


Implementation of QbD Approach to the Development of Chromatographic Methods for the Determination of Complete Impurity Profile of Substance on the Preclinical and Clinical Step of Drug Discovery Studies

 , , ,
, , ,
Abstract
:1. Introduction
2. Results and Discussion
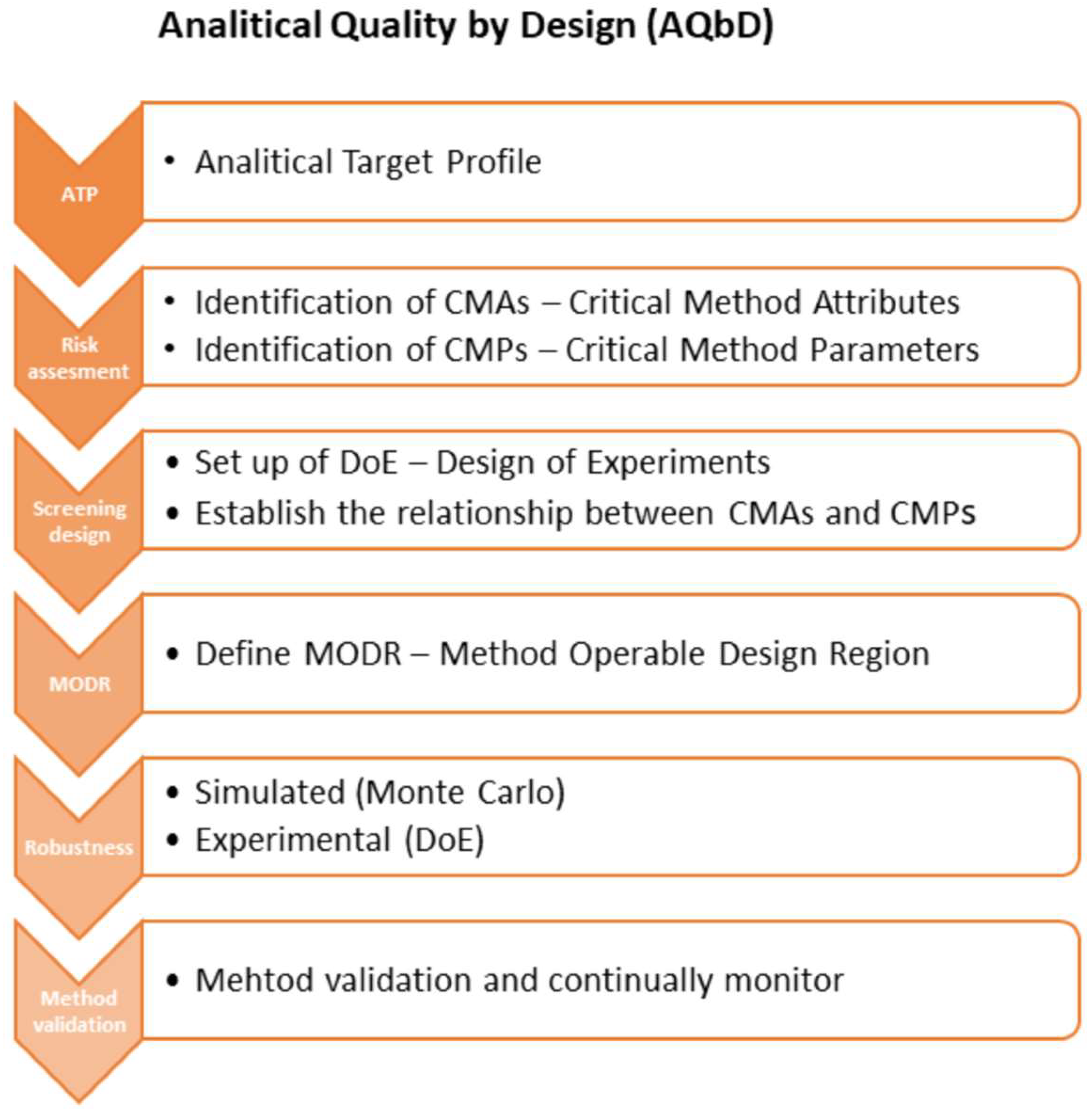
2.1. Analytical Target Profile (ATP)
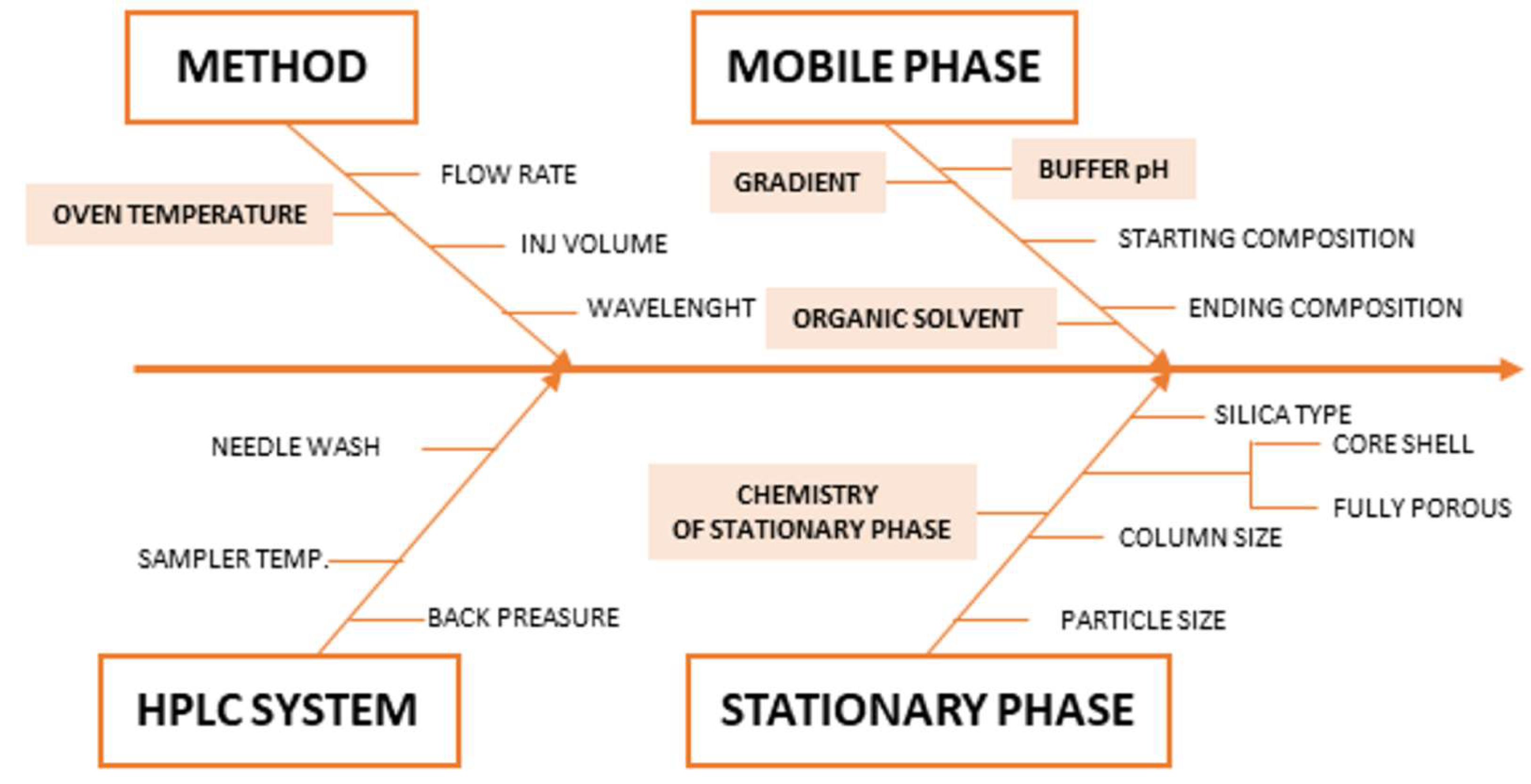
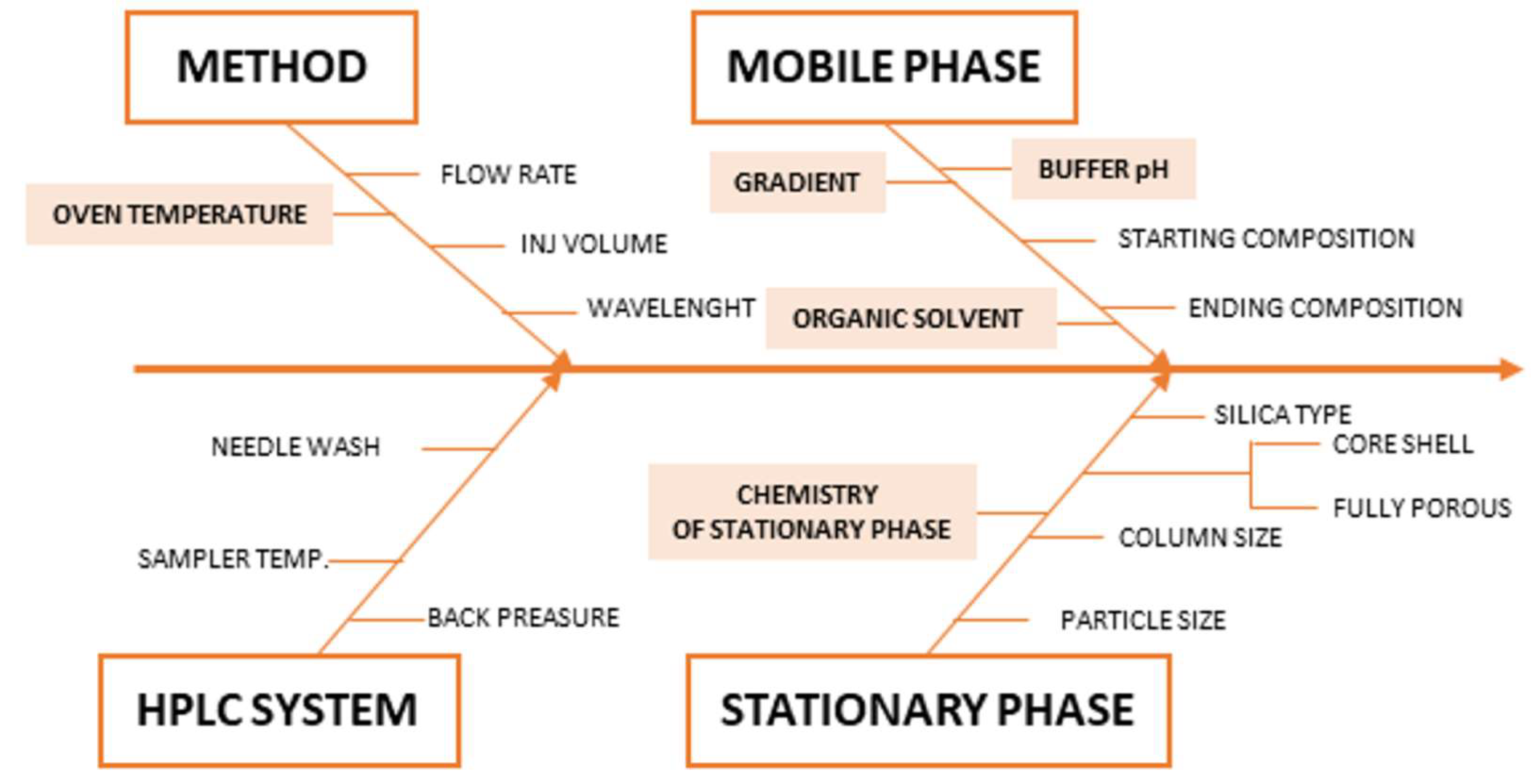
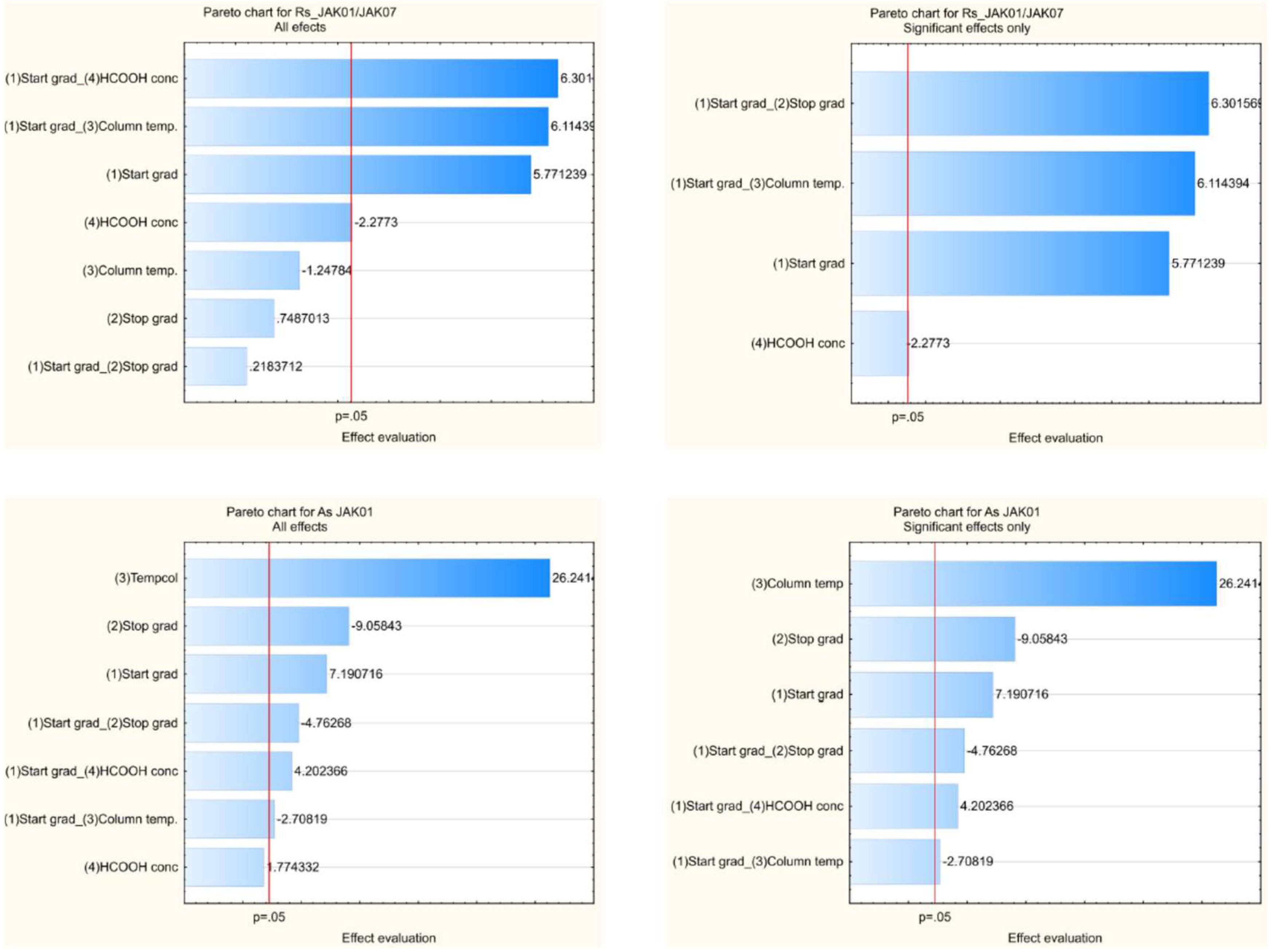
2.2. Risk Assessment, Critical Method Attributes CMAs, Critical Method Parameters CMPs
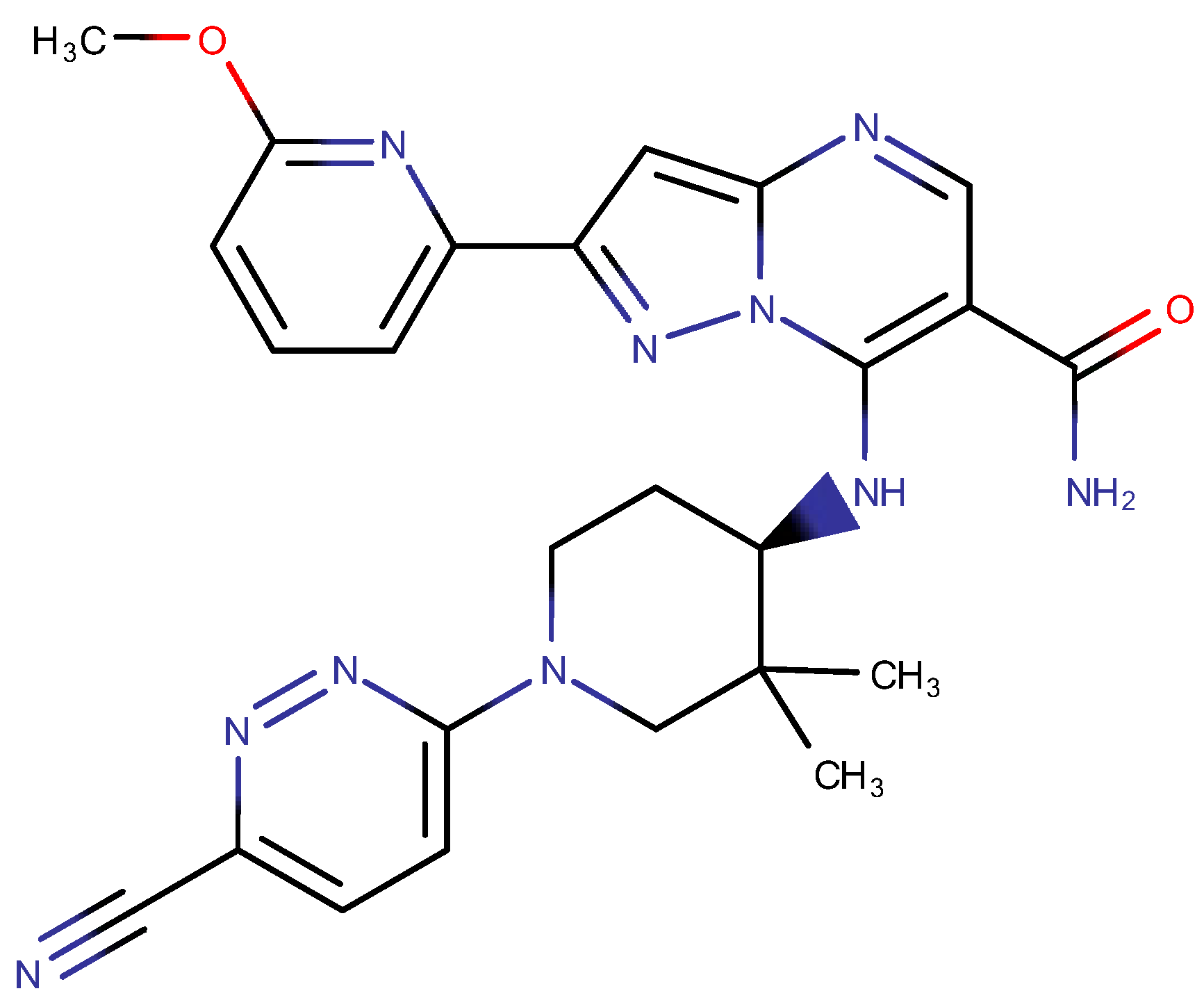
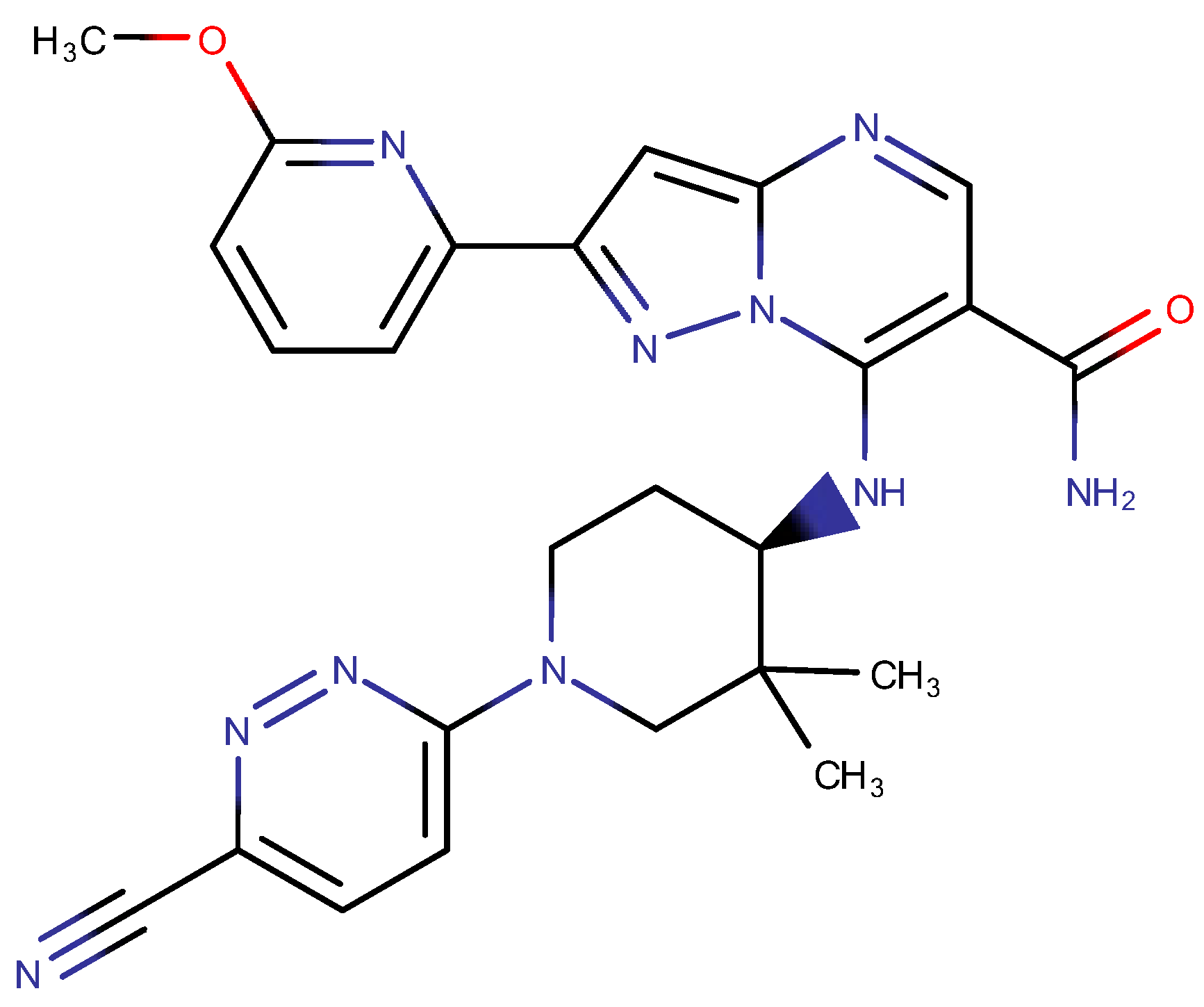
2.3. Structure-Based Pre-Development Study
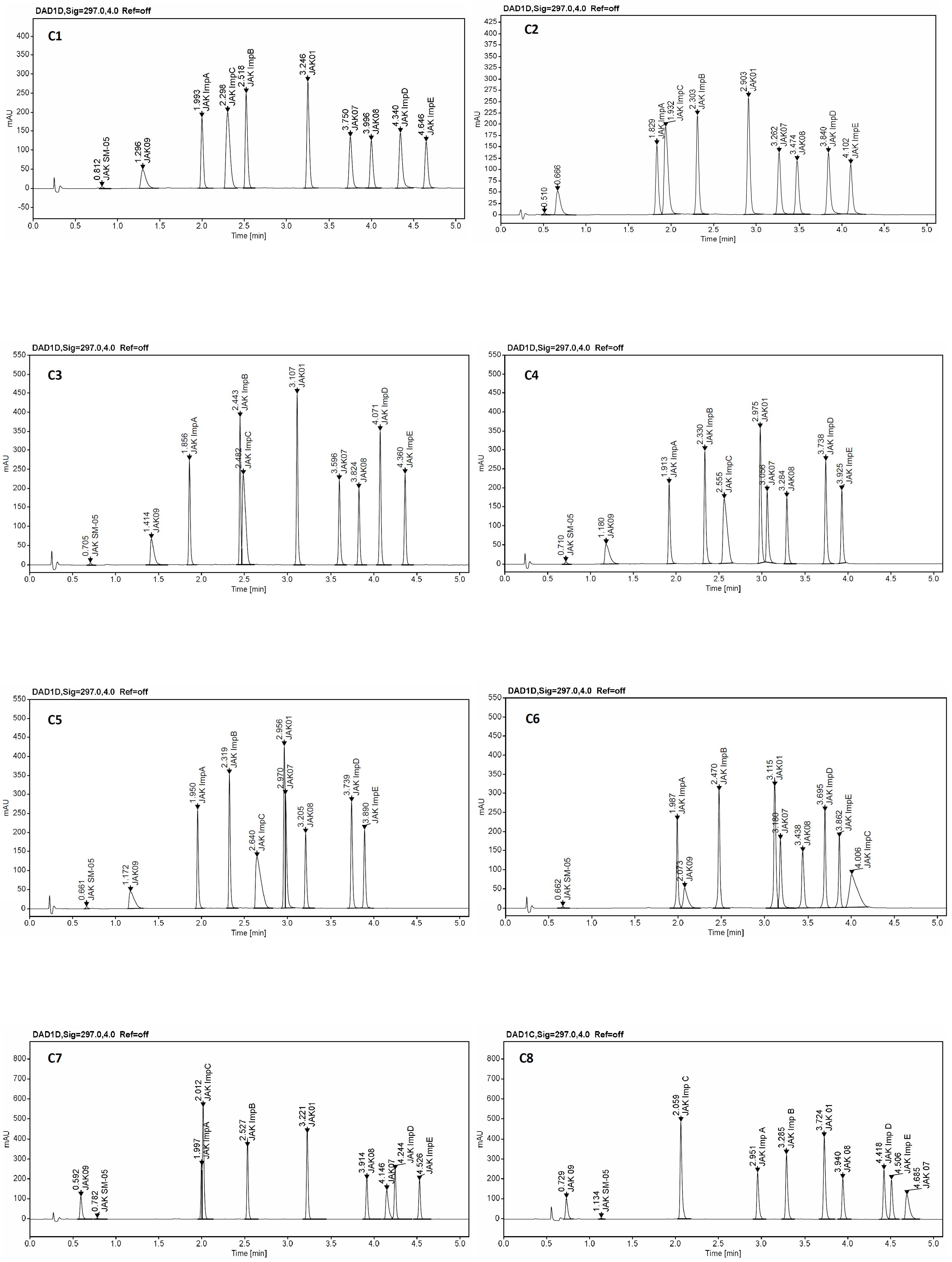
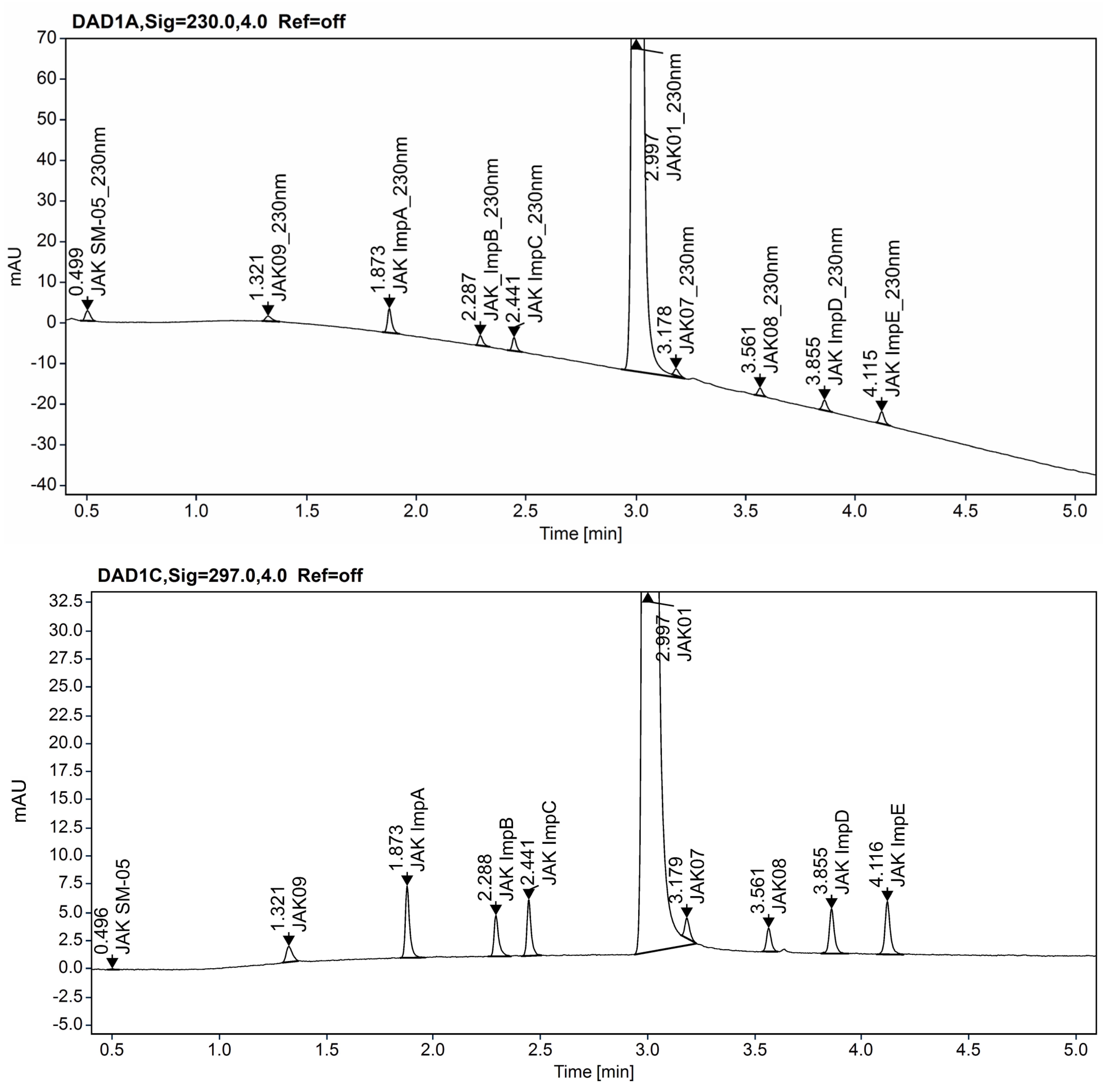
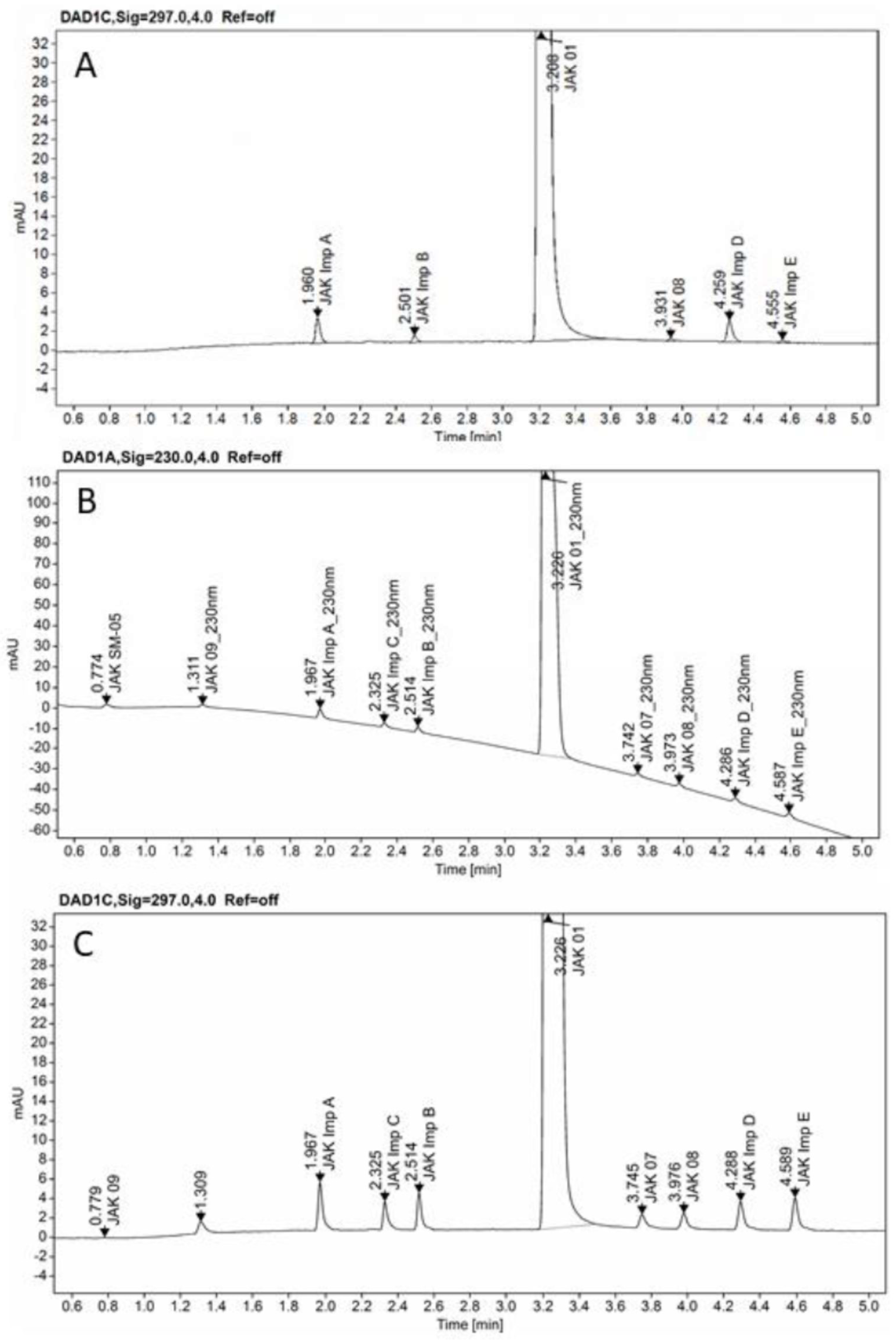
2.4. Screening Study
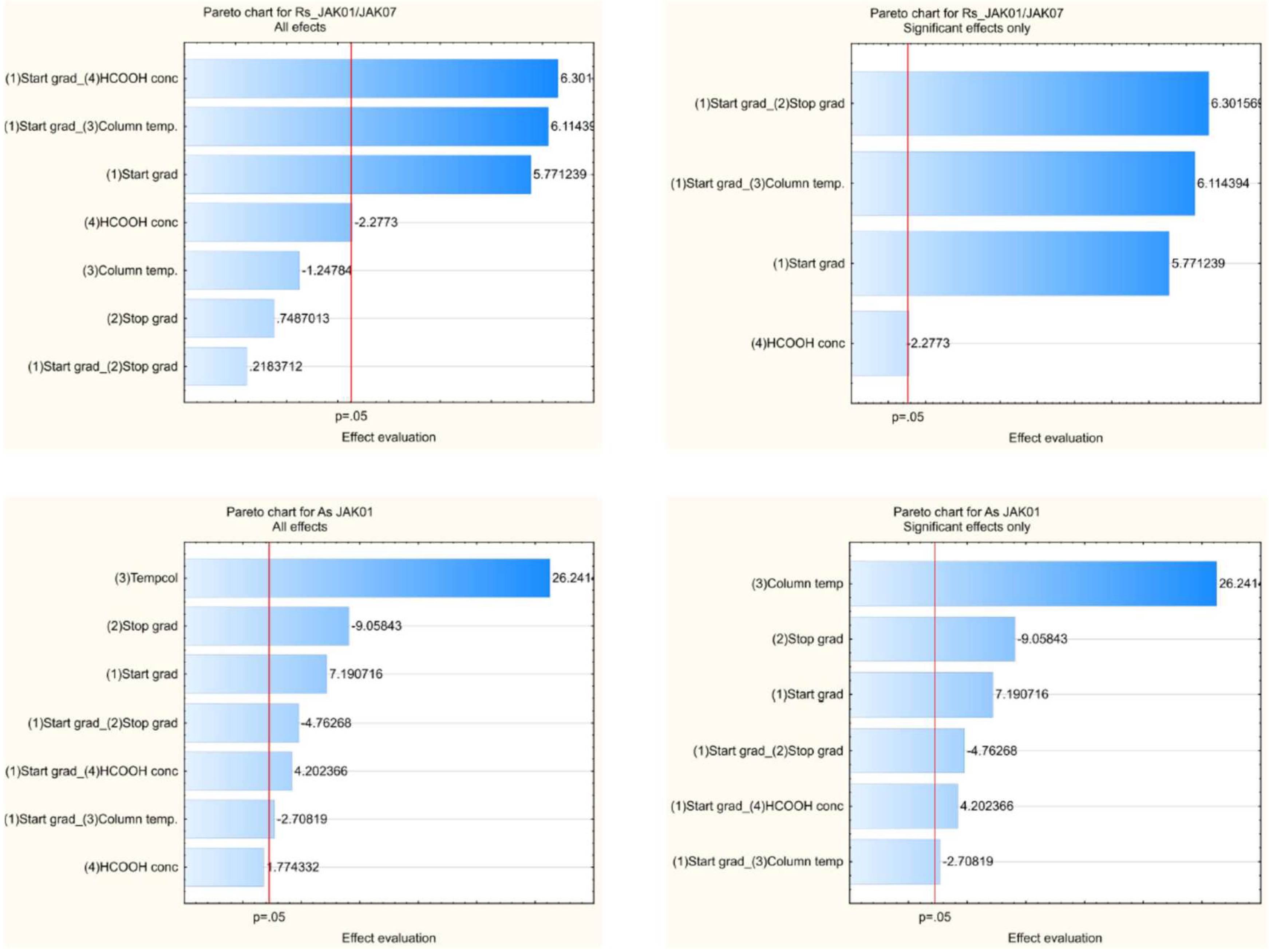
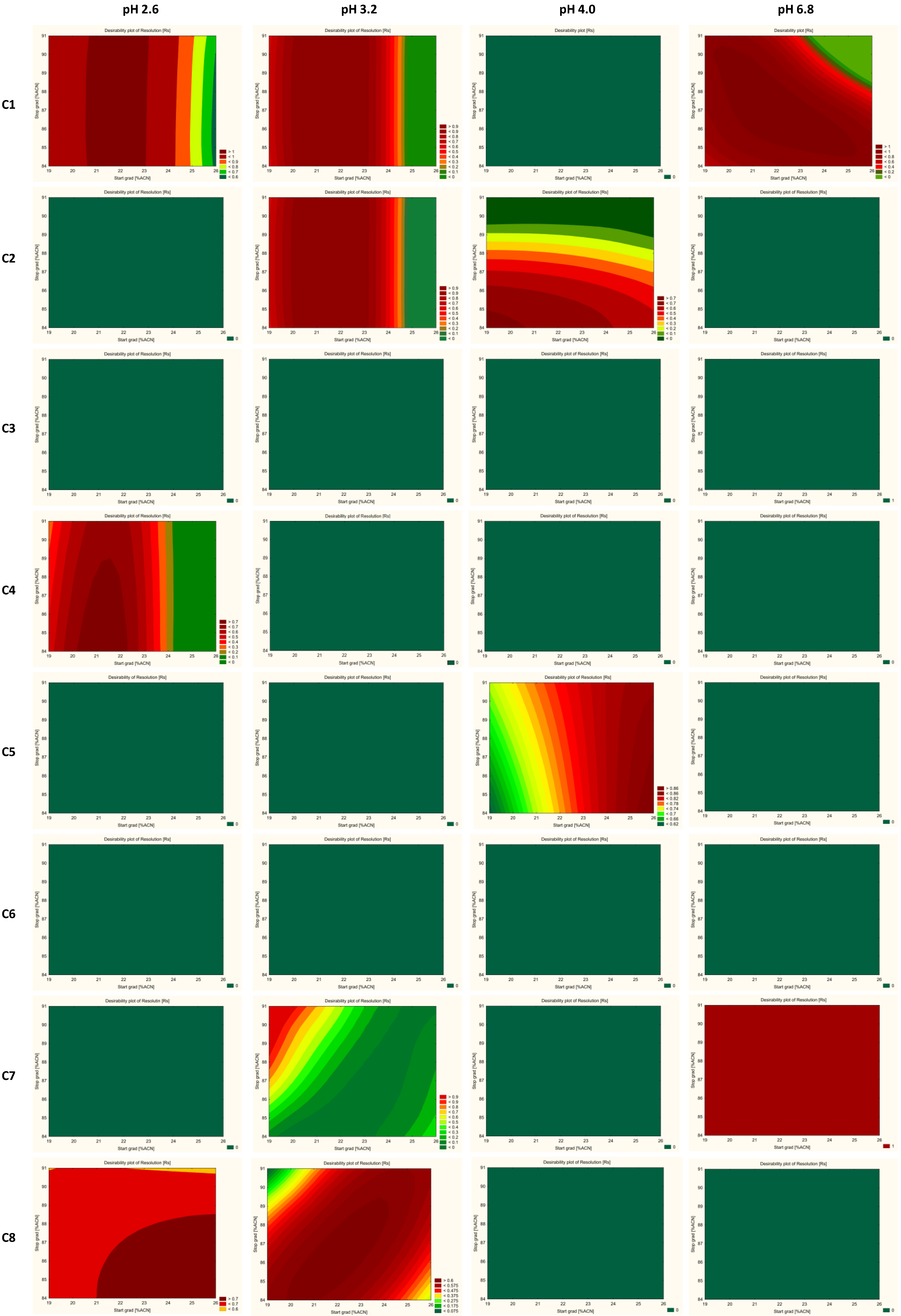
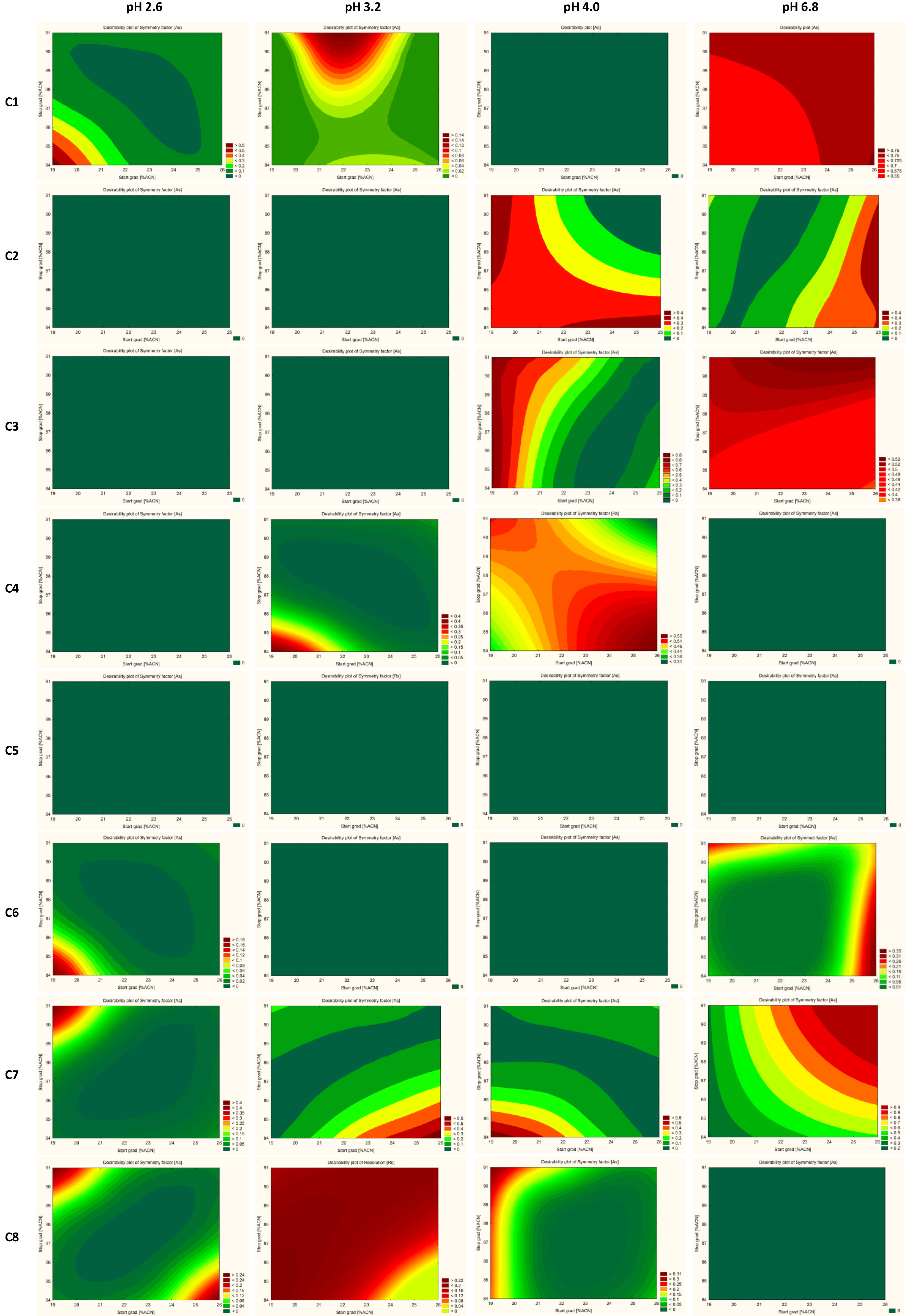
2.5. Optimization and Robustness Testing
2.6. Method Operable Design Region (MODR) and Control Strategy
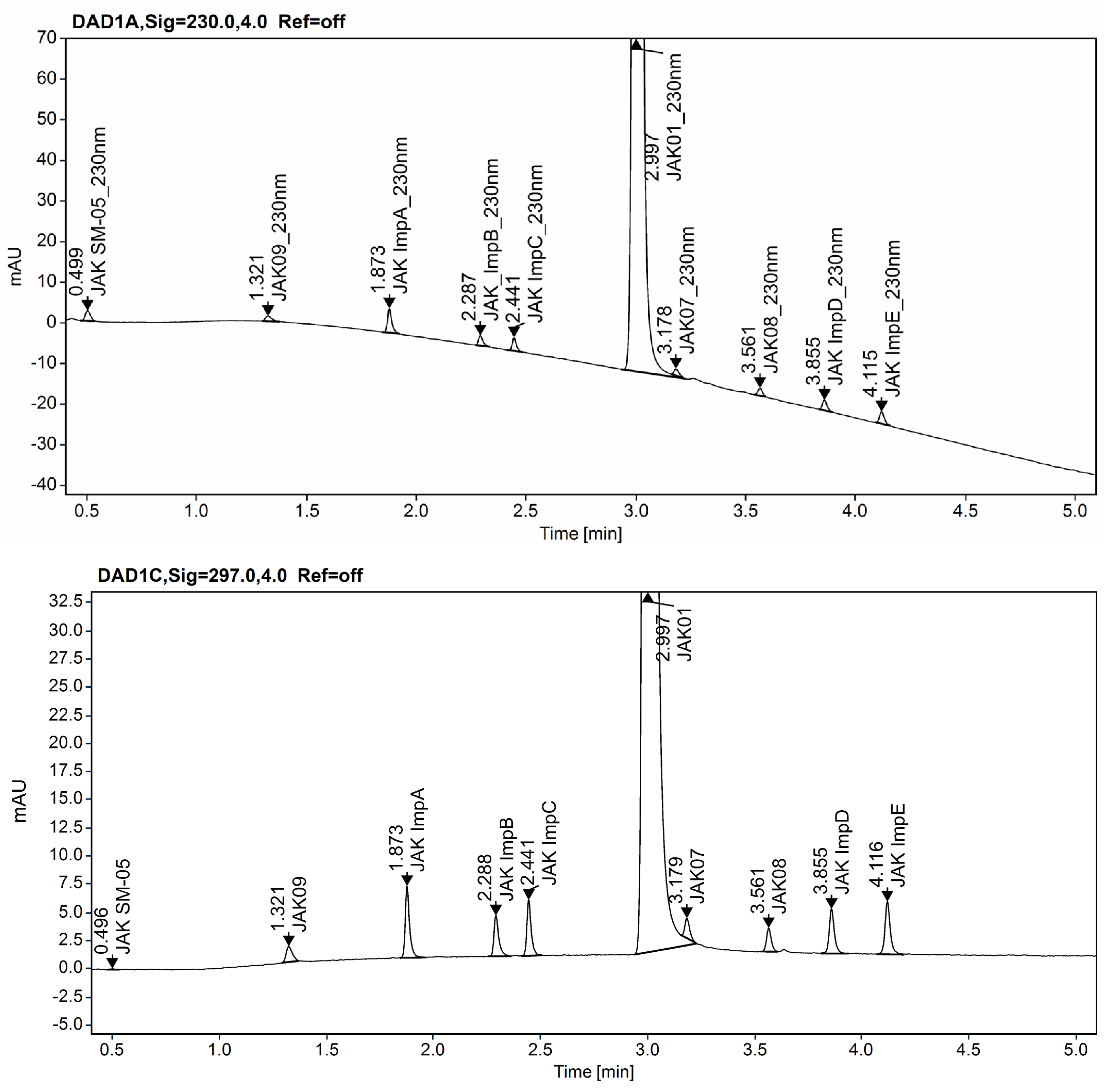
2.7. Validation
3. Material and Methods
3.1. Chemicals and Reagents
3.2. Solutions and Sample Preparations
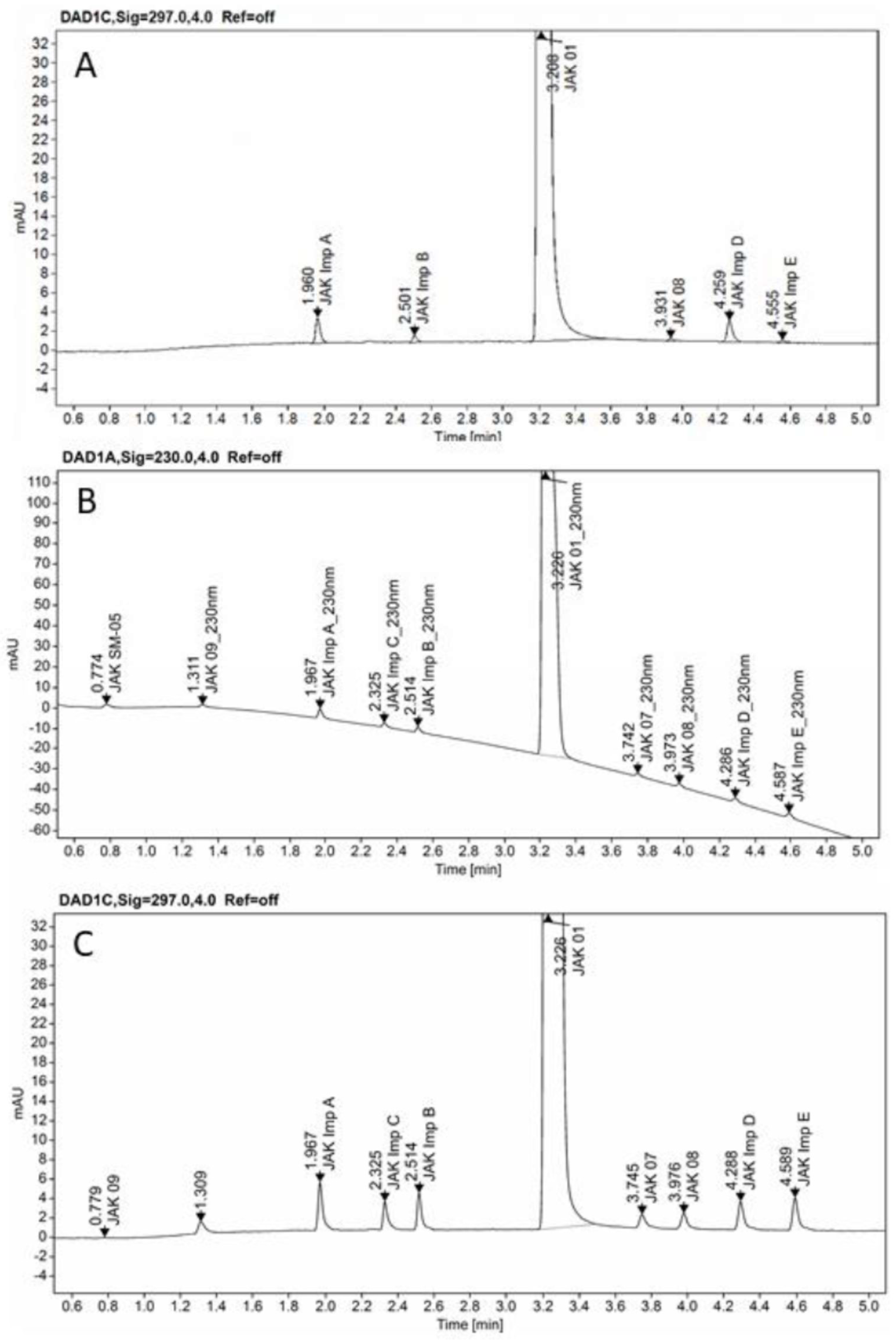
3.3. Instrumentations and Chromatographic Conditions
3.4. Calculations and Software
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Elder, D.; Teasdale, A. ICH, Q9, Quality Risk Management. In ICH Quality Guidelines: An Implementation Guide; Wiley: Hoboken, NJ, USA, 2017. [Google Scholar]
- Holm, P.; Allesø, M.; Bryder, M.C.; Holm, R. ICH, Q8(R2), Pharmaceutical Development. In ICH Quality Guidelines: An Implementation Guide; Wiley: Hoboken, NJ, USA, 2017. [Google Scholar]
- Borman, P.; Chatfield, M.; Nethercote, P.; Thompson, D.; Truman, K. The application of quality by design to analytical methods. Pharm. Technol. 2007, 31, 142–152. [Google Scholar]
- Schweitzer, M.; Pohl, M.; Brown, M.H.; Nethercote, P.; Borman, P.; Smith, K.; Larew, J. Implications and Opportunities of Applying QbD Principles to Analytical Measurements. Pharm. Technol. 2010, 34, 52–59. [Google Scholar]
- Vogt, F.G.; Kord, A.S. Development of quality-by-design analytical methods. J. Pharm. Sci. 2011, 100, 797–812. [Google Scholar] [CrossRef] [PubMed]
- Castle, B.C.; Forbes, R.A. Impact of Quality by Design in Process Development on the Analytical Control Strategy for a Small-Molecule Drug Substance. J. Pharm. Innov. 2013, 8, 247–264. [Google Scholar] [CrossRef]
- Reid, G.; Morgado, J.; Barnett, K.; Harrington, B.; Harwood, J.; Fortin, D. Analytical quality by design (AQbD) in pharmaceutical development. Am. Pharm. Rev. 2013, 16, 49–59. [Google Scholar]
- Parra, M.K.; Schmidt, A.H. Life Cycle Management of Analytical Methods. J. Pharm. Biomed. 2018, 147, 506–517. [Google Scholar] [CrossRef]
- Dispas, A.; Avohou, H.T.; Lebrun, P.; Hubert, P.; Hubert, C. Quality by design approach for the analysis of impurities in pharmaceutical drug products and drug substances. TrAC Trends Anal. Chem. 2018, 101, 24–33. [Google Scholar] [CrossRef]
- Basso, J.; Mendes, M.; Cova, T.F.; Sousa, J.J.; Pais, A.A.; Vitorino, C. Analytical Quality by Design (AQbD) as a multiaddressable platform for co-encapsulating drug assays. The Royal Society of Chemistry 2018. Anal. Methods 2018, 10, 5659. [Google Scholar] [CrossRef]
- Pasquini, B.; Orlandini, S.; Furlanetto, S.; Gotti, R.; Del Bubba, M.; Boscaro, F.; Bertaccini, B.; Douša, M.; Pieraccini, G. Qualit by Design as risk-based strategy in pharmaceutical analysis: Development of a liquid chromatography-tandem mass spectrometry method for determination of nintedanib and its impurities. J. Chromatogr. A 2020, 1611, 460615. [Google Scholar] [CrossRef]
- Pawar, A.; Pandita, N. Statistically Designed, Targeted Profile UPLC Method Development for Assay and Purity of Haloperidol in Haloperidol Drug Substance and Haloperidol 1 mg Tablets. Chromatographia 2020, 83, 725–737. [Google Scholar] [CrossRef]
- Hibbert, D.B. Experimental design in chromatography: A tutorial review. J. Chromatogr. B 2012, 910, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Kochling, J.; Wu, W.; Hua, Y.; Guan, Q.; Castaneda-Merced, J. A platform analytical quality by design (AQbD) approach for multiple UHPLC-UV and UHPLC–MS methods development for protein analysis. J. Pharm. Biomed. Anal. 2016, 125, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Gaudin, K.; Ferey, L. Quality by Design: A Tool for Separation Method Development in Pharmaceutical Laboratories. LCGC 2016, 29, 16–25. [Google Scholar]
- Rácz, N.; Molnár, I.; Zöldhegyi, A.; Rieger, H.-J.; Kormány, R. Simultaneous optimization of mobile phase composition and pH using retention modeling and experimental design. J. Pharm. Biomed. Anal. 2018, 160, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Orlandini, S.; Pinzauti, S.; Furlanetto, S. Application of quality by design to the development of analytical separation methods. Anal. Bioanal. Chem. 2013, 405, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Terfloth, G.J.; Kord, A.S. A systematic approach to RP-HPLC method development in a pharmaceutical QbD environment. Am. Pharm. Rev. 2009, 12, 87–95. [Google Scholar]
- Bhaskaran, N.A.; Kumar, L.; Reddy, M.S.; Pai, G.K. An analytical “quality by design” approach in RP-HPLC method development and validation for reliable and rapid estimation of irinotecan in an injectable formulation. Acta Pharm. 2021, 71, 57–79. [Google Scholar] [CrossRef]
- Jackson, P.; Borman, P.J.; Campa, C.; Chatfield, M.J.; Godfrey, M.; Hamilton, P.R.; Hoyer, W.; Norelli, F.; Orr, R.; Schofield, T. Using the analytical target profile to drive the analytical method lifecycle. Anal. Chem. 2019, 91, 2577–2585. [Google Scholar] [CrossRef]
- Rathore, A.S.; Winkle, H. Quality by design for biopharmaceuticals. Nat. Biotechnol. 2009, 27, 26–34. [Google Scholar] [CrossRef]
- Yu, L.X. Pharmaceutical quality by design: Product and process development, understanding, and control. Pharm. Res. 2007, 25, 781–791. [Google Scholar] [CrossRef]
- Csoka, I.; Pallagi, E.; Paal, T.L. Extension of quality-by-design concept to the early development phase of pharmaceutical R&D processes. Drug Discov. Today 2018, 23, 1340–1343. [Google Scholar] [PubMed]
- Deidda, R.; Orlandini, S.; Hubert, P.; Hubert, C. Risk-based approach for method development in pharmaceutical quality control context: A critical review. J. Pharm. Biomed. Anal. 2018, 161, 110–121. [Google Scholar] [CrossRef] [PubMed]
- ICH Harmonised Tripartite Guideline. Validation of Analytical Procedures: Text and Methodology Q2(R1). In Proceedings of the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, Geneva, Switzerland, 9 November 2005.
- Final Concept Paper: ICH Q14: Analytical Procedure Development and Revision of Q2(R1) Analytical Validation. Available online: https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q2_Q14/Q2R2Q14EWG_ConceptPaper_2018_1115.pdf (accessed on 25 May 2021).
- Tome, T.; Žigart, N.; Časar, Z.; Obreza, A. Development and Optimization of Liquid Chromatography Analytical Methods by Using AQbD Principles: Overview and Recent Advances. Org. Process Res. Dev. 2019, 23, 1784–1802. [Google Scholar] [CrossRef]
- Mroczkiewicz, M.; Stypik, B.; Bujak, A.; Szymczak, K.; Gunerka, P.; Dubiel, K.; Wieczorek, M.; Pieczykolan, J. Pyrazole[1,5-a]Pyrimidine Derivatives as Kinase Jak Inhibitors. W.O. Patent WO2018206739A1, 15 November 2018. [Google Scholar]
- Mroczkiewicz, M.; Stypik, B.; Bujak, A.; Szymczak, K.; Gunerka, P.; Dubiel, K.; Wieczorek, M.; Pieczykolan, J. Pyrazole[1,5-a]Pyrimidine Derivatives as Kinase Jak Inhibitors. E.P. Patent EP3621966 B1, 18 March 2020. [Google Scholar]
- Dulak-Lis, M.; Bujak, A.; Gala, K.; Banach, M.; Kędzierska, U.; Miszkiel, J.; Hucz-Kalitowska, J.; Mroczkiewicz, M.; Stypik, B.; Szymczak, K.; et al. A novel JAK/ROCK inhibitor, CPL409116, demonstrates potent efficacy in the mouse model of systemic lupus erythematosus. J. Pharmacol. Sci. 2021, 145, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Iuliani, P.; Carlucci, G.; Marrone, A. Investigation of the HPLC response of NSAIDs by fractional experimental design and multivariate regression analysis. Response optimization and new retention parameters. J. Pharm. Biomed. Anal. 2010, 51, 46–55. [Google Scholar] [CrossRef]
- Kishore, C.R.P.; Mohan, G.V.K. Development and Validation of Amlodipine Impurities in Amlodipine Tablets Using Design Space Computer Modeling. Am. J. Anal. Chem. 2016, 7, 918–926. [Google Scholar] [CrossRef]
- Prasad, S.S.; Mohan, G.V.K.; Babu, A.N. Orient. J. Chem. 2019, 35, 140–149. [Google Scholar]
- Sahu, P.K.; Ramisetti, N.R.; Cecchi, T.; Swain, S.; Patro, C.S.; Panda, J. An overview of experimental designs in HPLC method development and validation. J. Pharm. Biomed. Anal. 2018, 147, 590–611. [Google Scholar] [CrossRef]
- OECD. Test No. 107: Partition Coefficient (n-Octanol/Water): Shake Flask Method; OECD Publishing: Paris, France, 1995. [Google Scholar]
- Albert, A. Selective Toxicity: The Physicochemical Basis of Therapy; Chapman and Hall: London, UK, 1979. [Google Scholar]
- Klose, M.H.M.; Theiner, S.; Varbanov, H.P.; Hoefer, D.; Pichler, V.; Galanski, M.; Meier-Menches, S.M.; Keppler, B.K. Development and Validation of Liquid Chromatography-Based Methods to Assess the Lipophilicity of Cytotoxic Platinum(IV) Complexes. Inorganics 2018, 6, 130. [Google Scholar] [CrossRef]
- Dolan, J.W. Back to Basics: The Role of pH in Retention and Selectivity. LCGC N. Am. 2017, 35, 22–28. [Google Scholar]
- Lewis, J.A.; Lommen, D.C.; Raddatz, W.D.; Dolan, J.W.; Snyder, L.R.; Molnar, I. Computer simulation for the prediction of separation as a function of pH for reversed-phase high-performance liquid chromatography: I. Accuracy of a theory-based model. J. Chromatogr. A 1992, 592, 183–195. [Google Scholar] [CrossRef]
- ACD/Percepta; Version 2020.1.1; Advanced Chemistry Development, Inc.: Toronto, ON, Canada, 2020; Available online: http://www.acdlabs.com (accessed on 26 November 2020).
- Valkó, K.; Bevan, C.; Reynolds, D. Chromatographic Hydrophobicity Index by Fast-Gradient RP-HPLC: A High-Throughput Alternative to log P/log D. Anal. Chem. 1997, 69, 2022–2029. [Google Scholar] [CrossRef]
- Valko, K. Physicochemical and Biomimetic Properties in Drug Discovery—Chromatographic Techniques for Lead Optimization, 1st ed.; Wiley: Hoboken, NJ, USA, 2014. [Google Scholar]
- Snyder, L.R.; Dolan, J.W.; Carr, P.W. The hydrophobic-subtraction model of reversed-phase column selectivity. J. Chromatogr. A 2004, 1060, 77–116. [Google Scholar] [CrossRef]
- Snyder, L.R.; Dolan, J.W.; Marchand, D.H.; Carr, P.W. The hydrophobic-subtraction model of reversed-phase column selectivity. Adv. Chromatogr. 2012, 50, 297–376. [Google Scholar] [PubMed]
- Available online: http://www.hplccolumns.org (accessed on 29 January 2021).
- Available online: https://apps.usp.org/app/USPNF/columnsDB.html (accessed on 29 January 2021).
- Committee for Human Medicinal Products. ICH guideline M7(R1) on assessment and control of DNA reactive (mutagenic) impurities in pharmaceuticals to limit potential carcinogenic risk. Int. Conf. Harmon. 2015, 7, 1–110. [Google Scholar]
- Baertschi, S.; Olsen, B. Chapter 12—Mutagenic impurities. In Specification of Drug Substances and Products, 2nd ed.; Riley, C.M., Rosanske, T.W., Reid, G., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 321–344. [Google Scholar]
- Shaikh, T.; Gosar, A.; Sayyed, H. Nitrosamine Impurities in Drug Substances and Drug Products. J. Adv. Pharm. Pract. 2020, 2, 48–57. [Google Scholar]
- Orbitrium. DEREK Nexus—Trial licence. Available online: https://www.optibrium.com (accessed on 2 November 2020).
- Lhasa Limited. DEREK Nexus. Available online: https://www.lhasalimited.org (accessed on 2 November 2020).
- Marchant, C.A.; Briggs, K.A.; Long, A. In Silico Tools for Sharing Data and Knowledge on Toxicity and Metabolism: Derek for Windows, Meteor, and Vitic. Toxicol. Mech. Methods 2008, 18, 177–187. [Google Scholar] [CrossRef]
- US EPA Research. Toxicity Estimation Software Tool (TEST). Available online: https:/www.epa.gov/chemical-research/toxicity-estimation-software-tool-test (accessed on 30 October 2020).
- Martin, T. User’s Guide for T.E.S.T. (Version 4.2) (Toxicity Estimation Software Tool) A Program to Estimate Toxicity from Molecular Structure; EPA: Washington, DC, USA, 2016; Volume 63. [Google Scholar]
- StatSoft’s Electronic Statistics Textbook. 2006. Available online: http://www.statsoft.pl/textbook/stathome.html (accessed on 27 January 2021).
- TIBCO Software Inc. Data Science Textbook. 2020. Available online: https://docs.tibco.com/data-science/textbook (accessed on 27 January 2021).
- Stanisz, A. Przystępny kurs Statystyki z Zastosowaniem STATISTICA PL na Przykładach z Medycyny—Tom I–III; StatSoft: Kraków, Poland, 2006. [Google Scholar]
- Valko, K.L. Application of biomimetic HPLC to estimate in vivo behavior of early drug discovery compounds. Future Drug Discov. 2019, 1, FDD11. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name | logP | clogD 2.6 | clogD 4.5 | clogD 7.4 | clogD 10.5 | pKa (Acid) | pKa (Base) | CHILogD pH 2.6 | CHILogD pH 7.4 | CHILogD pH 10.5 |
|---|---|---|---|---|---|---|---|---|---|---|
| SM-05 | 0.46 | 0.46 | 0.46 | 0.46 | 0.46 | − | −2.50 | −0.12 | −0.03 | 0.99 |
| JAK-09 | 1.27 | −2.03 | −1.83 | −1.16 | 1.16 | 12.52 | −1.12 2.68 9.92 | 0.71 | 1.22 | 2.38 |
| JAK ImpC | −4.22 | −4.29 | −4.22 | −4.22 | −4.22 | 13.41 | −0.95 2.00 | 1.25 | 3.41 | 3.23 |
| JAK ImpA | 0.54 | 0.1 | 0.53 | 0.43 | −1.91 | 7.95 12.58 | −0.53 2.00 2.68 | 1.49 | 1.49 | 1.99 |
| JAK ImpB | 1.07 | −0.26 | 1.02 | 1.07 | 1.07 | 12.52 15.09 | −1.11 2.70 3.56 | 2.40 | 2.21 | 2.45 |
| CPL409116 (JAK01) | 1.44 | 0.98 | 1.43 | 1.44 | 1.44 | 12.52 | −1.11 2.00 2.70 | 3.51 | 3.09 | 3.05 |
| JAK-07 | 3.45 | 0.96 | 0.62 | 0.3 | 0.3 | − | −0.78 0.90 3.98 | 3.80 | 2.55 | 2.57 |
| JAK-08 | 2.55 | 2.3 | 2.55 | 2.55 | 2.54 | 12.52 | −1.12 0.79 2.70 | 4.63 | 4.04 | 3.61 |
| JAK ImpE | 3 | 2.71 | 3 | 3 | 3 | − | −1.20 1.98 2.40 | 4.86 | 4.82 | 4.07 |
| JAK ImpD | 2.65 | 2.36 | 2.65 | 2.65 | 2.65 | − | −1.20 1.99 2.40 | 5.12 | 4.46 | 3.83 |
| L.p. | Column Name | Fs | H | S | A | B | C (pH 2.8) | C (pH 7.0) | EB | USP Type | Phase Chemistry |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1. | ZORBAX Eclipse Plus C18 | 0 | 1.03 | 0 | −0.0700 | −0.0200 | 0 | 0.0200 | 7.8 | L1 | C18 |
| 2. | Kinetex EVO C18 | 9.65 | 1.01 | −0.00600 | −0.170 | −0.0240 | −0.110 | −0.0100 | 4.38 | L1 | C18 |
| 3. | ACQUITY UPLC BEH C18 | 15.51 | 1.00 | 0.0280 | −0.366 | 0.00700 | 0.142 | 0.0880 | 6.4 | L1 | C18 |
| 4. | InfinityLab Poroshell 120 Phenyl Hexyl | 18.23 | 0.752 | −0.0830 | −0.394 | 0.0180 | 0.136 | 0.140 | 3.59 | L11 | Phenyl−Hexyl |
| 5. | Kinetex Biphenyl | 26.75 | 0.697 | −0.173 | −0.583 | 0.0340 | 0.122 | 0.817 | 2.63 | L11 | Biphenyl |
| 6. | Kinetex PFP | 78.79 | 0.680 | 0.0800 | −0.270 | −0.0300 | 0.940 | 1.53 | 2.4 | L43 | Pentafluorophenyl |
| 7. | ACQUITY UPLC CSH C18 | 21.15 | 0.954 | −0.00200 | −0.179 | 0.118 | 0.0820 | 0.171 | 5.62 | L1 | C18 |
| 8. | ACQUITY UPLC CSH Fluoro−Phenyl | 25.63 | 0.708 | −0.0590 | −0.435 | 0.129 | −0.0680 | 0.223 | 2.865 | L11 | Fluoro−Phenyl |
| Compound Name | DEREK Nexus Results (Q)SAR Rule-Based | T.E.S.T Results (Q)SAR Statistic-Based | ICH M7 Class |
|---|---|---|---|
| CPL409116 (JAK-01) | Mutagenicity is INACTIVE | Mutagenicity NEGATIVE Predicted value p = 0.25 | Class 5 |
| JAK-07 | Mutagenicity is INACTIVE | Mutagenicity NEGATIVE Predicted value p = 0.26 | Class 5 |
| JAK-08 | Mutagenicity is INACTIVE | Mutagenicity NEGATIVE Predicted value p = 0.23 | Class 5 |
| JAK-09 | Mutagenicity is INACTIVE | Mutagenicity NEGATIVE Predicted value p = 0.39 | Class 5 |
| JAK SM-5 | Mutagenicity is INACTIVE | Mutagenicity NEGATIVE Predicted value p = 0.22 | Class 5 |
| JAK ImpA | Mutagenicity is INACTIVE | Mutagenicity NEGATIVE Predicted value p = 0.16 | Class 5 |
| JAK ImpB | Mutagenicity is INACTIVE | Mutagenicity NEGATIVE Predicted value p = 0.28 | Class 5 |
| JAK ImpC | Mutagenicity is INACTIVE | Mutagenicity NEGATIVE Predicted value p = 0.29 | Class 5 |
| JAK ImpD | Mutagenicity is INACTIVE | Mutagenicity NEGATIVE Predicted value p = 0.15 | Class 5 |
| JAK ImpE | Mutagenicity is INACTIVE | Mutagenicity NEGATIVE Predicted value p = 0.16 | Class 5 |
| Column | pH of Buffer | Critical Method Parameters (CMAs) | Optimal Conditions of CMPs | |
|---|---|---|---|---|
| Start Gradient [% ACN] | End Gradient [% ACN] | |||
| ZORBAX Eclipse Plus C18 (2.1 × 50 mm, 1.8 µm, Agilent Technologies) | 2.6 | Symmetry factor of JAK09 and JAK ImpD | 19–21 | 84–86 |
| 3.2 | Resolution of JAK SM-05/JAK09 Symmetry factor of JAK 09 Symmetry factor of JAK ImpC (>1.77) Symmetry factor of JAK ImpD | 21–23 | 88–90 | |
| 4.0 | Resolution of JAK ImpC/JAK ImpB (<1.5) Symmetry factor of JAK ImpD (>2.0) Symmetry factor of JAK ImpE (>1.9) | not found | not found | |
| 6.8 | Resolution of JAK ImpC—JAK Imp E | 20–23 | 85–90 | |
| Kinetex EVO C18 (2.1 × 50 mm, 1.7 µm, Phenomenex) | 2.6 | Resolution of JAK ImpA/JAK ImpB (<1.5) Symmetry factor of JAK09 (>1.8) Symmetry factor of JAK ImpC (>1.8) | not found | not found |
| 3.2 | Symmetry factor of JAK09 (>1.8) Symmetry factor of JAK ImpB (>1.8) Symmetry factor of JAK ImpD (>1.8) | not found | not found | |
| 4.0 | Symmetry factor of JAK09 (<1.7) Symmetry factor of JAK ImpC (<1.7) | 20–24 | 85–87 | |
| 6.8 | Resolution of JAK ImpB/JAK07 | not found | not found | |
| ACQUITY UPLC BEH C18 (2.1 × 50 mm, 1.7 µm Waters) | 2.6 | Resolution of JAK ImpB/JAK ImpC (<0.8) | not found | not found |
| 3.2 | Resolution of JAK ImpC/JAK ImpB (<0.5) Symmetry factor of JAK09 (>2.1) Symmetry factor of JAK ImpC (>1.9) | not found | not found | |
| 4.0 | Resolution of JAK ImpB/JAK ImpC (<0.8) | not found | not found | |
| 6.8 | Symmetry factor of JAK09 (>1.6) | 20–25 | 85–90 | |
| InfinityLab Poroshell 120 Phenyl Hexyl (2.1 × 50 mm, 1.9 µm, Agilent Technologies) | 2.6 | Symmetry factor of JAK ImpC (>2.0) | not found | not found |
| 3.2 | Resolution of JAK07/JAK08 (<0.5) Symmetry factor JAK07 (>1.8) | not found | not found | |
| 4.0 | Resolution of JAK08/JAK01 (<0.5) Symmetry factor JAK07 (<0.6) | not found | not found | |
| 6.8 | Symmetry factor JAK09 (>2.0) Symmetry factor JAK07 (>1.8) | not found | not found | |
| Kinetex Biphenyl (2.1 × 50 mm, 1.7 µm, Phenomenex) | 2.6 | Resolution of JAK07/JAK01 (<0.6) Symmetry factor of JAK09 (>2.3) Symmetry factor of JAK ImpB (>2.4) Symmetry factor of JAK07 (>2.3) | not found | not found |
| 3.2 | Resolution of JAK07/JAK01 (<1.1) Symmetry factor of JAK09 (>1.9) Symmetry factor of JAK ImpB (>1.9) | not found | not found | |
| 4.0 | Symmetry factor of JAK01 (>2.5) | not found | not found | |
| 6.8 | Resolution of JAK07/JAK ImpB (<0.8) Symmetry factor of JAK07 (>2.4) | not found | not found | |
| Kinetex PFP (2.1 × 50 mm, 1.7 µm, Phenomenex) | 2.6 | Resolution of JAK09/JAK ImpA (<1.6) Symmetry factor of JAK09 (>1.8) Symmetry factor of JAK ImpC (>1.8) | not found | not found |
| 3.2 | Resolution of JAK08/JAK07 (<0.5) Symmetry factor of JAK09 (>1.8) Symmetry factor of JAK ImpC (>2.1) | not found | not found | |
| 4.0 | Resolution of JAK ImpE/JAK ImpD (<0.8) Symmetry factor of JAK ImpC (>1.9) | not found | not found | |
| 6.8 | Resolution of JAK08/JAK07 (<0.85) | not found | not found | |
| ACQUITY UPLC CSH C18 (2.1 × 50 mm, 1.7 µm, Waters) | 2.6 | Resolution of JAK ImpC/JAK ImpA (<1.8) Symmetry factor of JAK07 (>1.8) | not found | not found |
| 3.2 | Resolution of JAK07/JAK08 (<2.0) Symmetry factor of JAK07 (>1.8) | not found | not found | |
| 4.0 | Resolution of JAK ImpB/JAK ImpC (<0.6) | not found | not found | |
| 6.8 | Symmetry factor of JAK09 Symmetry factor of JAK ImpB Symmetry factor of JAK07 | 20–23 | 85–87 | |
| ACQUITY UPLC CSH Fluoro-Phenyl (2.1 × 100 mm, 1.7 µm, Waters) | 2.6 | Resolution JAK ImpE/JAK ImpD (<2.28, >2.00) Symmetry factor of JAK07 (<1.8) | 20–21 | 89–90 |
| 3.2 | Resolution JAK ImpE/JAK ImpD (<2.3, >2.00) Symmetry factor of JAK07 (<1.8) | 20–23 | 85–90 | |
| 4.0 | Resolution JAK07/JAK ImpE (<1.3) | not found | not found | |
| 6.8 | Resolution JAK08/JAK01 Symmetry factor of JAK01 (<0.8) | not found | not found | |
| Start Gradient [%ACN] | End Gradient [%ACN] | Column Temperature [°C] | Concentration of HCOOH Solution [mM] | Results of Rs | Results of As | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| JAK 09 | JAK Imp A | JAK Imp C | JAK Imp B | JAK 01 | JAK 07 | JAK 08 | JAK Imp D | JAK Imp E | JAK SM-05 | JAK 09 | JAK Imp A | JAK Imp C | JAK Imp B | JAK 01 | JAK 07 | JAK 08 | JAK Imp D | JAK Imp E | ||||
| 19.0 | 84.0 | 28.0 | 9.0 | 11.20 | 7.83 | 5.01 | 2.79 | 11.70 | 7.42 | 4.20 | 4.2 | 4.36 | 1.57 | 1.35 | 1.28 | 1.40 | 1.25 | 1.20 | 1.26 | 1.17 | 1.22 | 1.23 |
| 21.0 | 86.0 | 28.0 | 9.0 | 3.02 | 8.27 | 3.95 | 3.14 | 12.15 | 7.61 | 3.60 | 4.57 | 4.19 | 1.56 | 1.79 | 1.31 | 1.46 | 1.26 | 1.18 | 1.22 | 1.18 | 1.16 | 1.20 |
| 19.0 | 86.0 | 28.0 | 11.0 | 6.88 | 7.04 | 3.63 | 2.87 | 10.20 | 6.75 | 2.83 | 3.74 | 3.54 | 1.56 | 1.43 | 1.15 | 1.44 | 1.14 | 1.16 | 1.21 | 1.20 | 1.20 | 1.20 |
| 21.0 | 84.0 | 28.0 | 11.0 | 2.70 | 7.00 | 4.23 | 2.96 | 10.75 | 6.96 | 2.96 | 3.80 | 3.75 | 1.60 | 1.65 | 1.37 | 1.45 | 1.49 | 1.43 | 1.39 | 1.39 | 1.50 | 1.51 |
| 19.0 | 86.0 | 32.0 | 9.0 | 6.43 | 6.71 | 3.28 | 3.08 | 9.50 | 6.44 | 2.62 | 3.40 | 3.51 | 1.54 | 1.47 | 1.55 | 1.38 | 1.55 | 1.59 | 1.44 | 1.48 | 1.51 | 1.52 |
| 21.0 | 84.0 | 32.0 | 9.0 | 2.57 | 8.32 | 3.77 | 3.37 | 11.92 | 7.52 | 3.11 | 4.16 | 4.31 | 1.46 | 1.65 | 1.71 | 1.57 | 1.73 | 1.69 | 1.60 | 1.59 | 1.60 | 1.65 |
| 19.0 | 84.0 | 32.0 | 11.0 | 6.09 | 7.22 | 3.38 | 3.02 | 12.00 | 6.90 | 3.18 | 4.25 | 4.28 | 1.42 | 1.47 | 1.78 | 1.49 | 1.68 | 1.59 | 1.51 | 1.46 | 1.52 | 1.57 |
| 21.0 | 86.0 | 32.0 | 11.0 | 2.35 | 8.28 | 3.75 | 3.32 | 11.69 | 7.78 | 3.06 | 4.14 | 4.18 | 1.60 | 1.64 | 1.77 | 1.53 | 1.63 | 1.56 | 1.49 | 1.45 | 1.54 | 1.54 |
| 20.0 | 85.0 | 30.0 | 10.0 | 5.73 | 9.32 | 4.54 | 3.24 | 14.04 | 8.53 | 3.98 | 5.43 | 4.76 | 1.39 | 1.72 | 1.31 | 1.38 | 1.41 | 1.32 | 1.28 | 1.23 | 1.40 | 1.24 |
| 19.0 | 84.0 | 28.0 | 9.0 | 11.6 | 7.07 | 5.38 | 2.84 | 11.51 | 7.61 | 4.28 | 4.09 | 4.30 | 1.55 | 1.34 | 1.33 | 1.38 | 1.27 | 1.25 | 1.25 | 1.22 | 1.26 | 1.29 |
| 21.0 | 86.0 | 28.0 | 9.0 | 3.07 | 8.15 | 4.01 | 3.18 | 12.05 | 7.45 | 3.45 | 4.85 | 4.49 | 1.56 | 1.79 | 1.27 | 1.46 | 1.23 | 1.18 | 1.18 | 1.15 | 1.18 | 1.23 |
| 19.0 | 86.0 | 28.0 | 11.0 | 7.34 | 7.28 | 3.81 | 2.91 | 10.91 | 6.99 | 3.02 | 3.88 | 3.61 | 1.53 | 1.43 | 1.12 | 1.48 | 1.10 | 1.11 | 1.15 | 1.12 | 1.18 | 1.22 |
| 21.0 | 84.0 | 28.0 | 11.0 | 2.65 | 7.73 | 4.02 | 2.88 | 10.32 | 6.64 | 2.78 | 4.06 | 3.93 | 1.63 | 1.62 | 1.34 | 1.42 | 1.41 | 1.46 | 1.40 | 1.41 | 1.46 | 1.50 |
| 19.0 | 86.0 | 32.0 | 9.0 | 6.36 | 6.79 | 3.29 | 3.17 | 9.51 | 6.52 | 2.66 | 3.39 | 3.50 | 1.54 | 1.44 | 1.52 | 1.36 | 1.51 | 1.52 | 1.41 | 1.42 | 1.51 | 1.51 |
| 21.0 | 84.0 | 32.0 | 9.0 | 2.54 | 8.37 | 3.75 | 3.36 | 11.27 | 7.19 | 2.98 | 4.02 | 4.20 | 1.45 | 1.66 | 1.73 | 1.61 | 1.77 | 1.67 | 1.54 | 1.54 | 1.58 | 1.61 |
| 19.0 | 84.0 | 32.0 | 11.0 | 6.37 | 7.38 | 3.70 | 3.18 | 12.33 | 6.55 | 3.20 | 4.00 | 4.32 | 1.40 | 1.50 | 1.78 | 1.52 | 1.69 | 1.57 | 1.48 | 1.45 | 1.53 | 1.55 |
| 21.0 | 86.0 | 32.0 | 11.0 | 2.34 | 8.20 | 3.77 | 3.34 | 11.77 | 7.73 | 3.02 | 4.10 | 4.14 | 1.62 | 1.66 | 1.72 | 1.53 | 1.65 | 1.59 | 1.49 | 1.46 | 1.59 | 1.56 |
| 20.0 | 85.0 | 30.0 | 10.0 | 5.75 | 9.26 | 4.53 | 3.27 | 14.10 | 8.51 | 3.99 | 5.41 | 4.70 | 1.39 | 1.72 | 1.35 | 1.41 | 1.38 | 1.30 | 1.32 | 1.23 | 1.55 | 1.28 |
| min | 2.34 | 6.71 | 3.28 | 2.79 | 9.50 | 6.44 | 2.62 | 3.39 | 3.50 | 1.39 | 1.34 | 1.12 | 1.36 | 1.10 | 1.11 | 1.15 | 1.12 | 1.16 | 1.20 | |||
| max | 11.60 | 9.32 | 5.38 | 3.37 | 14.10 | 8.53 | 4.28 | 5.43 | 4.76 | 1.63 | 1.79 | 1.78 | 1.61 | 1.77 | 1.69 | 1.60 | 1.59 | 1.60 | 1.65 | |||
| Parameter | Acceptance Criteria | JAK SM-05 | JAK 09 | JAK Imp A | JAK Imp C | JAK Imp B | JAK 07 | JAK 08 | JAK Imp D | JAK Imp E | Compliance with Acceptance Criteria (Yes/No) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Linearity, R | LOQ—120% of the specification limit (for each impurity) Concentration levels n = 5, R ≥ 0.99 | 0.99 | 0.99 | 0.99 | 0.99 | 0.99 | 0.99 | 0.99 | 0.99 | 0.99 | Yes |
| Repeatability, RSD (%) | LOQ—%RSD (area) ≤ 15 % 100% of the specification limit—%RSD (area) ≤ 15 % | 6.8 | 8.5 | 2.7 | 4.7 | 4.2 | 9.1 | 3.4 | 2.5 | 2.0 | Yes |
| 4.5 | 5.2 | 0.8 | 1.8 | 2.0 | 5.3 | 1.8 | 3.3 | 1.5 | Yes | ||
| Limit of Detection (LOD), | S/N ≥ 3 (n = 3) | 6 | 3 | 8 | 9 | 11 | 5 | 5 | 4 | 11 | Yes |
| Limit of Quantitation (LOQ) | S/N ≥ 10 (n = 6) | 21 | 12 | 22 | 29 | 31 | 14 | 16 | 8 | 32 | Yes |
| Specificity | Resolution between two neighboring peaks, Rs ≥ 2.0 | - | 9.6 | 12.0 | 8.4 | 4.6 | 9.9 | 4.1 | 5.8 | 5.6 | Yes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gurba-Bryśkiewicz, L.; Dawid, U.; Smuga, D.A.; Maruszak, W.; Delis, M.; Szymczak, K.; Stypik, B.; Moroz, A.; Błocka, A.; Mroczkiewicz, M.; et al. Implementation of QbD Approach to the Development of Chromatographic Methods for the Determination of Complete Impurity Profile of Substance on the Preclinical and Clinical Step of Drug Discovery Studies. Int. J. Mol. Sci. 2022, 23, 10720. https://doi.org/10.3390/ijms231810720
Gurba-Bryśkiewicz L, Dawid U, Smuga DA, Maruszak W, Delis M, Szymczak K, Stypik B, Moroz A, Błocka A, Mroczkiewicz M, et al. Implementation of QbD Approach to the Development of Chromatographic Methods for the Determination of Complete Impurity Profile of Substance on the Preclinical and Clinical Step of Drug Discovery Studies. International Journal of Molecular Sciences. 2022; 23(18):10720. https://doi.org/10.3390/ijms231810720
Chicago/Turabian StyleGurba-Bryśkiewicz, Lidia, Urszula Dawid, Damian A. Smuga, Wioleta Maruszak, Monika Delis, Krzysztof Szymczak, Bartosz Stypik, Aleksandra Moroz, Aleksandra Błocka, Michał Mroczkiewicz, and et al. 2022. "Implementation of QbD Approach to the Development of Chromatographic Methods for the Determination of Complete Impurity Profile of Substance on the Preclinical and Clinical Step of Drug Discovery Studies" International Journal of Molecular Sciences 23, no. 18: 10720. https://doi.org/10.3390/ijms231810720
APA StyleGurba-Bryśkiewicz, L., Dawid, U., Smuga, D. A., Maruszak, W., Delis, M., Szymczak, K., Stypik, B., Moroz, A., Błocka, A., Mroczkiewicz, M., Dubiel, K., & Wieczorek, M. (2022). Implementation of QbD Approach to the Development of Chromatographic Methods for the Determination of Complete Impurity Profile of Substance on the Preclinical and Clinical Step of Drug Discovery Studies. International Journal of Molecular Sciences, 23(18), 10720. https://doi.org/10.3390/ijms231810720