
Plasma Small Extracellular Vesicle Cathepsin D Dysregulation in GRN/C9orf72 and Sporadic Frontotemporal Lobar Degeneration
 , , , , , , , ,
, , , , , , , ,  ,
,  and
and
Abstract
1. Introduction
2. Results
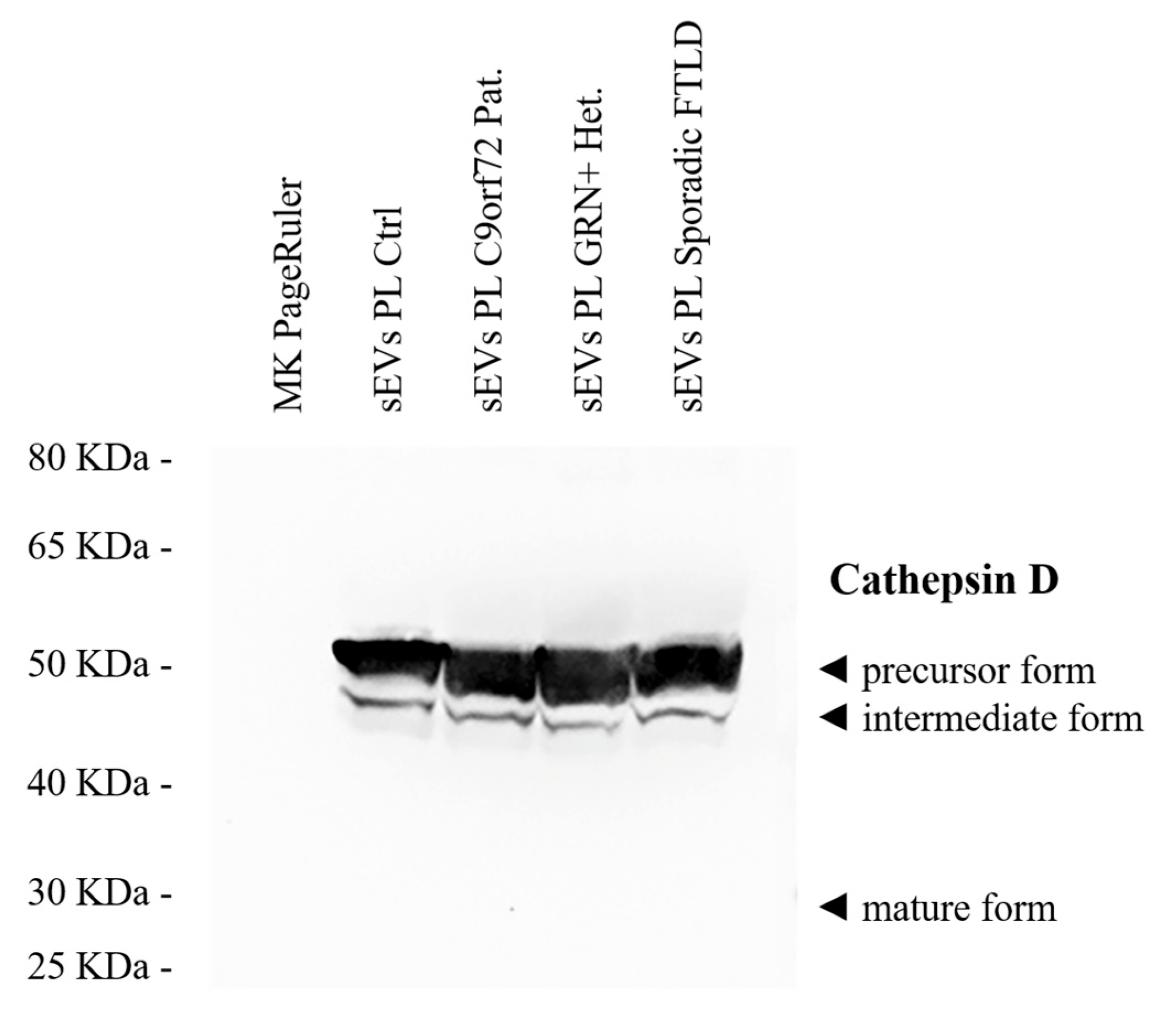
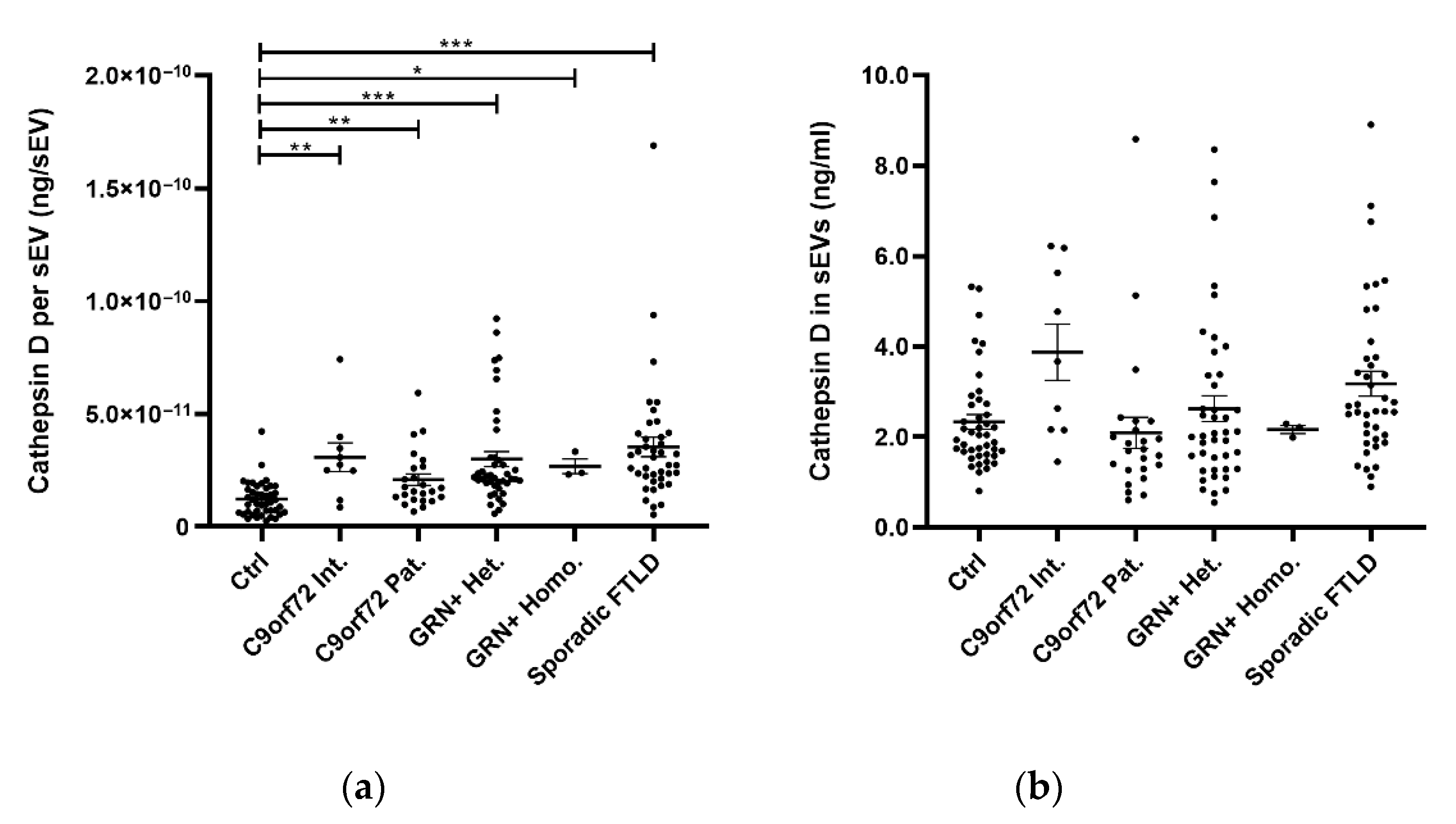
2.1. sEV Cathepsin D Dysregulation in Genetic and Sporadic FTLD
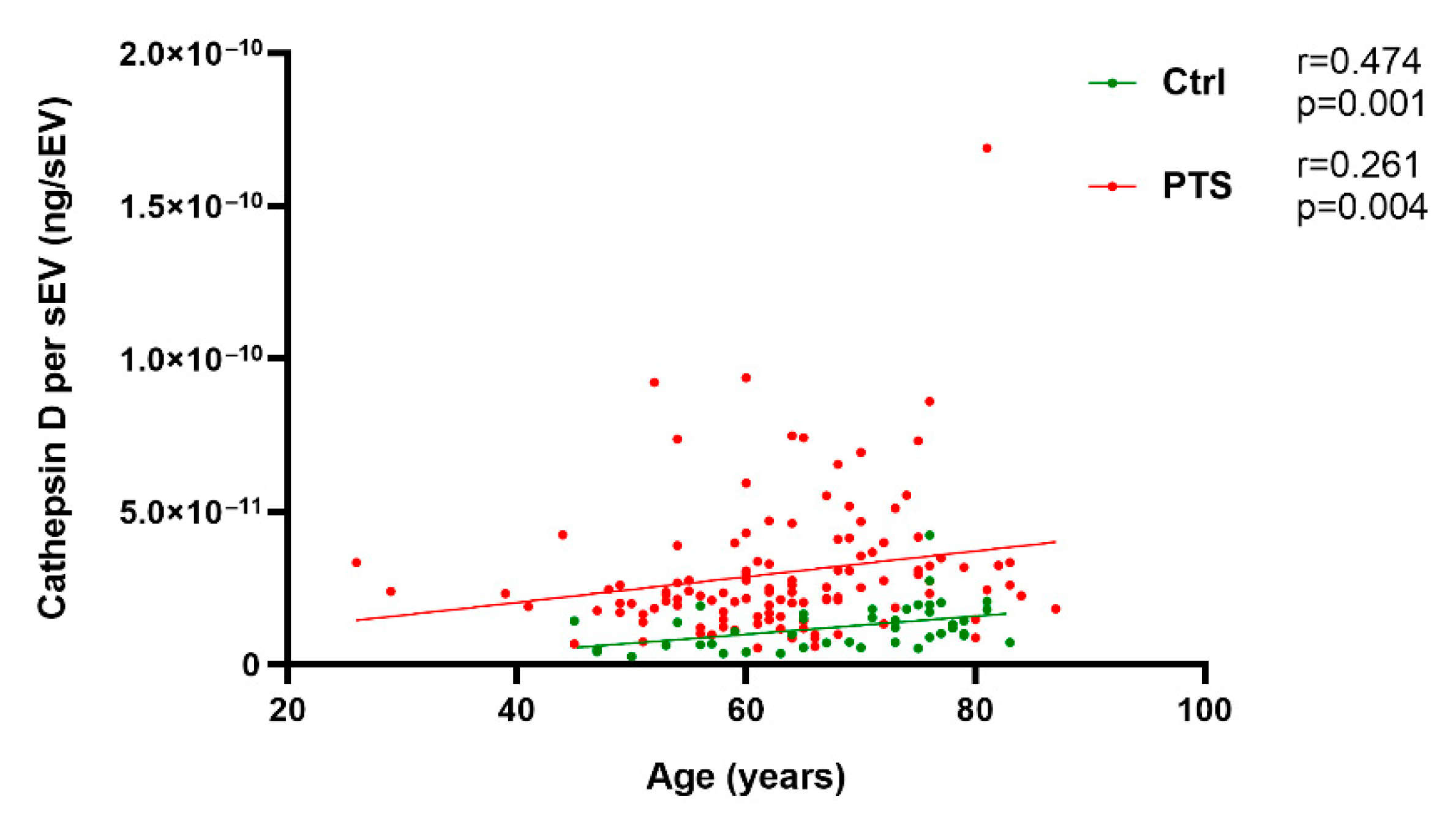
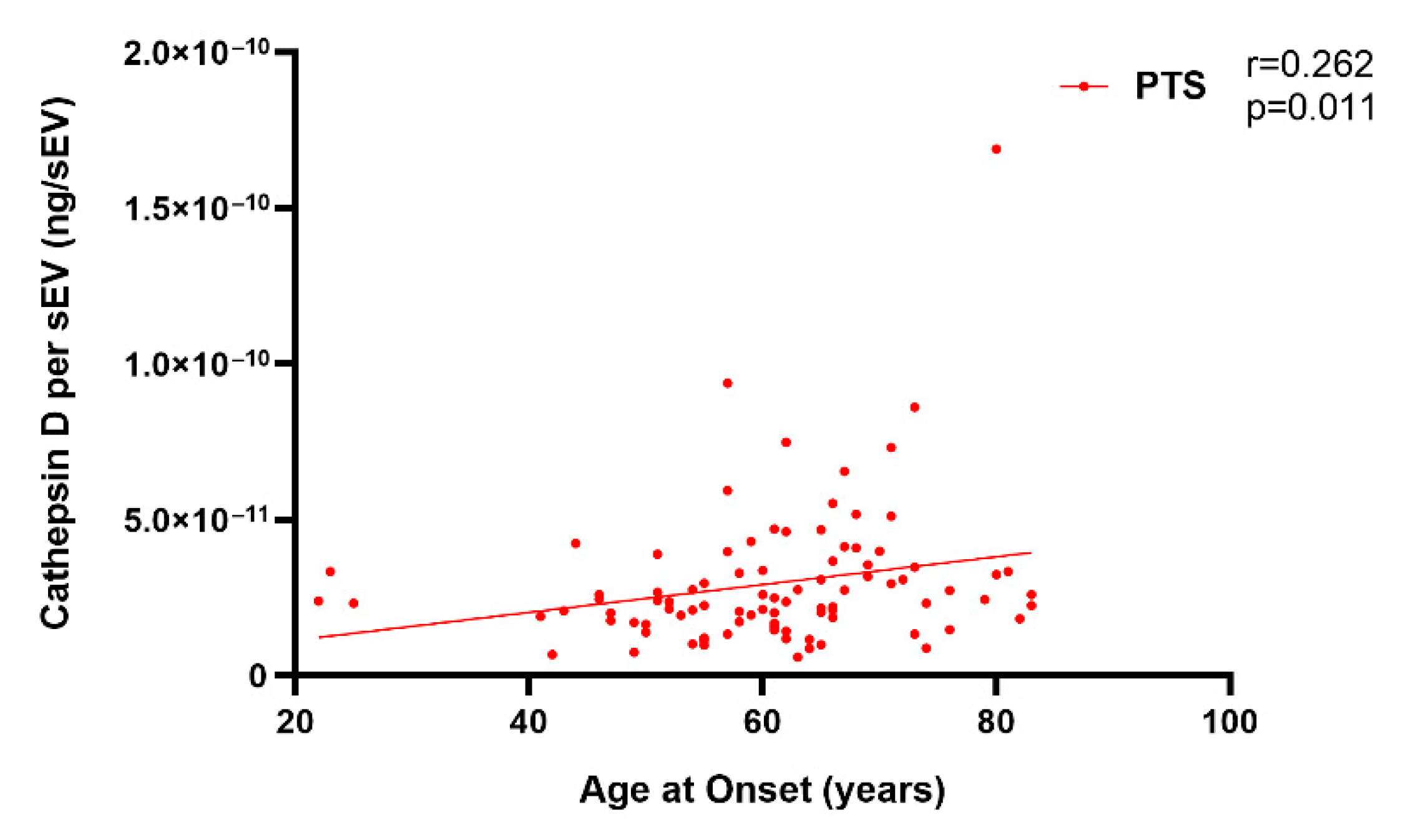
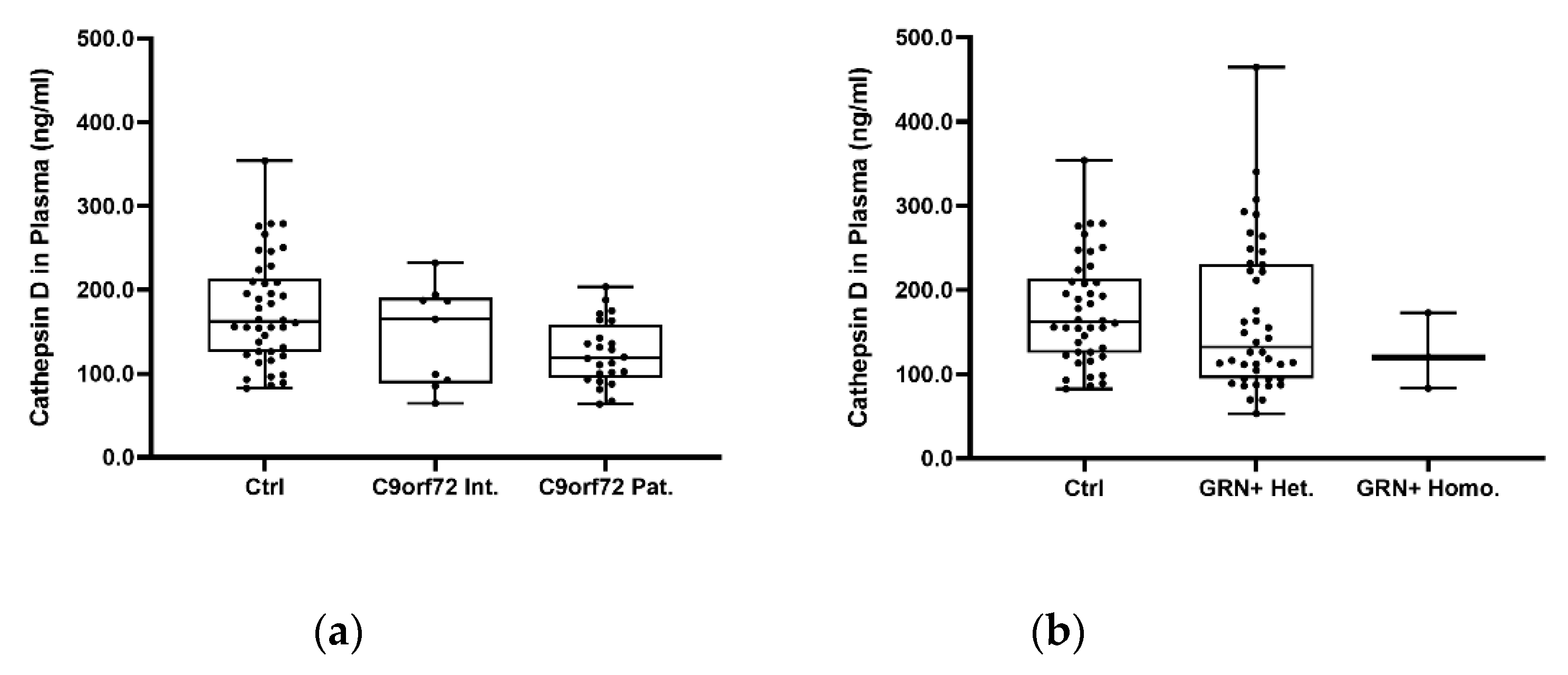
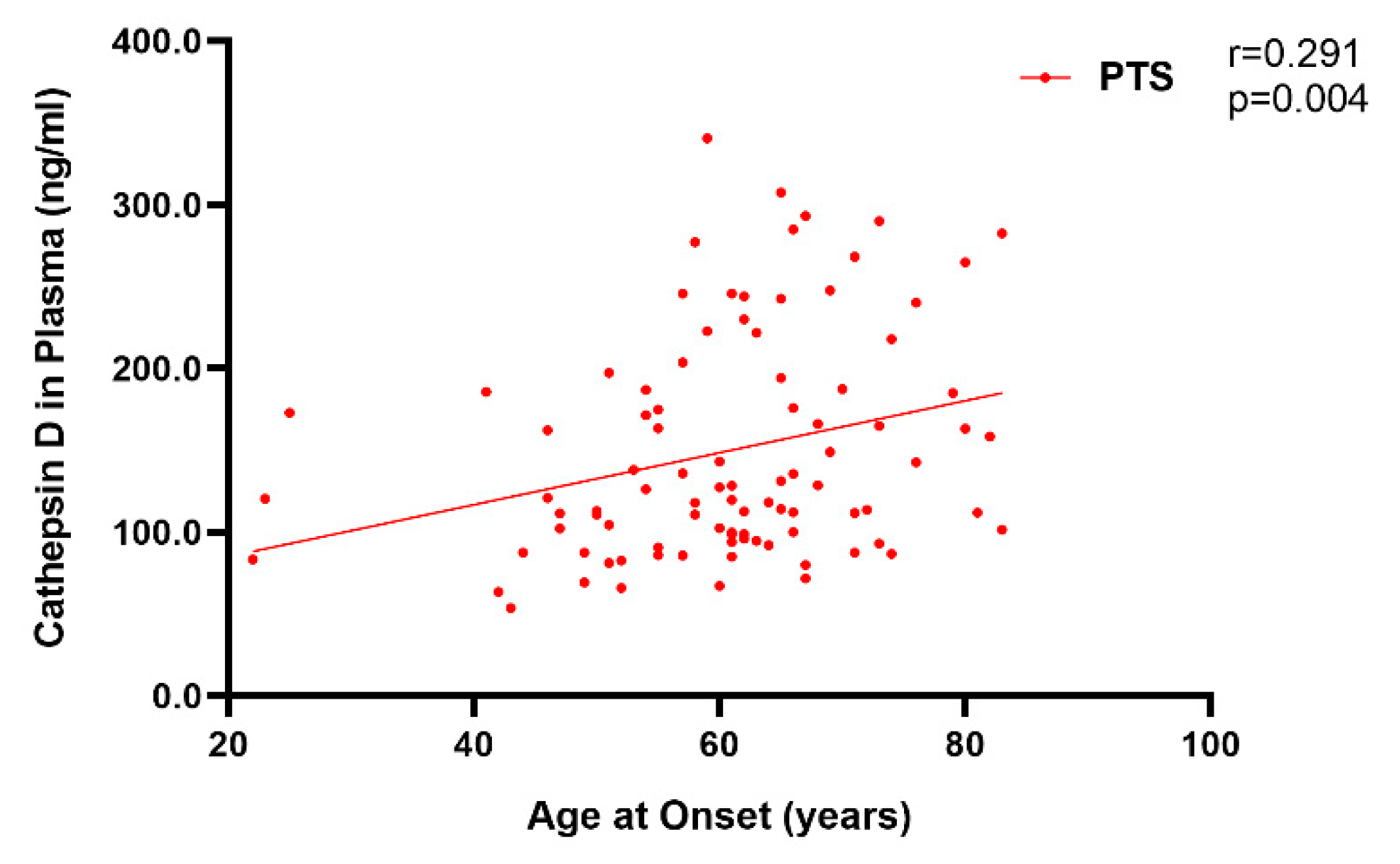
2.2. Plasma Cathepsin D in Sporadic and Genetic FTLD
3. Discussion
4. Materials and Methods
4.1. Subjects
4.2. Small EV Isolation and Characterization
4.3. Biochemical Analyses
4.4. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ratnavalli, E.; Brayne, C.; Dawson, K.; Hodges, J.R. The prevalence of frontotemporal dementia. Neurology 2002, 58, 1615–1621. [Google Scholar] [CrossRef] [PubMed]
- Bang, J.; Spina, S.; Miller, B.L. Frontotemporal dementia. Lancet 2015, 386, 1672–1682. [Google Scholar] [CrossRef]
- Mackenzie, I.R.; Neumann, M.; Bigio, E.H.; Cairns, N.J.; Alafuzoff, I.; Kril, J.; Kovacs, G.G.; Ghetti, B.; Halliday, G.; Holm, I.E.; et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: An update. Acta Neuropathol. 2010, 119, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R.; Neumann, M. Molecular neuropathology of frontotemporal dementia: Insights into disease mechanisms from postmortem studies. J. Neurochem. 2016, 138 (Suppl. 1), 54–70. [Google Scholar] [CrossRef]
- Ferrari, R.; Manzoni, C.; Hardy, J. Genetics and molecular mechanisms of frontotemporal lobar degeneration: An update and future avenues. Neurobiol. Aging 2019, 78, 98–110. [Google Scholar] [CrossRef]
- Rademakers, R.; Neumann, M.; Mackenzie, I.R. Advances in understanding the molecular basis of frontotemporal dementia. Nat. Rev. Neurol. 2012, 8, 423–434. [Google Scholar] [CrossRef]
- Rohrer, J.D.; Guerreiro, R.; Vandrovcova, J.; Uphill, J.; Reiman, D.; Beck, J.; Isaacs, A.M.; Authier, A.; Ferrari, R.; Fox, N.C.; et al. The heritability and genetics of frontotemporal lobar degeneration. Neurology 2009, 73, 1451–1456. [Google Scholar] [CrossRef]
- Fostinelli, S.; Ciani, M.; Zanardini, R.; Zanetti, O.; Binetti, G.; Ghidoni, R.; Benussi, L. The Heritability of Frontotemporal Lobar Degeneration: Validation of Pedigree Classification Criteria in a Northern Italy Cohort. J. Alzheimers Dis. 2018, 61, 753–760. [Google Scholar] [CrossRef]
- Hutton, M.; Lendon, C.L.; Rizzu, P.; Baker, M.; Froelich, S.; Houlden, H.; Pickering-Brown, S.; Chakraverty, S.; Isaacs, A.; Grover, A.; et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998, 393, 702–705. [Google Scholar] [CrossRef]
- Poorkaj, P.; Bird, T.D.; Wijsman, E.; Nemens, E.; Garruto, R.M.; Anderson, L.; Andreadis, A.; Wiederholt, W.C.; Raskind, M.; Schellenberg, G.D. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann. Neurol. 1998, 43, 815–825. [Google Scholar] [CrossRef]
- Baker, M.; Mackenzie, I.R.; Pickering-Brown, S.M.; Gass, J.; Rademakers, R.; Lindholm, C.; Snowden, J.; Adamson, J.; Sadovnick, A.D.; Rollinson, S.; et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 2006, 442, 916–919. [Google Scholar] [CrossRef]
- Cruts, M.; Gijselinck, I.; van der Zee, J.; Engelborghs, S.; Wils, H.; Pirici, D.; Rademakers, R.; Vandenberghe, R.; Dermaut, B.; Martin, J.J.; et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 2006, 442, 920–924. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef]
- Ghidoni, R.; Benussi, L.; Glionna, M.; Franzoni, M.; Binetti, G. Low plasma progranulin levels predict progranulin mutations in frontotemporal lobar degeneration. Neurology 2008, 71, 1235–1239. [Google Scholar] [CrossRef]
- Wang, J.; Van Damme, P.; Cruchaga, C.; Gitcho, M.A.; Vidal, J.M.; Seijo-Martínez, M.; Wang, L.; Wu, J.Y.; Robberecht, W.; Goate, A. Pathogenic cysteine mutations affect progranulin function and production of mature granulins. J. Neurochem. 2010, 112, 1305–1315. [Google Scholar] [CrossRef]
- Karch, C.M.; Ezerskiy, L.; Redaelli, V.; Giovagnoli, A.R.; Tiraboschi, P.; Pelliccioni, G.; Pelliccioni, P.; Kapetis, D.; D’Amato, I.; Piccoli, E.; et al. Missense mutations in progranulin gene associated with frontotemporal lobar degeneration: Study of pathogenetic features. Neurobiol. Aging 2016, 38, 215.e1–215.e12. [Google Scholar] [CrossRef]
- Smith, K.R.; Damiano, J.; Franceschetti, S.; Carpenter, S.; Canafoglia, L.; Morbin, M.; Rossi, G.; Pareyson, D.; Mole, S.E.; Staropoli, J.F.; et al. Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am. J. Hum. Genet. 2012, 90, 1102–1107. [Google Scholar] [CrossRef]
- Benussi, L.; Rossi, G.; Glionna, M.; Tonoli, E.; Piccoli, E.; Fostinelli, S.; Paterlini, A.; Flocco, R.; Albani, D.; Pantieri, R.; et al. C9ORF72 hexanucleotide repeat number in frontotemporal lobar degeneration: A genotype-phenotype correlation study. J. Alzheimers Dis. 2014, 38, 799–808. [Google Scholar] [CrossRef]
- Conner, G.E.; Richo, G. Isolation and characterization of a stable activation intermediate of the lysosomal aspartyl protease cathepsin D. Biochemistry 1992, 31, 1142–1147. [Google Scholar] [CrossRef]
- Benes, P.; Vetvicka, V.; Fusek, M. Cathepsin D—Many functions of one aspartic protease. Crit. Rev. Oncol. Hematol. 2008, 68, 12–28. [Google Scholar] [CrossRef]
- Nakanishi, H. Neuronal and microglial cathepsins in aging and age-related diseases. Ageing Res. Rev. 2003, 2, 367–381. [Google Scholar] [CrossRef]
- Ladror, U.S.; Snyder, S.W.; Wang, G.T.; Holzman, T.F.; Krafft, G.A. Cleavage at the amino and carboxyl termini of Alzheimer’s amyloid-beta by cathepsin D. J. Biol. Chem. 1994, 269, 18422–18428. [Google Scholar] [CrossRef]
- Khurana, V.; Elson-Schwab, I.; Fulga, T.A.; Sharp, K.A.; Loewen, C.A.; Mulkearns, E.; Tyynelä, J.; Scherzer, C.R.; Feany, M.B. Lysosomal dysfunction promotes cleavage and neurotoxicity of tau in vivo. PLoS Genet. 2010, 6, e1001026. [Google Scholar] [CrossRef]
- Sevlever, D.; Jiang, P.; Yen, S.H. Cathepsin D is the main lysosomal enzyme involved in the degradation of alpha-synuclein and generation of its carboxy-terminally truncated species. Biochemistry 2008, 47, 9678–9687. [Google Scholar] [CrossRef]
- Koike, M.; Nakanishi, H.; Saftig, P.; Ezaki, J.; Isahara, K.; Ohsawa, Y.; Schulz-Schaeffer, W.; Watanabe, T.; Waguri, S.; Kametaka, S.; et al. Cathepsin D deficiency induces lysosomal storage with ceroid lipofuscin in mouse CNS neurons. J. Neurosci. 2000, 20, 6898–6906. [Google Scholar] [CrossRef]
- Siintola, E.; Partanen, S.; Strömme, P.; Haapanen, A.; Haltia, M.; Maehlen, J.; Lehesjoki, A.E.; Tyynelä, J. Cathepsin D deficiency underlies congenital human neuronal ceroid-lipofuscinosis. Brain 2006, 129, 1438–1445. [Google Scholar] [CrossRef]
- Fritchie, K.; Siintola, E.; Armao, D.; Lehesjoki, A.E.; Marino, T.; Powell, C.; Tennison, M.; Booker, J.M.; Koch, S.; Partanen, S.; et al. Novel mutation and the first prenatal screening of cathepsin D deficiency (CLN10). Acta Neuropathol. 2009, 117, 201–208. [Google Scholar] [CrossRef]
- Davidson, Y.; Gibbons, L.; Pritchard, A.; Hardicre, J.; Wren, J.; Tian, J.; Shi, J.; Stopford, C.; Julien, C.; Thompson, J.; et al. Genetic associations between cathepsin D exon 2 C-->T polymorphism and Alzheimer’s disease, and pathological correlations with genotype. J. Neurol. Neurosurg. Psychiatry 2006, 77, 515–517. [Google Scholar] [CrossRef]
- Robak, L.A.; Jansen, I.E.; van Rooij, J.; Uitterlinden, A.G.; Kraaij, R.; Jankovic, J.; International Parkinson’s Disease Genomics Consortium (IPDGC); Heutink, P.; Shulman, J.M. Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain 2017, 140, 3191–3203. [Google Scholar] [CrossRef]
- Ward, M.E.; Chen, R.; Huang, H.Y.; Ludwig, C.; Telpoukhovskaia, M.; Taubes, A.; Boudin, H.; Minami, S.S.; Reichert, M.; Albrecht, P.; et al. Individuals with progranulin haploinsufficiency exhibit features of neuronal ceroid lipofuscinosis. Sci. Transl. Med. 2017, 9, eaah5642. [Google Scholar] [CrossRef] [PubMed]
- Valdez, C.; Wong, Y.C.; Schwake, M.; Bu, G.; Wszolek, Z.K.; Krainc, D. Progranulin-mediated deficiency of cathepsin D results in FTD and NCL-like phenotypes in neurons derived from FTD patients. Hum. Mol. Genet. 2017, 26, 4861–4872. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Paushter, D.H.; Feng, T.; Pardon, C.M.; Mendoza, C.S.; Hu, F. Regulation of cathepsin D activity by the FTLD protein progranulin. Acta Neuropathol. 2017, 134, 151–153. [Google Scholar] [CrossRef] [PubMed]
- Beel, S.; Moisse, M.; Damme, M.; De Muynck, L.; Robberecht, W.; Van Den Bosch, L.; Saftig, P.; Van Damme, P. Progranulin functions as a cathepsin D chaperone to stimulate axonal outgrowth in vivo. Hum. Mol. Genet. 2017, 26, 2850–2863. [Google Scholar] [CrossRef] [PubMed]
- Shao, Q.; Yang, M.; Liang, C.; Ma, L.; Zhang, W.; Jiang, Z.; Luo, J.; Lee, J.K.; Liang, C.; Chen, J.F. C9orf72 and smcr8 mutant mice reveal MTORC1 activation due to impaired lysosomal degradation and exocytosis. Autophagy 2020, 16, 1635–1650. [Google Scholar] [CrossRef] [PubMed]
- Arrant, A.E.; Onyilo, V.C.; Unger, D.E.; Roberson, E.D. Progranulin Gene Therapy Improves Lysosomal Dysfunction and Microglial Pathology Associated with Frontotemporal Dementia and Neuronal Ceroid Lipofuscinosis. J. Neurosci. 2018, 38, 2341–2358. [Google Scholar] [CrossRef] [PubMed]
- Telpoukhovskaia, M.A.; Liu, K.; Sayed, F.A.; Etchegaray, J.I.; Xie, M.; Zhan, L.; Li, Y.; Zhou, Y.; Le, D.; Bahr, B.A.; et al. Discovery of small molecules that normalize the transcriptome and enhance cysteine cathepsin activity in progranulin-deficient microglia. Sci. Rep. 2020, 10, 13688. [Google Scholar] [CrossRef]
- Wils, H.; Kleinberger, G.; Pereson, S.; Janssens, J.; Capell, A.; Van Dam, D.; Cuijt, I.; Joris, G.; De Deyn, P.P.; Haass, C.; et al. Cellular ageing, increased mortality and FTLD-TDP-associated neuropathology in progranulin knockout mice. J. Pathol. 2012, 228, 67–76. [Google Scholar] [CrossRef]
- Götzl, J.K.; Mori, K.; Damme, M.; Fellerer, K.; Tahirovic, S.; Kleinberger, G.; Janssens, J.; van der Zee, J.; Lang, C.M.; Kremmer, E.; et al. Common pathobiochemical hallmarks of progranulin-associated frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis. Acta Neuropathol. 2014, 127, 845–860. [Google Scholar] [CrossRef]
- Huang, M.; Modeste, E.; Dammer, E.; Merino, P.; Taylor, G.; Duong, D.M.; Deng, Q.; Holler, C.J.; Gearing, M.; Dickson, D.; et al. Network analysis of the progranulin-deficient mouse brain proteome reveals pathogenic mechanisms shared in human frontotemporal dementia caused by GRN mutations. Acta Neuropathol. Commun. 2020, 8, 163. [Google Scholar] [CrossRef]
- Wu, Y.; Shao, W.; Todd, T.W.; Tong, J.; Yue, M.; Koga, S.; Castanedes-Casey, M.; Librero, A.L.; Lee, C.W.; Mackenzie, I.R.; et al. Microglial lysosome dysfunction contributes to white matter pathology and TDP-43 proteinopathy in GRN-associated FTD. Cell Rep. 2021, 36, 109581. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Boxer, A.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Miller, B.L.; Kapogiannis, D. Altered lysosomal proteins in neural-derived plasma exosomes in preclinical Alzheimer disease. Neurology 2015, 85, 40–47. [Google Scholar] [CrossRef]
- Lui, H.; Zhang, J.; Makinson, S.R.; Cahill, M.K.; Kelley, K.W.; Huang, H.Y.; Shang, Y.; Oldham, M.C.; Martens, L.H.; Gao, F.; et al. Progranulin Deficiency Promotes Circuit-Specific Synaptic Pruning by Microglia via Complement Activation. Cell 2016, 165, 921–935. [Google Scholar] [CrossRef]
- O’Rourke, J.G.; Bogdanik, L.; Yáñez, A.; Lall, D.; Wolf, A.J.; Muhammad, A.K.; Ho, R.; Carmona, S.; Vit, J.P.; Zarrow, J.; et al. C9orf72 is required for proper macrophage and microglial function in mice. Science 2016, 351, 1324–1329. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Mathews, P.M.; Levy, E. Exosome Production Is Key to Neuronal Endosomal Pathway Integrity in Neurodegenerative Diseases. Front. Neurosci. 2019, 13, 1347. [Google Scholar] [CrossRef]
- Surgucheva, I.; Sharov, V.S.; Surguchov, A. γ-Synuclein: Seeding of α-synuclein aggregation and transmission between cells. Biochemistry 2012, 51, 4743–4754. [Google Scholar] [CrossRef]
- Bellini, S.; Saraceno, C.; Benussi, L.; Squitti, R.; Cimini, S.; Ricci, M.; Canafoglia, L.; Coppola, C.; Puoti, G.; Ferrari, C.; et al. Plasma Small Extracellular Vesicles with Complement Alterations in GRN/C9orf72 and Sporadic Frontotemporal Lobar Degeneration. Cells 2022, 11, 488. [Google Scholar] [CrossRef]
- Stoka, V.; Turk, V.; Turk, B. Lysosomal cathepsins and their regulation in aging and neurodegeneration. Ageing Res. Rev. 2016, 32, 22–37. [Google Scholar] [CrossRef]
- Kim, J.W.; Jung, S.Y.; Kim, Y.; Heo, H.; Hong, C.H.; Seo, S.W.; Choi, S.H.; Son, S.J.; Lee, S.; Chang, J. Identification of Cathepsin D as a Plasma Biomarker for Alzheimer’s Disease. Cells 2021, 10, 138. [Google Scholar] [CrossRef]
- Kang, J.; Kim, J.W.; Heo, H.; Lee, J.; Park, K.Y.; Yoon, J.H.; Chang, J. Identification of BAG2 and Cathepsin D as Plasma Biomarkers for Parkinson’s Disease. Clin. Transl. Sci. 2021, 14, 606–616. [Google Scholar] [CrossRef]
- Morena, F.; Argentati, C.; Trotta, R.; Crispoltoni, L.; Stabile, A.; Pistilli, A.; di Baldassarre, A.; Calafiore, R.; Montanucci, P.; Basta, G.; et al. A Comparison of Lysosomal Enzymes Expression Levels in Peripheral Blood of Mild- and Severe-Alzheimer’s Disease and MCI Patients: Implications for Regenerative Medicine Approaches. Int. J. Mol. Sci. 2017, 18, 1806. [Google Scholar] [CrossRef]
- Nakanishi, H.; Tominaga, K.; Amano, T.; Hirotsu, I.; Inoue, T.; Yamamoto, K. Age-related changes in activities and localizations of cathepsins D, E, B, and L in the rat brain tissues. Exp. Neurol. 1994, 126, 119–128. [Google Scholar] [CrossRef]
- Nakanishi, H.; Amano, T.; Sastradipura, D.F.; Yoshimine, Y.; Tsukuba, T.; Tanabe, K.; Hirotsu, I.; Ohono, T.; Yamamoto, K. Increased expression of cathepsins E and D in neurons of the aged rat brain and their colocalization with lipofuscin and carboxy-terminal fragments of Alzheimer amyloid precursor protein. J. Neurochem. 1997, 68, 739–749. [Google Scholar] [CrossRef]
- Sato, Y.; Suzuki, Y.; Ito, E.; Shimazaki, S.; Ishida, M.; Yamamoto, T.; Yamamoto, H.; Toda, T.; Suzuki, M.; Suzuki, A.; et al. Identification and characterization of an increased glycoprotein in aging: Age-associated translocation of cathepsin D. Mech. Ageing Dev. 2006, 127, 771–778. [Google Scholar] [CrossRef]
- Augereau, P.; Miralles, F.; Cavaillès, V.; Gaudelet, C.; Parker, M.; Rochefort, H. Characterization of the proximal estrogen-responsive element of human cathepsin D gene. Mol. Endocrinol. 1994, 8, 693–703. [Google Scholar] [CrossRef][Green Version]
- Benussi, L.; Longobardi, A.; Kocoglu, C.; Carrara, M.; Bellini, S.; Ferrari, C.; Nicsanu, R.; Saraceno, C.; Bonvicini, C.; Fostinelli, S.; et al. Investigating the Endo-Lysosomal System in Major Neurocognitive Disorders Due to Alzheimer’s Disease, Frontotemporal Lobar Degeneration and Lewy Body Disease: Evidence for SORL1 as a Cross-Disease Gene. Int. J. Mol. Sci. 2021, 22, 13633. [Google Scholar] [CrossRef]
- Desnick, R.J.; Schuchman, E.H. Enzyme replacement therapy for lysosomal diseases: Lessons from 20 years of experience and remaining challenges. Annu. Rev. Genom. Hum. Genet. 2012, 13, 307–335. [Google Scholar] [CrossRef]
- Marques, A.R.A.; Di Spiezio, A.; Thießen, N.; Schmidt, L.; Grötzinger, J.; Lüllmann-Rauch, R.; Damme, M.; Storck, S.E.; Pietrzik, C.U.; Fogh, J.; et al. Enzyme replacement therapy with recombinant pro-CTSD (cathepsin D) corrects defective proteolysis and autophagy in neuronal ceroid lipofuscinosis. Autophagy 2020, 16, 811–825. [Google Scholar] [CrossRef]
- Drobny, A.; Prieto Huarcaya, S.; Dobert, J.; Kluge, A.; Bunk, J.; Schlothauer, T.; Zunke, F. The role of lysosomal cathepsins in neurodegeneration: Mechanistic insights, diagnostic potential and therapeutic approaches. Biochim. Biophys. Acta Mol. Cell Res. 2022, 1869, 119243. [Google Scholar] [CrossRef]
- Neary, D.; Snowden, J.S.; Gustafson, L.; Passant, U.; Stuss, D.; Black, S.; Freedman, M.; Kertesz, A.; Robert, P.H.; Albert, M.; et al. Frontotemporal lobar degeneration: A consensus on clinical diagnostic criteria. Neurology 1998, 51, 1546–1554. [Google Scholar] [CrossRef] [PubMed]
- Rascovsky, K.; Hodges, J.R.; Knopman, D.; Mendez, M.F.; Kramer, J.H.; Neuhaus, J.; van Swieten, J.C.; Seelaar, H.; Dopper, E.G.; Onyike, C.U.; et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011, 134, 2456–2477. [Google Scholar] [CrossRef] [PubMed]
- Benussi, L.; Ghidoni, R.; Pegoiani, E.; Moretti, D.V.; Zanetti, O.; Binetti, G. Progranulin Leu271LeufsX10 is one of the most common FTLD and CBS associated mutations worldwide. Neurobiol. Dis. 2009, 33, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.; Piccoli, E.; Benussi, L.; Caso, F.; Redaelli, V.; Magnani, G.; Binetti, G.; Ghidoni, R.; Perani, D.; Giaccone, G.; et al. A novel progranulin mutation causing frontotemporal lobar degeneration with heterogeneous phenotypic expression. J. Alzheimers Dis. 2011, 23, 7–12. [Google Scholar] [CrossRef]
- Ghidoni, R. RawData_CathepsinD_NTA_FTLD [Dataset]. Zenodo 2022. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ctrl | C9orf72 Int. | C9orf72 Pat. | GRN+ Het. | GRN+ Homo | Sporadic FTLD | p Value | |
|---|---|---|---|---|---|---|---|
| N. | 43 | 9 | 24 | 42 | 3 | 40 | |
| Sex (% female) | 72.1 | 44.4 | 45.8 | 42.9 | 33.3 | 65.0 | 0.053 a |
| Age, years | 68.1 ± 10.6 | 67.2 ± 5.2 | 62.7 ± 10.4 | 60.1 ± 7.6 | 31.3 ± 6.8 | 67.7 ± 10.4 | <0.001 b |
| Age at onset, years | / | 64.1 ± 6.2 | 60.6 ± 10.6 | 58.9 ± 8.1 | 23.3 ± 1.5 | 64.4 ± 10.8 | <0.001 b |
| EV conc., EVs/ml | 2.3 × 1011 ± 1.1 × 1011 | 1.5 × 1011 ± 7.1 × 1010 | 1.1 × 1011 ± 5.3 × 1010 | 9.7 × 1010 ± 4.3 × 1010 | 8.3 × 1010 ± 1.6 × 1010 | 1.1 × 1011 ± 6.3 × 1010 | <0.001 c |
| Cathepsin D per sEV, ng/sEV # | 1.2 × 10−11 ± 7.6 × 10−12 | 3.1 × 10−11 ± 1.9 × 10−11 | 2.1 × 10−11 ± 1.3 × 10−11 | 3.0 × 10−11 ± 2.2 × 10−11 | 2.7 × 10−11 ± 5.6 × 10−12 | 3.5 × 10−11 ± 2.8 × 10−11 | <0.001 c |
| Cathepsin D in sEVs, ng/mL # | 2.3 ± 1.1 | 3.9 ± 1.9 | 2.1 ± 1.7 | 2.6 ± 1.8 | 2.2 ± 0.2 | 3.2 ± 1.8 | 0.019 d |
| Cathepsin D plasma, ng/mL # | 175.5 ± 63.5 | 145.1 ± 60.2 | 124.5 ± 38.0 | 166.5 ± 89.3 | 125.5 ± 45.0 | 157.1 ± 69.2 | 0.058 d |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bellini, S.; Saraceno, C.; Benussi, L.; Geviti, A.; Longobardi, A.; Nicsanu, R.; Cimini, S.; Ricci, M.; Canafoglia, L.; Coppola, C.; et al. Plasma Small Extracellular Vesicle Cathepsin D Dysregulation in GRN/C9orf72 and Sporadic Frontotemporal Lobar Degeneration. Int. J. Mol. Sci. 2022, 23, 10693. https://doi.org/10.3390/ijms231810693
Bellini S, Saraceno C, Benussi L, Geviti A, Longobardi A, Nicsanu R, Cimini S, Ricci M, Canafoglia L, Coppola C, et al. Plasma Small Extracellular Vesicle Cathepsin D Dysregulation in GRN/C9orf72 and Sporadic Frontotemporal Lobar Degeneration. International Journal of Molecular Sciences. 2022; 23(18):10693. https://doi.org/10.3390/ijms231810693
Chicago/Turabian StyleBellini, Sonia, Claudia Saraceno, Luisa Benussi, Andrea Geviti, Antonio Longobardi, Roland Nicsanu, Sara Cimini, Martina Ricci, Laura Canafoglia, Cinzia Coppola, and et al. 2022. "Plasma Small Extracellular Vesicle Cathepsin D Dysregulation in GRN/C9orf72 and Sporadic Frontotemporal Lobar Degeneration" International Journal of Molecular Sciences 23, no. 18: 10693. https://doi.org/10.3390/ijms231810693
APA StyleBellini, S., Saraceno, C., Benussi, L., Geviti, A., Longobardi, A., Nicsanu, R., Cimini, S., Ricci, M., Canafoglia, L., Coppola, C., Puoti, G., Binetti, G., Rossi, G., & Ghidoni, R. (2022). Plasma Small Extracellular Vesicle Cathepsin D Dysregulation in GRN/C9orf72 and Sporadic Frontotemporal Lobar Degeneration. International Journal of Molecular Sciences, 23(18), 10693. https://doi.org/10.3390/ijms231810693