

Novel Roles of RNA m6A Methylation Regulators in the Occurrence of Alzheimer’s Disease and the Subtype Classification


Abstract
:1. Introduction
2. Results
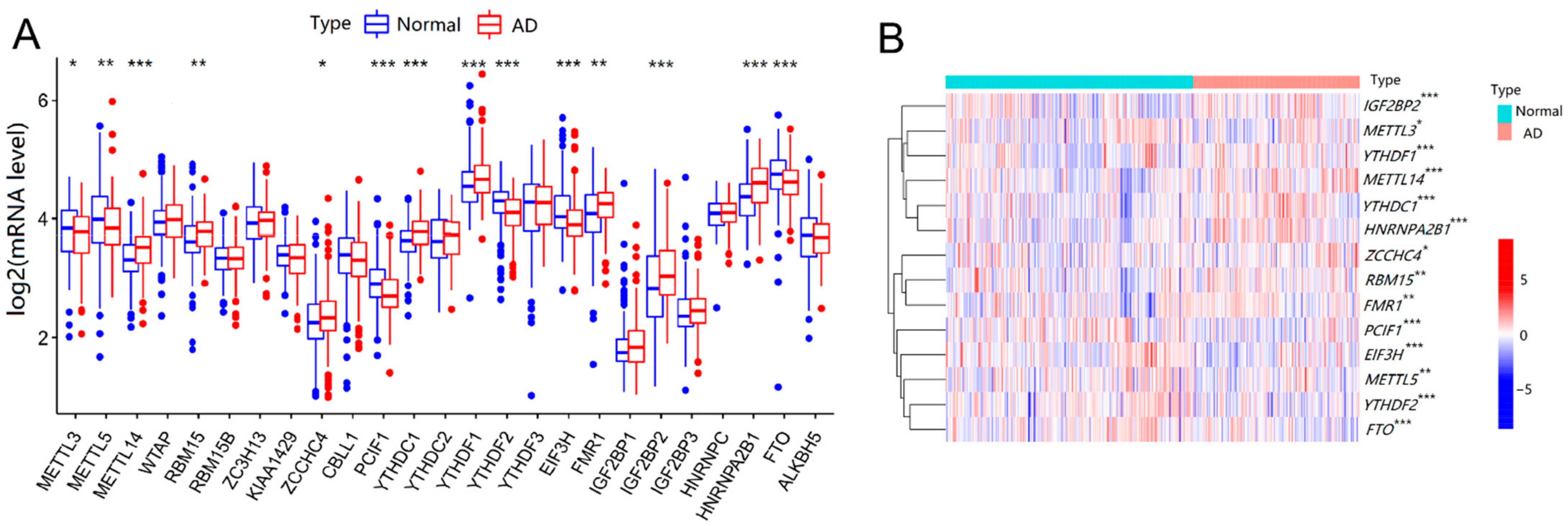
2.1. Expression and Correlation of 25 m6A RNA Methylation Regulators in AD
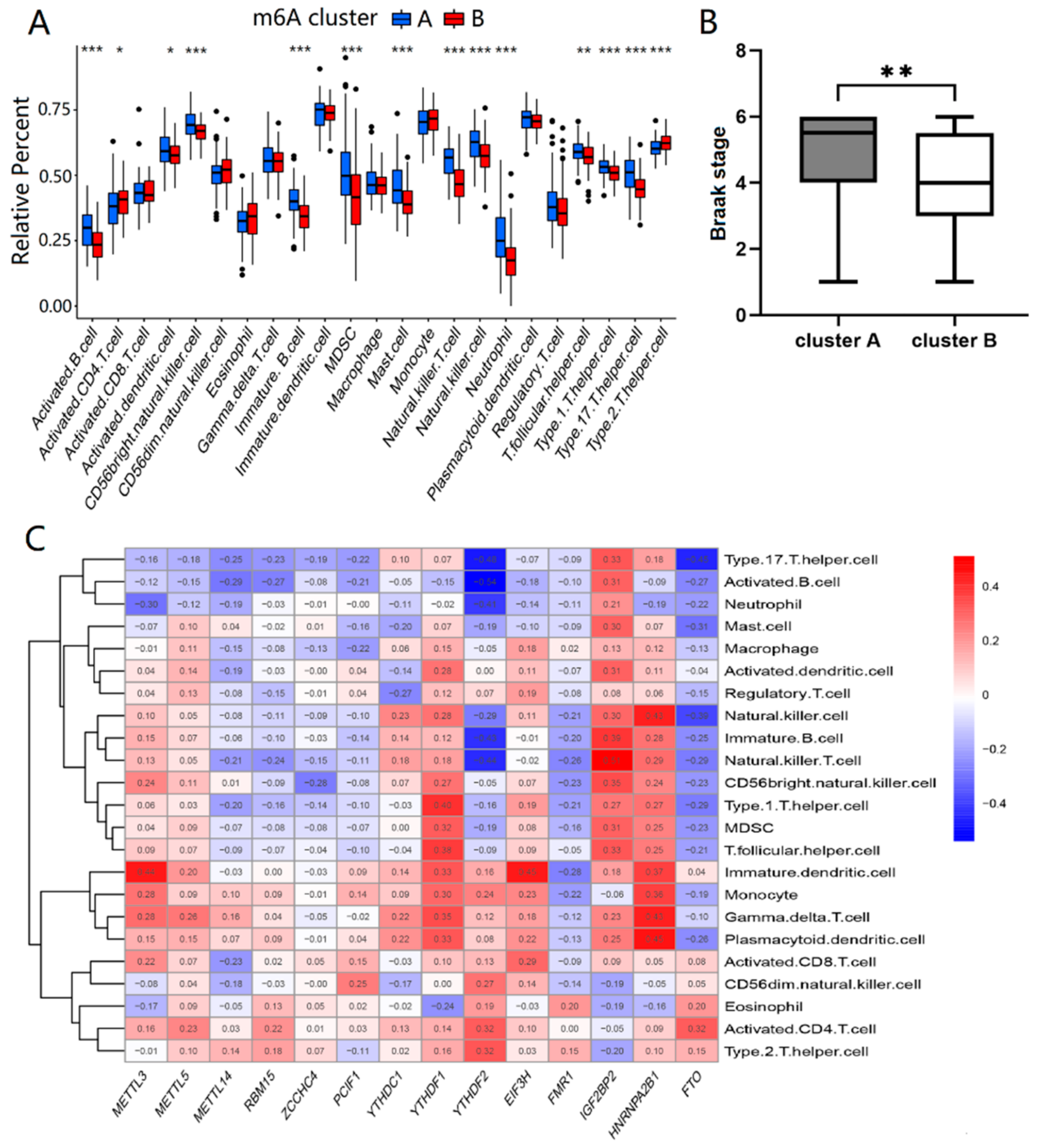
2.2. Two Distinct m6A Patterns Identified by Significant m6A Regulators
2.3. Screening and Functional Enrichment Analysis of DEGs
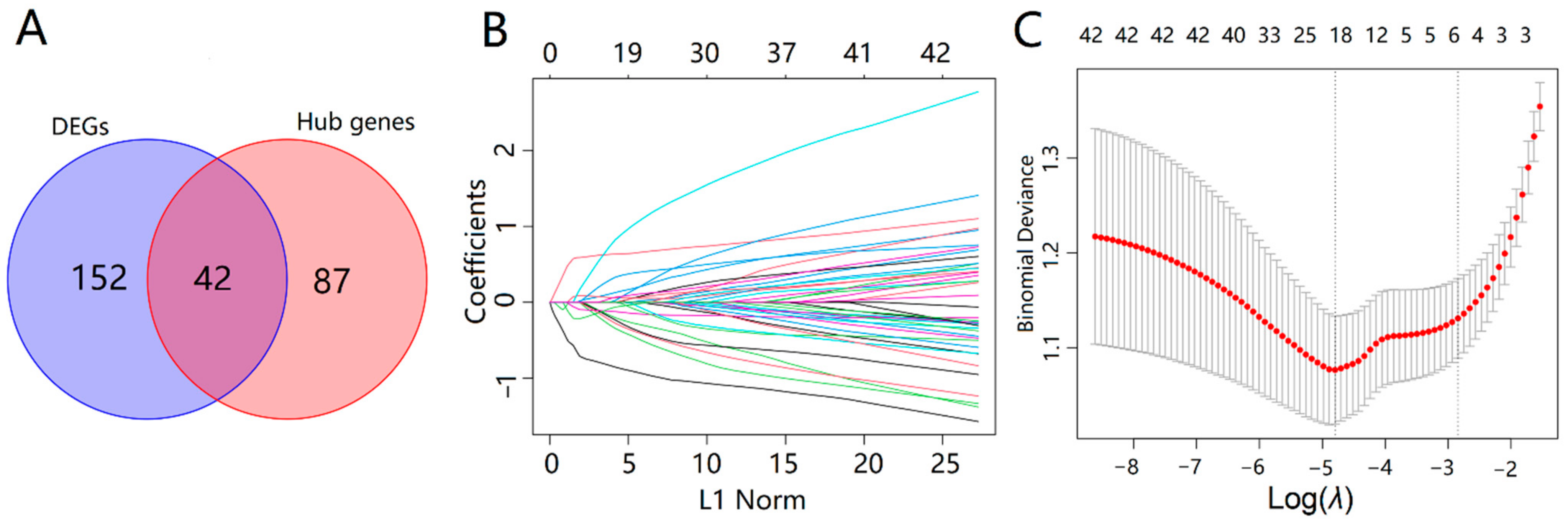
2.4. Identification of Key AD Genes
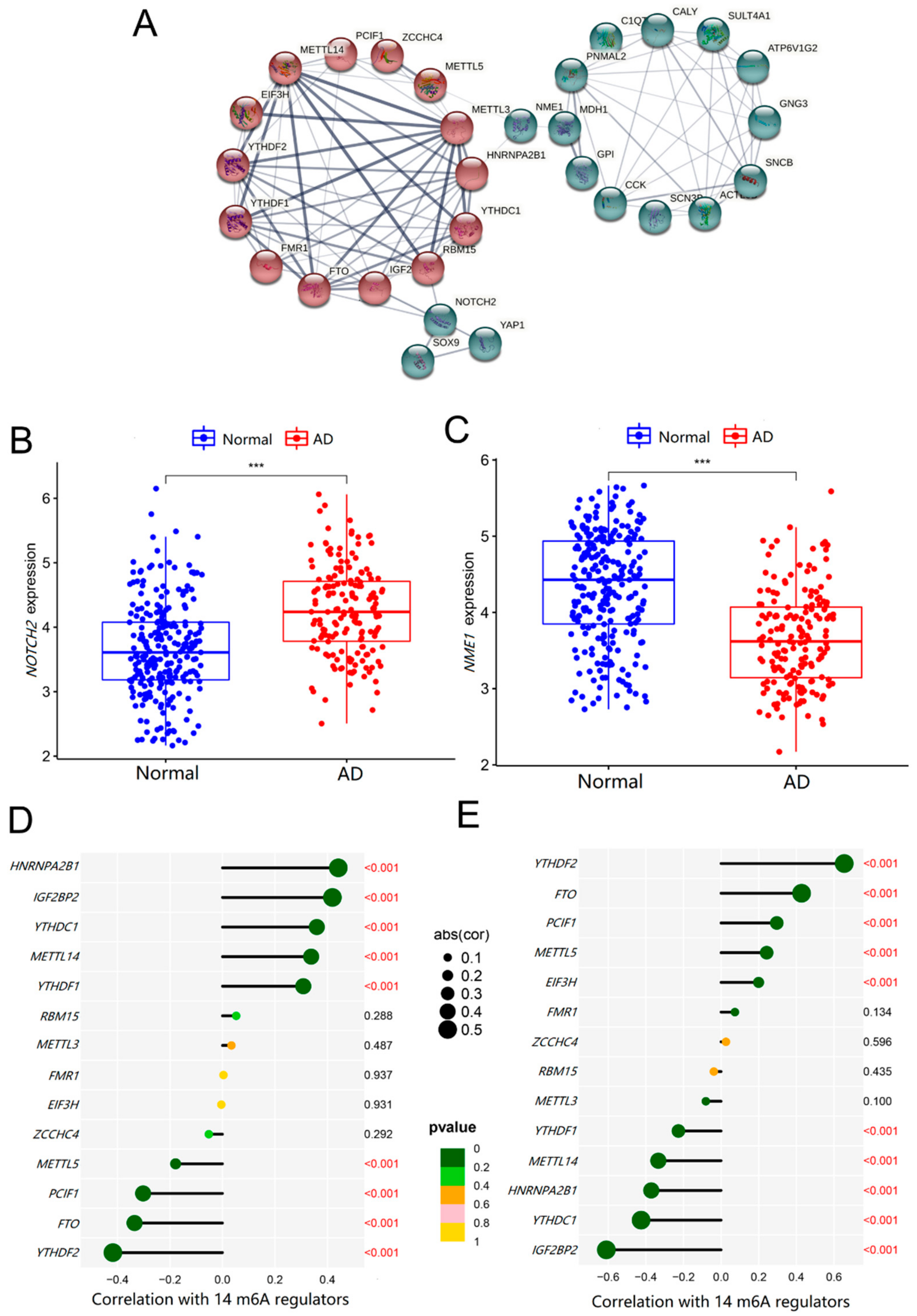
2.5. Co-Expression Relationship of the 14 Significant m6A Regulators and the 19 Key AD Genes
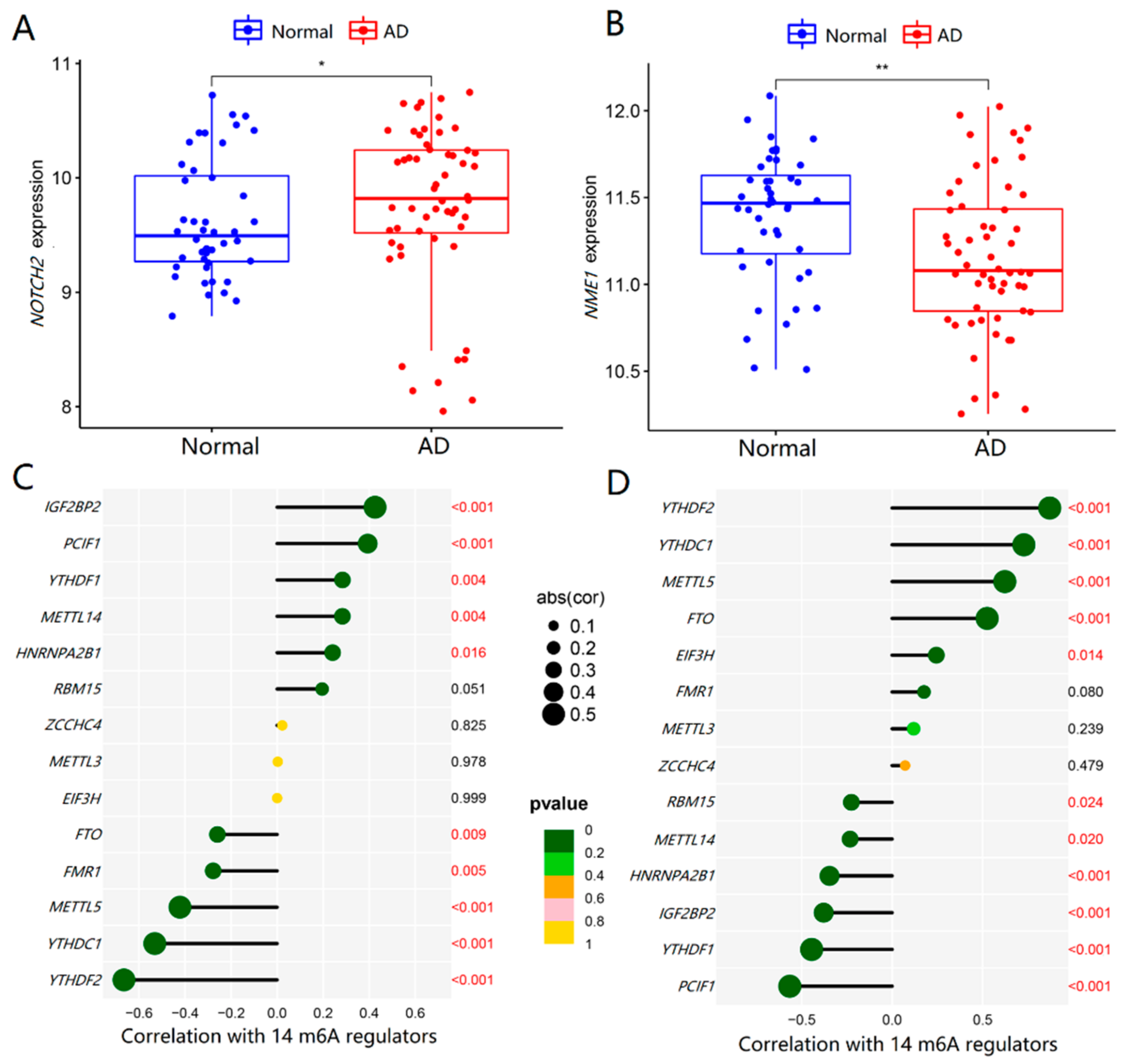
2.6. Roles of NOTCH2 and NME1 in AD and Their Association with m6A Regulators
3. Discussion
4. Materials and Methods
4.1. Data Acquisition and Preprocessing
4.2. Selection of m6A RNA Methylation Regulators
4.3. Identification of Molecular Subtypes Based on the Significant m6A Regulators
4.4. Estimation of Immune Cell Infiltration
4.5. Screening and Functional Enrichment Analysis of DEGs
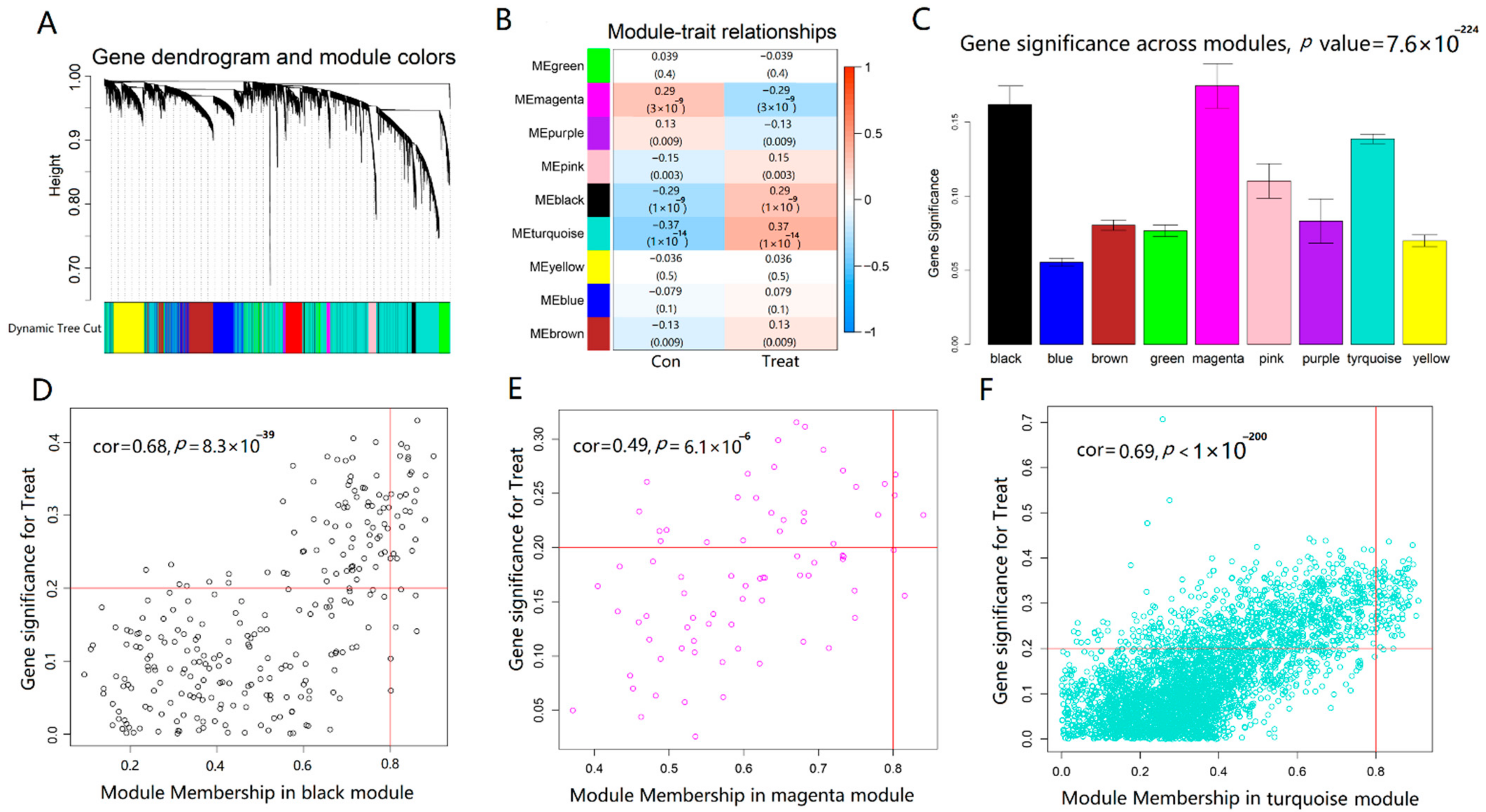
4.6. Identification of Co-Expression Modules and Key AD Genes
4.7. PPI Network Construction
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Scheltens, P.; de Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef]
- Gatz, M.; Reynolds, C.A.; Fratiglioni, L.; Johansson, B.; Mortimer, J.A.; Berg, S.; Fiske, A.; Pedersen, N.L. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 2006, 63, 168–174. [Google Scholar] [CrossRef]
- Wang, X.; Lu, Z.; Gomez, A.; Hon, G.C.; Yue, Y.; Han, D.; Fu, Y.; Parisien, M.; Dai, Q.; Jia, G.; et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 2014, 505, 117–120. [Google Scholar] [CrossRef]
- Hsu, P.J.; Shi, H.; Zhu, A.C.; Lu, Z.; Miller, N.; Edens, B.M.; Ma, Y.C.; He, C. The RNA-binding protein FMRP facilitates the nuclear export of N (6)-methyladenosine-containing mRNAs. J. Biol. Chem. 2019, 294, 19889–19895. [Google Scholar] [CrossRef]
- Liu, Y.; You, Y.; Lu, Z.; Yang, J.; Li, P.; Liu, L.; Xu, H.; Niu, Y.; Cao, X. N (6)-methyladenosine RNA modification-mediated cellular metabolism rewiring inhibits viral replication. Science 2019, 365, 1171–1176. [Google Scholar] [CrossRef]
- Wang, X.; Feng, J.; Xue, Y.; Guan, Z.; Zhang, D.; Liu, Z.; Gong, Z.; Wang, Q.; Huang, J.; Tang, C.; et al. Structural basis of N (6)-adenosine methylation by the METTL3-METTL14 complex. Nature 2016, 534, 575–578. [Google Scholar] [CrossRef]
- Liu, J.; Yue, Y.; Han, D.; Wang, X.; Fu, Y.; Zhang, L.; Jia, G.; Yu, M.; Lu, Z.; Deng, X.; et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2014, 10, 93–95. [Google Scholar] [CrossRef]
- Pendleton, K.E.; Chen, B.; Liu, K.; Hunter, O.V.; Xie, Y.; Tu, B.P.; Conrad, N.K. The U6 snRNA m(6)A Methyltransferase METTL16 Regulates SAM Synthetase Intron Retention. Cell 2017, 169, 824–835. [Google Scholar] [CrossRef]
- van Tran, N.; Ernst, F.G.M.; Hawley, B.R.; Zorbas, C.; Ulryck, N.; Hackert, P.; Bohnsack, K.E.; Bohnsack, M.T.; Jaffrey, S.R.; Graille, M.; et al. The human 18S rRNA m6A methyltransferase METTL5 is stabilized by TRMT112. Nucleic Acids Res. 2019, 47, 7719–7733. [Google Scholar] [CrossRef] [Green Version]
- Ping, X.L.; Sun, B.F.; Wang, L.; Xiao, W.; Yang, X.; Wang, W.J.; Adhikari, S.; Shi, Y.; Lv, Y.; Chen, Y.S.; et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014, 24, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Knuckles, P.; Lence, T.; Haussmann, I.U.; Jacob, D.; Kreim, N.; Carl, S.H.; Masiello, I.; Hares, T.; Villaseñor, R.; Hess, D.; et al. Zc3h13/Flacc is required for adenosine methylation by bridging the mRNA-binding factor Rbm15/Spenito to the m(6)A machinery component Wtap/Fl(2)d. Genes Dev. 2018, 32, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Patil, D.P.; Chen, C.K.; Pickering, B.F.; Chow, A.; Jackson, C.; Guttman, M.; Jaffrey, S.R. m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature 2016, 537, 369–373. [Google Scholar] [CrossRef]
- Wen, J.; Lv, R.; Ma, H.; Shen, H.; He, C.; Wang, J.; Jiao, F.; Liu, H.; Yang, P.; Tan, L.; et al. Zc3h13 Regulates Nuclear RNA m(6)A Methylation and Mouse Embryonic Stem Cell Self-Renewal. Mol. Cell 2018, 69, 1028–1038. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Liu, J.; Cui, X.; Cao, J.; Luo, G.; Zhang, Z.; Cheng, T.; Gao, M.; Shu, X.; Ma, H.; et al. VIRMA mediates preferential m(6)A mRNA methylation in 3’UTR and near stop codon and associates with alternative polyadenylation. Cell Discov. 2018, 4, 10. [Google Scholar] [CrossRef] [PubMed]
- Bawankar, P.; Lence, T.; Paolantoni, C.; Haussmann, I.U.; Kazlauskiene, M.; Jacob, D.; Heidelberger, J.B.; Richter, F.M.; Nallasivan, M.P.; Morin, V.; et al. Hakai is required for stabilization of core components of the m(6)A mRNA methylation machinery. Nat. Commun. 2021, 12, 3778. [Google Scholar] [CrossRef]
- Ma, H.; Wang, X.; Cai, J.; Dai, Q.; Natchiar, S.K.; Lv, R.; Chen, K.; Lu, Z.; Chen, H.; Shi, Y.G.; et al. N(6-)Methyladenosine methyltransferase ZCCHC4 mediates ribosomal RNA methylation. Nat. Chem. Biol. 2019, 15, 88–94. [Google Scholar] [CrossRef]
- Boulias, K.; Toczydłowska-Socha, D.; Hawley, B.R.; Liberman, N.; Takashima, K.; Zaccara, S.; Guez, T.; Vasseur, J.J.; Debart, F.; Aravind, L.; et al. Identification of the m(6)Am Methyltransferase PCIF1 Reveals the Location and Functions of m(6)Am in the Transcriptome. Mol. Cell 2019, 75, 631–643. [Google Scholar] [CrossRef]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.G.; et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887. [Google Scholar] [CrossRef]
- Zheng, G.; Dahl, J.A.; Niu, Y.; Fedorcsak, P.; Huang, C.M.; Li, C.J.; Vågbø, C.B.; Shi, Y.; Wang, W.L.; Song, S.H.; et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 2013, 49, 18–29. [Google Scholar] [CrossRef] [Green Version]
- Xiao, W.; Adhikari, S.; Dahal, U.; Chen, Y.S.; Hao, Y.J.; Sun, B.F.; Sun, H.Y.; Li, A.; Ping, X.L.; Lai, W.Y.; et al. Nuclear m(6)A Reader YTHDC1 Regulates mRNA Splicing. Mol. Cell 2016, 61, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.J.; Zhu, Y.; Ma, H.; Guo, Y.; Shi, X.; Liu, Y.; Qi, M.; Lu, Z.; Shi, H.; Wang, J.; et al. Ythdc2 is an N(6)-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res. 2017, 27, 1115–1127. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhao, B.S.; Roundtree, I.A.; Lu, Z.; Han, D.; Ma, H.; Weng, X.; Chen, K.; Shi, H.; He, C. N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell 2015, 161, 1388–1399. [Google Scholar] [CrossRef]
- Shi, H.; Wang, X.; Lu, Z.; Zhao, B.S.; Ma, H.; Hsu, P.J.; Liu, C.; He, C. YTHDF3 facilitates translation and decay of N(6)-methyladenosine-modified RNA. Cell Res. 2017, 27, 315–328. [Google Scholar] [CrossRef]
- Du, H.; Zhao, Y.; He, J.; Zhang, Y.; Xi, H.; Liu, M.; Ma, J.; Wu, L. YTHDF2 destabilizes m(6)A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat. Commun. 2016, 7, 12626. [Google Scholar] [CrossRef] [PubMed]
- Choe, J.; Lin, S.; Zhang, W.; Liu, Q.; Wang, L.; Ramirez-Moya, J.; Du, P.; Kim, W.; Tang, S.; Sliz, P.; et al. mRNA circularization by METTL3-eIF3h enhances translation and promotes oncogenesis. Nature 2018, 561, 556–560. [Google Scholar] [CrossRef]
- Edupuganti, R.R.; Geiger, S.; Lindeboom, R.G.H.; Shi, H.; Hsu, P.J.; Lu, Z.; Wang, S.Y.; Baltissen, M.P.A.; Jansen, P.; Rossa, M.; et al. N(6)-methyladenosine (m(6)A) recruits and repels proteins to regulate mRNA homeostasis. Nat. Struct. Mol. Biol. 2017, 24, 870–878. [Google Scholar] [CrossRef]
- Huang, H.; Weng, H.; Sun, W.; Qin, X.; Shi, H.; Wu, H.; Zhao, B.S.; Mesquita, A.; Liu, C.; Yuan, C.L.; et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol. 2018, 20, 285–295. [Google Scholar] [CrossRef]
- Zarnack, K.; König, J.; Tajnik, M.; Martincorena, I.; Eustermann, S.; Stévant, I.; Reyes, A.; Anders, S.; Luscombe, N.M.; Ule, J. Direct competition between hnRNP C and U2AF65 protects the transcriptome from the exonization of Alu elements. Cell 2013, 152, 453–466. [Google Scholar] [CrossRef]
- Alarcón, C.R.; Goodarzi, H.; Lee, H.; Liu, X.; Tavazoie, S.; Tavazoie, S.F. HNRNPA2B1 Is a Mediator of m(6)A-Dependent Nuclear RNA Processing Events. Cell 2015, 162, 1299–1308. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Ren, Y.; Mao, K.; Hua, F.; Yang, Y.; Wei, N.; Yue, C.; Li, D.; Zhang, H. FTO is involved in Alzheimer’s disease by targeting TSC1-mTOR-Tau signaling. Biochem. Biophys. Res. Commun. 2018, 498, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Shafik, A.M.; Zhang, F.; Guo, Z.; Dai, Q.; Pajdzik, K.; Li, Y.; Kang, Y.; Yao, B.; Wu, H.; He, C.; et al. N6-methyladenosine dynamics in neurodevelopment and aging, and its potential role in Alzheimer’s disease. Genome Biol. 2021, 22, 17. [Google Scholar] [CrossRef]
- Luo, X.; Xu, S.; Zhong, Y.; Tu, T.; Xu, Y.; Li, X.; Wang, B.; Yang, F. High gene expression levels of VEGFA and CXCL8 in the peritumoral brain zone are associated with the recurrence of glioblastoma: A bioinformatics analysis. Oncol. Lett. 2019, 18, 6171–6179. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, Y.; Guo, Y.; Liu, R.; Yu, Q.; Gong, L.; Liu, Z.; Xie, W.; Wang, C. ALKBH1 promotes lung cancer by regulating m6A RNA demethylation. Biochem. Pharmacol. 2021, 189, 114284. [Google Scholar] [CrossRef]
- Yang, Y.; Wu, J.; Liu, F.; He, J.; Wu, F.; Chen, J.; Jiang, Z. IGF2BP1 Promotes the Liver Cancer Stem Cell Phenotype by Regulating MGAT5 mRNA Stability by m6A RNA Methylation. Stem Cells Dev. 2021, 30, 1115–1125. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Yu, X.; He, G. m6A-Mediated Tumor Invasion and Methylation Modification in Breast Cancer Microenvironment. J. Oncol. 2021, 2021, 9987376. [Google Scholar] [CrossRef]
- Sun, T.; Wu, R.; Ming, L. The role of m6A RNA methylation in cancer. Biomed. Pharmacother. 2019, 112, 108613. [Google Scholar] [CrossRef]
- Li, W.; Gao, Y.; Jin, X.; Wang, H.; Lan, T.; Wei, M.; Yan, W.; Wang, G.; Li, Z.; Zhao, Z.; et al. Comprehensive analysis of N6-methylandenosine regulators and m6A-related RNAs as prognosis factors in colorectal cancer. Mol. Ther. Nucleic Acids 2022, 27, 598–610. [Google Scholar] [CrossRef]
- Chen, M.; Nie, Z.Y.; Wen, X.H.; Gao, Y.H.; Cao, H.; Zhang, S.F. m6A RNA methylation regulators can contribute to malignant progression and impact the prognosis of bladder cancer. Biosci. Rep. 2019, 39, BSR20192892. [Google Scholar] [CrossRef]
- Weng, Y.L.; Wang, X.; An, R.; Cassin, J.; Vissers, C.; Liu, Y.; Liu, Y.; Xu, T.; Wang, X.; Wong, S.Z.H.; et al. Epitranscriptomic m(6)A Regulation of Axon Regeneration in the Adult Mammalian Nervous System. Neuron 2018, 97, 313–325. [Google Scholar] [CrossRef] [Green Version]
- Henstridge, C.M.; Pickett, E.; Spires-Jones, T.L. Synaptic pathology: A shared mechanism in neurological disease. Ageing Res. Rev. 2016, 28, 72–84. [Google Scholar] [CrossRef]
- González, H.; Pacheco, R. T-cell-mediated regulation of neuroinflammation involved in neurodegenerative diseases. J. Neuroinflammation 2014, 11, 201. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Liu, Z.; Shen, Y.; Zhu, H. A Novel Perspective Linkage between Kidney Function and Alzheimer’s Disease. Front. Cell. Neurosci. 2018, 12, 384. [Google Scholar] [CrossRef] [PubMed]
- Delhaye, S.; Bardoni, B. Role of phosphodiesterases in the pathophysiology of neurodevelopmental disorders. Mol. Psychiatry 2021, 26, 4570–4582. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Sharma, V.; Singh, V.P.; Kaundal, M.; Gupta, M.K.; Bariwal, J.; Deshmukh, R. Herbs to curb cyclic nucleotide phosphodiesterase and their potential role in Alzheimer’s disease. Mech. Ageing Dev. 2015, 149, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Boareto, M.; Engler, A.; Louvi, A.; Giachino, C.; Iber, D.; Taylor, V. Id4 Downstream of Notch2 Maintains Neural Stem Cell Quiescence in the Adult Hippocampus. Cell Rep. 2019, 28, 1485–1498. [Google Scholar] [CrossRef]
- Chen, C.D.; Oh, S.Y.; Hinman, J.D.; Abraham, C.R. Visualization of APP dimerization and APP-Notch2 heterodimerization in living cells using bimolecular fluorescence complementation. J. Neurochem. 2006, 97, 30–43. [Google Scholar] [CrossRef]
- Oh, S.Y.; Ellenstein, A.; Chen, C.D.; Hinman, J.D.; Berg, E.A.; Costello, C.E.; Yamin, R.; Neve, R.L.; Abraham, C.R. Amyloid precursor protein interacts with notch receptors. J. Neurosci. Res. 2005, 82, 32–42. [Google Scholar] [CrossRef]
- Atwood, C.S.; Bowen, R.L. A Unified Hypothesis of Early- and Late-Onset Alzheimer’s Disease Pathogenesis. J. Alzheimers Dis. 2015, 47, 33–47. [Google Scholar] [CrossRef]
- Anantha, J.; Goulding, S.R.; Wyatt, S.L.; Concannon, R.M.; Collins, L.M.; Sullivan, A.M.; O’Keeffe, G.W. STRAP and NME1 Mediate the Neurite Growth-Promoting Effects of the Neurotrophic Factor GDF5. iScience 2020, 23, 101457. [Google Scholar] [CrossRef]
- Owlanj, H.; Yang, H.J.; Feng, Z.W. Nucleoside diphosphate kinase Nm23-M1 involves in oligodendroglial versus neuronal cell fate decision in vitro. Differentiation 2012, 84, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets-update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Categories | m6A Regulators | Mechanisms | References |
|---|---|---|---|
| m6A writers | METTL3 | Catalyzes m6A modification | [7] |
| METTL5 | Affects the selection of methylation sites | [10] | |
| METTL14 | Assists METTL3 in recognizing the substrates substraction | [8] | |
| WTAP | Promotes METTL3-METTL14 heterodimer to the nuclear speckle | [11] | |
| RBM15/15B | Binds the m6A complex and recruits it to special RNA site | [12,13] | |
| ZC3H13 | Bridges WTAP to the mRNA-binding factor Nito | [14] | |
| KIAA1429 | Guides methyltransferase components to specific RNA regions | [15] | |
| ZCCHC4 | Adds m6A to18S and 28S ribosomal RNAs | [17] | |
| CBLL1 | Binds the m6A evolutionary conserved protein complex | [16] | |
| PCIF1 | Catalyzes m6A methylation on 2-O-methylated adenine | [18] | |
| m6A erasers | FTO | Removes m6A modification | [19] |
| ALKBH5 | Removes m6A modification | [20] | |
| m6A readers | YTHDC1 | Promotes RNA splicing and translocation | [21] |
| YTHDC2 | Enhances the translation of target RNA | [22] | |
| YTHDF1 | Promotes mRNA translation | [23] | |
| YTHDF2 | Reduces mRNA stability | [25] | |
| YTHDF3 | Mediates the translation or degradation | [24] | |
| EIF3H | Interacts with METTL3 and enhances translation | [26] | |
| FMR1 | Affects the nuclear export of m6A-modified RNA targets | [27] | |
| IGF2BP1/2/3 | Enhances mRNA stability | [28] | |
| HNRNPC | Mediates mRNA splicing | [29] | |
| HNRNPA2B1 | Promotes primary microRNA processing | [30] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, M.; Cheng, W.; Zhang, L.; Zhou, C.; Peng, X.; Yu, S.; Zhang, W. Novel Roles of RNA m6A Methylation Regulators in the Occurrence of Alzheimer’s Disease and the Subtype Classification. Int. J. Mol. Sci. 2022, 23, 10766. https://doi.org/10.3390/ijms231810766
Li M, Cheng W, Zhang L, Zhou C, Peng X, Yu S, Zhang W. Novel Roles of RNA m6A Methylation Regulators in the Occurrence of Alzheimer’s Disease and the Subtype Classification. International Journal of Molecular Sciences. 2022; 23(18):10766. https://doi.org/10.3390/ijms231810766
Chicago/Turabian StyleLi, Min, Wenli Cheng, Luyun Zhang, Cheng Zhou, Xinyue Peng, Susu Yu, and Wenjuan Zhang. 2022. "Novel Roles of RNA m6A Methylation Regulators in the Occurrence of Alzheimer’s Disease and the Subtype Classification" International Journal of Molecular Sciences 23, no. 18: 10766. https://doi.org/10.3390/ijms231810766
APA StyleLi, M., Cheng, W., Zhang, L., Zhou, C., Peng, X., Yu, S., & Zhang, W. (2022). Novel Roles of RNA m6A Methylation Regulators in the Occurrence of Alzheimer’s Disease and the Subtype Classification. International Journal of Molecular Sciences, 23(18), 10766. https://doi.org/10.3390/ijms231810766