. RUNX1 and RUNX3 protect against YAP-mediated EMT, stemness and shorter survival outcomes in breast cancer. Oncotarget 2018, 9, 14175–14192. [Google Scholar] [CrossRef]
- Meng, G.; Wei, J.; Wang, Y.; Qu, D.; Zhang, J. MiR-21 regulates immunosuppression mediated by myeloid-derived suppressor cells by impairing RUNX1-YAP interaction in lung cancer. Cancer Cell Int. 2020, 20, 495. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Mei, T.; Zeng, Y. HDAC2 promotes the migration and invasion of non-small cell lung cancer cells via upregulation of fibronectin. Biomed. Pharmacother. 2016, 84, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Werbajh, S.E.; Urtreger, A.J.; Puricelli, L.I.; De Lustig, E.S.; Joffé, E.B.D.K.; Kornblihtt, A.R. Downregulation of fibronectin transcription in highly metastatic adenocarcinoma cells. FEBS Lett. 1998, 440, 277–281. [Google Scholar] [CrossRef]
- Michalik, L.; Wahli, W. PPARs Mediate Lipid Signaling in Inflammation and Cancer. PPAR Res. 2008, 2008, 134059. [Google Scholar] [CrossRef]
- Theocharis, S.; Kanelli, H.; Politi, E.; Margeli, A.; Karkandaris, C.; Philippides, T.; Koutselinis, A. Expression of peroxisome proliferator activated receptor-gamma in non-small cell lung carcinoma: Correlation with histological type and grade. Lung Cancer 2002, 36, 249–255. [Google Scholar] [CrossRef]
- Sabatino, L.; Fucci, A.; Pancione, M.; Colantuoni, V. PPARG Epigenetic Deregulation and Its Role in Colorectal Tumorigenesis. PPAR Res. 2012, 2012, 687492. [Google Scholar] [CrossRef]
- Platta, C.S.; Greenblatt, D.Y.; Kunnimalaiyaan, M.; Chen, H. Valproic Acid Induces Notch1 Signaling in Small Cell Lung Cancer Cells. J. Surg. Res. 2008, 148, 31–37. [Google Scholar] [CrossRef]
- Park, H.K.; Han, B.R.; Park, W.H. Combination of arsenic trioxide and valproic acid efficiently inhibits growth of lung cancer cells via G2/M-phase arrest and apoptotic cell death. Int. J. Mol. Sci. 2020, 21, 2649. [Google Scholar] [CrossRef]
- Hubaux, R.; Vandermeers, F.; Cosse, J.-P.; Crisanti, C.; Kapoor, V.; Albelda, S.M.; Mascaux, C.; Delvenne, P.; Hubert, P.; Willems, L. Valproic acid improves second-line regimen of small cell lung carcinoma in preclinical models. ERJ Open Res. 2015, 1, 00028-2015. [Google Scholar] [CrossRef] [PubMed]
- Shirsath, N.; Rathos, M.; Chaudhari, U.; Sivaramakrishnan, H.; Joshi, K. Potentiation of anticancer effect of valproic acid, an antiepileptic agent with histone deacetylase inhibitory activity, by the cyclin-dependent kinase inhibitor P276-00 in human non-small-cell lung cancer cell lines. Lung Cancer 2013, 82, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Rakba, N.; Melhaoui, A.; Loyer, P.; Delcros, J.G.; Morel, I.; Lescoat, G. Bgugaine, a pyrrolidine alkaloid from Arisarum vulgare, is a strong hepatotoxin in rat and human liver cell cultures. Toxicol. Lett. 1999, 104, 239–248. [Google Scholar] [CrossRef]
- Rakba, N.; Melhaoui, A.; Rissel, M.; Morel, I.; Loyer, P.; Lescoat, G. Irniine, a pyrrolidine alkaloid, isolated from Arisarum vulgare can induce apoptosis and/or necrosis in rat hepatocyte cultures. Toxicon 2000, 38, 1389–1402. [Google Scholar] [CrossRef]
- Garcia-Quiroz, J.; Camacho, J. Astemizole: An old anti-histamine as a new promising anti-cancer drug. Anti-Cancer Agents Med. Chem. 2012, 11, 307–314. [Google Scholar] [CrossRef]
- Gray, K.A.; Daugherty, L.C.; Gordon, S.M.; Seal, R.L.; Wright, M.W.; Bruford, E.A. Genenames.org: The HGNC resources in 2013. Nucleic Acids Res. 2012, 41, D545–D552. [Google Scholar] [CrossRef]
- Gundersen, G.W.; Jones, M.R.; Rouillard, A.; Kou, Y.; Monteiro, C.D.; Feldmann, A.S.; Hu, K.S.; Ma’Ayan, A. GEO2Enrichr: Browser extension and server app to extract gene sets from GEO and analyze them for biological functions. Bioinformatics 2015, 31, 3060–3062. [Google Scholar] [CrossRef]
- Wang, Z.; Monteiro, C.D.; Jagodnik, K.M.; Fernandez, N.F.; Gundersen, G.W.; Rouillard, A.D.; Jenkins, S.L.; Feldmann, A.S.; Hu, K.S.; McDermott, M.G.; et al. Extraction and analysis of signatures from the Gene Expression Omnibus by the crowd. Nat. Commun. 2016, 7, 12846. [Google Scholar] [CrossRef]
- Kibbe, W.A.; Arze, C.; Felix, V.; Mitraka, E.; Bolton, E.; Fu, G.; Mungall, C.J.; Binder, J.X.; Malone, J.; Vasant, D.; et al. Disease Ontology 2015 update: An expanded and updated database of Human diseases for linking bio-medical knowledge through disease data. Nucleic Acids Res. 2015, 43, D1071–D1078. [Google Scholar] [CrossRef]
- Law, V.; Knox, C.; Djoumbou, Y.; Jewison, T.; Guo, A.C.; Liu, Y.; Maciejewski, A.; Arndt, D.; Wilson, M.; Neveu, V.; et al. DrugBank 4.0: Shedding new light on drug metabolism. Nucleic Acids Res. 2013, 42, D1091–D1097. [Google Scholar] [CrossRef]
- Boedigheimer, M.J.; Wolfinger, R.D.; Bass, M.B.; Bushel, P.R.; Chou, J.W.; Cooper, M.; Corton, J.C.; Fostel, J.; Hester, S.; Lee, J.S.; et al. Sources of variation in baseline gene expression levels from toxicogenomics study control animals across multiple laboratories. BMC Genom. 2008, 9, 285. [Google Scholar] [CrossRef] [PubMed]
- Leek, J.T.; Storey, J.D. Capturing Heterogeneity in Gene Expression Studies by Surrogate Variable Analysis. PLoS Genet. 2007, 3, 1724–1735. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; Xu, H.; Gordonov, S.; Lim, M.P.; Perkins, M.H.; Ma’Ayan, A. Expression2Kinases: mRNA profiling linked to multiple upstream regulatory layers. Bioinformatics 2011, 28, 105–111. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019, 28, 1947–1951. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 29–34. [Google Scholar] [CrossRef]
- Musa, A.; Ghoraie, L.S.; Zhang, S.-D.; Galzko, G.; Yli-Harja, O.; Dehmer, M.; Haibe-Kains, B.; Emmert-Streib, F. A review of connectivity map and computational approaches in pharmacogenomics. Briefings Bioinform. 2017, 19, 506–523. [Google Scholar] [CrossRef]
- Zaravinos, A.; Roufas, C.; Nagara, M.; de Lucas Moreno, B.; Oblovatskaya, M.; Efstathiades, C.; Ayiomamitis, G.D. Cytolytic activity correlates with the mutational burden and deregulated expression of immune checkpoints in colorectal cancer. J. Exp. Clin. Cancer Res. 2019, 38, 364. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Belinda, P.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chatziantoniou, A.; Zaravinos, A. Signatures of Co-Deregulated Genes and Their Transcriptional Regulators in Lung Cancer. Int. J. Mol. Sci. 2022, 23, 10933. https://doi.org/10.3390/ijms231810933
Chatziantoniou A, Zaravinos A. Signatures of Co-Deregulated Genes and Their Transcriptional Regulators in Lung Cancer. International Journal of Molecular Sciences. 2022; 23(18):10933. https://doi.org/10.3390/ijms231810933
Chicago/Turabian StyleChatziantoniou, Angeliki, and Apostolos Zaravinos. 2022. "Signatures of Co-Deregulated Genes and Their Transcriptional Regulators in Lung Cancer" International Journal of Molecular Sciences 23, no. 18: 10933. https://doi.org/10.3390/ijms231810933
APA StyleChatziantoniou, A., & Zaravinos, A. (2022). Signatures of Co-Deregulated Genes and Their Transcriptional Regulators in Lung Cancer. International Journal of Molecular Sciences, 23(18), 10933. https://doi.org/10.3390/ijms231810933