
Integrating Genetic and Chromosome Maps of Allium cepa: From Markers Visualization to Genome Assembly Verification

 , , ,
, , ,  , and
, and
Abstract
1. Introduction
2. Results
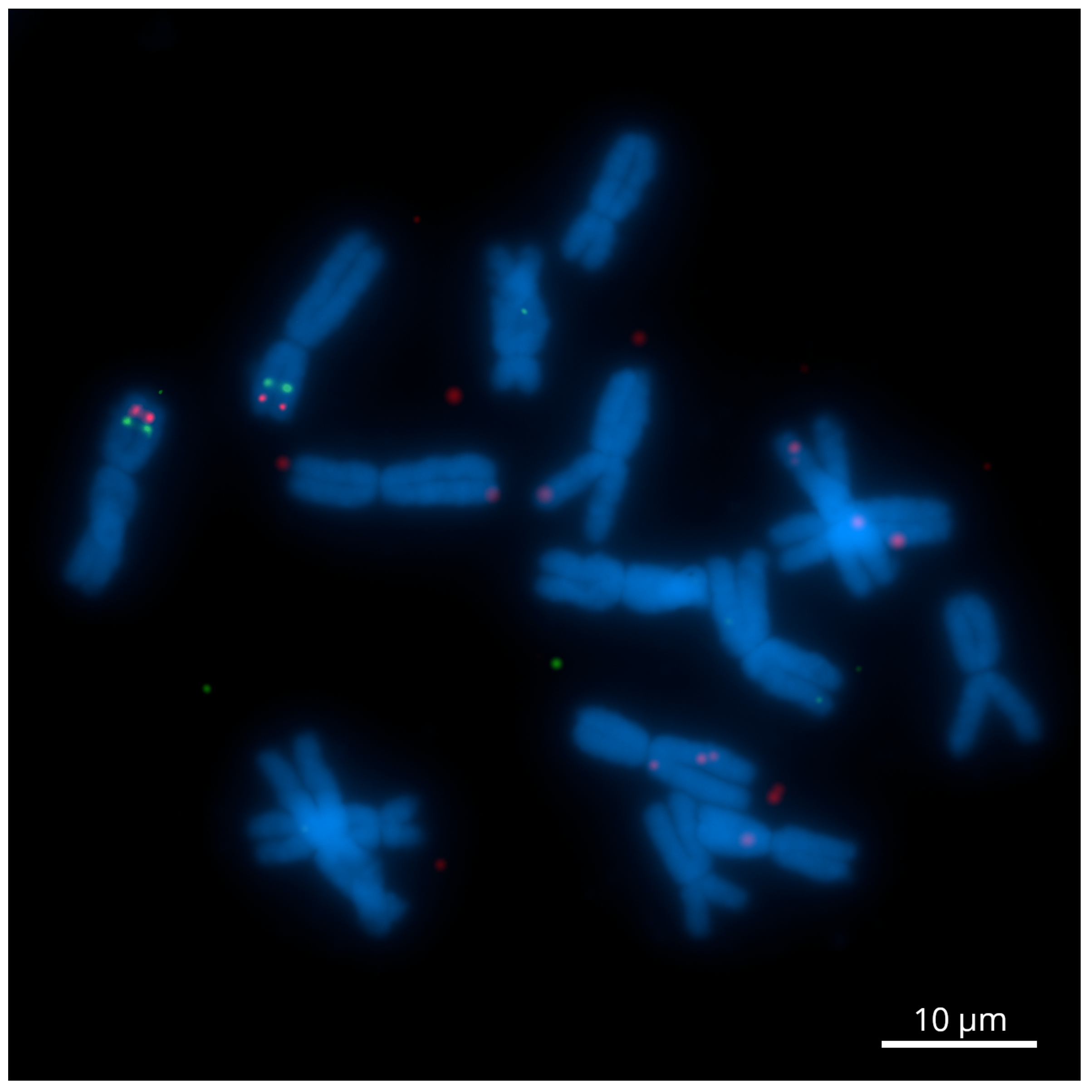
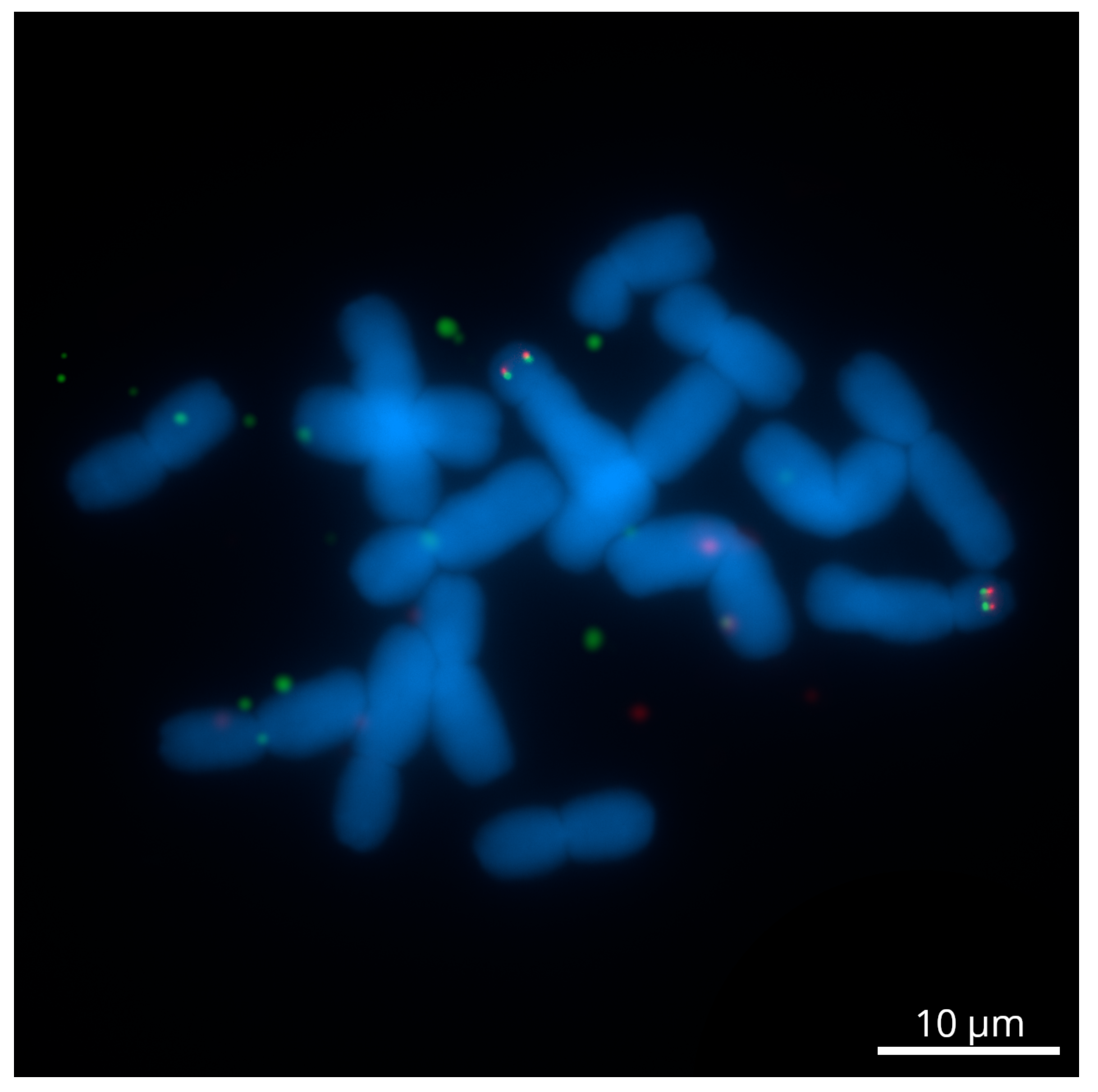
2.1. The Development of Genome-Wide Specific Tyr-FISH Probes
2.2. A Dual-Color Tyr-FISH Visualization of Marker/Gene
2.2.1. Chromosome 2
2.2.2. Chromosome 6
2.3. The Integration of Genetic, Cytogenetic and Pseudochromosome Maps
2.3.1. Chromosome 2
2.3.2. Chromosome 6
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Tyr-FISH Probe Preparation
4.3. Chromosome Preparation
4.4. A Dual-Color Sequential Tyr-FISH
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| FISH | Fluorescent in situ hybridization |
| TSA | Transcriptome Shotgun Assembly |
| RPHC | Relative Position of Hybridization sites on Chromosome arm |
| SD | Standard Deviation |
| EST | Expressed Sequence Tag |
| FL | Fractional Length |
References
- Almogy, G.; Pratt, M.; Oberstrass, F.; Lee, L.; Mazur, D.; Beckett, N.; Barad, O.; Soifer, I.; Perelman, E.; Etzioni, Y.; et al. Cost-efficient whole genome-sequencing using novel mostly natural sequencing-by-synthesis chemistry and open fluidics platform. bioRxiv 2022. [Google Scholar] [CrossRef]
- Shirasawa, K.; Harada, D.; Hirakawa, H.; Isobe, S.; Kole, C. Chromosome-level de novo genome assemblies of over 100 plant species. Breed. Sci. 2021, 71, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Appels, R.; Eversole, K.; Feuille, C.; Keller, B.; Rogers, J.; Stein, N. The International Wheat Genome Sequencing Consortium (IWGSC). Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 2018, 361, 10–1126. [Google Scholar]
- Sun, X.; Zhu, S.; Li, N.; Cheng, Y.; Zhao, J.; Qiao, X.; Lu, L.; Liu, S.; Wang, Y.; Liu, C.; et al. A chromosome-level genome assembly of garlic (Allium sativum) provides insights into genome evolution and allicin biosynthesis. Mol. Plant 2020, 13, 1328–1339. [Google Scholar] [CrossRef]
- Ghurye, J.; Pop, M. Modern technologies and algorithms for scaffolding assembled genomes. PLoS Comput. Biol. 2019, 15, e1006994. [Google Scholar] [CrossRef]
- Pop, M.; Salzberg, S.L. Bioinformatics challenges of new sequencing technology. Trends Genet. 2008, 24, 142–149. [Google Scholar] [CrossRef]
- Alkan, C.; Sajjadian, S.; Eichler, E.E. Limitations of next-generation genome sequence assembly. Nat. Methods 2011, 8, 61–65. [Google Scholar] [CrossRef]
- The International Wheat Genome Sequencing Consortium (IWGSC); Mayer, K.F.; Rogers, J.; Doležel, J.; Pozniak, C.; Eversole, K.; Feuillet, C.; Gill, B.; Friebe, B.; Lukaszewski, A.J.; et al. A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 2014, 345, 1251788. [Google Scholar]
- Chapman, J.A.; Mascher, M.; Buluç, A.; Barry, K.; Georganas, E.; Session, A.; Strnadova, V.; Jenkins, J.; Sehgal, S.; Oliker, L.; et al. A whole-genome shotgun approach for assembling and anchoring the hexaploid bread wheat genome. Genome Biol. 2015, 16, 1–17. [Google Scholar] [CrossRef]
- Fierst, J.L. Using linkage maps to correct and scaffold de novo genome assemblies: Methods, challenges, and computational tools. Front. Genet. 2015, 6, 220. [Google Scholar] [CrossRef]
- Maccaferri, M.; Harris, N.S.; Twardziok, S.O.; Pasam, R.K.; Gundlach, H.; Spannagl, M.; Ormanbekova, D.; Lux, T.; Prade, V.M.; Milner, S.G.; et al. Durum wheat genome highlights past domestication signatures and future improvement targets. Nat. Genet. 2019, 51, 885–895. [Google Scholar] [CrossRef]
- Walve, R.; Rastas, P.; Salmela, L. Kermit: Linkage map guided long read assembly. Algorithms Mol. Biol. 2019, 14, 1–10. [Google Scholar] [CrossRef]
- Wu, H.; Yao, D.; Chen, Y.; Yang, W.; Zhao, W.; Gao, H.; Tong, C. De novo genome assembly of Populus simonii further supports that Populus simonii and Populus trichocarpa belong to different sections. G3 Genes Genomes Genet. 2020, 10, 455–466. [Google Scholar] [CrossRef]
- Koo, D.H.; Jo, S.H.; Bang, J.W.; Park, H.M.; Lee, S.; Choi, D. Integration of cytogenetic and genetic linkage maps unveils the physical architecture of tomato chromosome 2. Genetics 2008, 179, 1211–1220. [Google Scholar] [CrossRef][Green Version]
- Szinay, D.; Chang, S.B.; Khrustaleva, L.; Peters, S.; Schijlen, E.; Bai, Y.; Stiekema, W.J.; Van Ham, R.C.; De Jong, H.; Klein Lankhorst, R.M. High-resolution chromosome mapping of BACs using multi-colour FISH and pooled-BAC FISH as a backbone for sequencing tomato chromosome 6. Plant J. 2008, 56, 627–637. [Google Scholar] [CrossRef]
- Ren, Y.; Zhao, H.; Kou, Q.; Jiang, J.; Guo, S.; Zhang, H.; Hou, W.; Zou, X.; Sun, H.; Gong, G.; et al. A high resolution genetic map anchoring scaffolds of the sequenced watermelon genome. PLoS ONE 2012, 7, e29453. [Google Scholar]
- Burton, J.N.; Adey, A.; Patwardhan, R.P.; Qiu, R.; Kitzman, J.O.; Shendure, J. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 2013, 31, 1119–1125. [Google Scholar] [CrossRef]
- Yuan, Y.; Chung, C.Y.L.; Chan, T.F. Advances in optical mapping for genomic research. Comput. Struct. Biotechnol. J. 2020, 18, 2051–2062. [Google Scholar] [CrossRef]
- Mascher, M.; Wicker, T.; Jenkins, J.; Plott, C.; Lux, T.; Koh, C.S.; Ens, J.; Gundlach, H.; Boston, L.B.; Tulpová, Z.; et al. Long-read sequence assembly: A technical evaluation in barley. Plant Cell 2021, 33, 1888–1906. [Google Scholar] [CrossRef]
- Dudchenko, O.; Batra, S.S.; Omer, A.D.; Nyquist, S.K.; Hoeger, M.; Durand, N.C.; Shamim, M.S.; Machol, I.; Lander, E.S.; Aiden, A.P.; et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 2017, 356, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Kudryavtseva, N.; Ermolaev, A.; Karlov, G.; Kirov, I.; Shigyo, M.; Sato, S.; Khrustaleva, L. A dual-color tyr-FISH method for visualizing genes/markers on plant chromosomes to create integrated genetic and cytogenetic maps. Int. J. Mol. Sci. 2021, 22, 5860. [Google Scholar] [CrossRef] [PubMed]
- Kalkman, E. Analysis of the C-banded karyotype of Allium cepa L. Standard system of nomenclature and polymorphism. Genetica 1984, 65, 141–148. [Google Scholar] [CrossRef]
- Sagar, N.A.; Pareek, S.; Benkeblia, N.; Xiao, J. Onion (Allium cepa L.) bioactives: Chemistry, pharmacotherapeutic functions, and industrial applications. Food Front. 2022, 3, 1–33. [Google Scholar] [CrossRef]
- Ricroch, A.; Yockteng, R.; Brown, S.; Nadot, S. Evolution of genome size across some cultivated Allium species. Genome 2005, 48, 511–520. [Google Scholar] [CrossRef]
- Finkers, R.; van Kaauwen, M.; Ament, K.; Burger-Meijer, K.; Egging, R.; Huits, H.; Kodde, L.; Kroon, L.; Shigyo, M.; Sato, S.; et al. Insights from the first genome assembly of Onion (Allium cepa). G3 2021, 11, jkab243. [Google Scholar] [CrossRef]
- Duangjit, J.; Bohanec, B.; Chan, A.P.; Town, C.; Havey, M.J. Transcriptome sequencing to produce SNP-based genetic maps of onion. Theor. Appl. Genet. 2013, 126, 2093–2101. [Google Scholar] [CrossRef]
- Scholten, O.E.; van Kaauwen, M.P.; Shahin, A.; Hendrickx, P.M.; Keizer, L.; Burger, K.; van Heusden, A.W.; van der Linden, C.G.; Vosman, B. SNP-markers in Allium species to facilitate introgression breeding in onion. BMC Plant Biol. 2016, 16, 1–10. [Google Scholar] [CrossRef]
- Choi, Y.; Kim, S.; Lee, J. Construction of an onion (Allium cepa L.) genetic linkage map using genotyping-by-sequencing analysis with a reference gene set and identification of QTLs controlling anthocyanin synthesis and content. Plants 2020, 9, 616. [Google Scholar] [CrossRef]
- Fujito, S.; Akyol, T.Y.; Mukae, T.; Wako, T.; Yamashita, K.i.; Tsukazaki, H.; Hirakawa, H.; Tanaka, K.; Mine, Y.; Sato, S.; et al. Construction of a high-density linkage map and graphical representation of the arrangement of transcriptome-based unigene markers on the chromosomes of onion, Allium cepa L. BMC Genom. 2021, 22, 1–8. [Google Scholar] [CrossRef]
- Schwarzacher, T.; Heslop-Harrison, P. Practical in situ Hybridization; BIOS Scientific Publishers Ltd.: Oxford, UK, 2000. [Google Scholar]
- Green, M.R.; Sambrook, J. Labeling of DNA probes by nick translation. Cold Spring Harb. Protoc. 2020, 2020, pdb–prot100602. [Google Scholar] [CrossRef]
- Zhang, N.; Zeng, L.; Shan, H.; Ma, H. Highly conserved low-copy nuclear genes as effective markers for phylogenetic analyses in angiosperms. New Phytol. 2012, 195, 923–937. [Google Scholar] [CrossRef]
- Jurgen, S.S. Cytogenetics, Plants, Animals, Humans. In Proceedings of the Library of Congres Catologing in Publication Datay; Springer: Berlin/Heidelberg, Germany, 1980; Volume 38, p. 39. [Google Scholar]
- Pearce, S.R.; Pich, U.; Harrison, G.; Flavell, A.J.; Heslop-Harrison, J.; Schubert, I.; Kumar, A. TheTy1-copia group retrotransposons ofAllium cepa are distributed throughout the chromosomes but are enriched in the terminal heterochromatin. Chromosome Res. 1996, 4, 357–364. [Google Scholar] [CrossRef]
- Pich, U.; Schubert, I. Terminal heterochromatin and alternative telomeric sequences in Allium cepa. Chromosome Res. Int. J. Mol. Supramol. Evol. Asp. Chromosome Biol. 1998, 6, 315–321. [Google Scholar] [CrossRef]
- Barnes, S.R.; James, A.M.; Jamieson, G. The organisation, nucleotide sequence, and chromosomal distribution of a satellite DNA from Allium cepa. Chromosoma 1985, 92, 185–192. [Google Scholar] [CrossRef]
- Stevenson, M.; Armstrong, S.; Jones, G.; Ford-Lloyd, B. Distribution of a 375 bp repeat sequence inAllium (Alliaceae) as revealed by FISH. Plant Syst. Evol. 1999, 217, 31–42. [Google Scholar] [CrossRef]
- Do, G.S.; Seo, B.B.; Yamamoto, M.; Suzuki, G.; Mukai, Y. Identification and chromosomal location of tandemly repeated DNA sequences in Allium cepa. Genes Genet. Syst. 2001, 76, 53–60. [Google Scholar] [CrossRef]
- Copenhaver, G.P.; Nickel, K.; Kuromori, T.; Benito, M.I.; Kaul, S.; Lin, X.; Bevan, M.; Murphy, G.; Harris, B.; Parnell, L.D.; et al. Genetic definition and sequence analysis of Arabidopsis centromeres. Science 1999, 286, 2468–2474. [Google Scholar] [CrossRef] [PubMed]
- Mayer, K.F.; Waugh, R.; Langridge, P.; Close, T.J.; Wise, R.P.; Graner, A.; Matsumoto, T.; Sato, K.; Schulman, A.; Ariyadasa, R.; et al. A physical, genetic and functional sequence assembly of the barley genome. Nature 2012, 491, 711–716. [Google Scholar]
- Vosa, C.G. Heterochromatic patterns in Allium. Heredity 1976, 36, 383–392. [Google Scholar] [CrossRef]
- Castiglione, M.R.; Giraldi, E.; Frediana, M. The DNA-Methylation Pattern of Allium cepa Metaphase-Chromosomes. Biol. Zentralblatt 1995, 114, 57–66. [Google Scholar]
- Boideau, F.; Richard, G.; Coriton, O.; Huteau, V.; Belser, C.; Deniot, G.; Eber, F.; Falentin, C.; Ferreira de Carvalho, J.; Gilet, M.; et al. Epigenomic and structural events preclude recombination in Brassica napus. New Phytol. 2022, 234, 545–559. [Google Scholar] [CrossRef]
- Yelina, N.E.; Lambing, C.; Hardcastle, T.J.; Zhao, X.; Santos, B.; Henderson, I.R. DNA methylation epigenetically silences crossover hot spots and controls chromosomal domains of meiotic recombination in Arabidopsis. Genes Dev. 2015, 29, 2183–2202. [Google Scholar] [CrossRef]
- Albini, S.; Jones, G. Synaptonemal complex spreading in Allium cepa and Allium fistulosum. II. Pachytene observations: The SC karyotype and the correspondence of late recombination nodules and chiasmata. Genome 1988, 30, 399–410. [Google Scholar] [CrossRef]
- Pucker, B.; Irisarri, I.; de Vries, J.; Xu, B. Plant genome sequence assembly in the era of long reads: Progress, challenges and future directions. Quant. Plant Biol. 2022, 3, E5. [Google Scholar] [CrossRef]
- Scalzitti, N.; Jeannin-Girardon, A.; Collet, P.; Poch, O.; Thompson, J.D. A benchmark study of ab initio gene prediction methods in diverse eukaryotic organisms. BMC Genom. 2020, 21, 1–20. [Google Scholar] [CrossRef]
- Salzberg, S.L. Next-generation genome annotation: We still struggle to get it right. Genome Biol. 2019, 20, 92. [Google Scholar] [CrossRef]
- Tang, H.; Zhang, X.; Miao, C.; Zhang, J.; Ming, R.; Schnable, J.C.; Schnable, P.S.; Lyons, E.; Lu, J. ALLMAPS: Robust scaffold ordering based on multiple maps. Genome Biol. 2015, 16, 1–15. [Google Scholar] [CrossRef]
- Afshinfard, A.; Jackman, S.D.; Wong, J.; Coombe, L.; Chu, J.; Nikolic, V.; Dilek, G.; Malkoç, Y.; Warren, R.L.; Birol, I. Physlr: Next-Generation Physical Maps. DNA 2022, 2, 116–130. [Google Scholar] [CrossRef]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The complete sequence of a human genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef]
- Shearer, L.A.; Anderson, L.K.; de Jong, H.; Smit, S.; Goicoechea, J.L.; Roe, B.A.; Hua, A.; Giovannoni, J.J.; Stack, S.M. Fluorescence in situ hybridization and optical mapping to correct scaffold arrangement in the tomato genome. G3 Genes Genomes Genet. 2014, 4, 1395–1405. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Wu, T.D.; Watanabe, C.K. GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2005, 21, 1859–1875. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef]
- Kirov, I.; Divashuk, M.; Van Laere, K.; Soloviev, A.; Khrustaleva, L. An easy SteamDrop method for high quality plant chromosome preparation. Mol. Cytogenet. 2014, 7, 1–10. [Google Scholar] [CrossRef]
- Kirov, I.; Khrustaleva, L.; Van Laere, K.; Soloviev, A.; Meeus, S.; Romanov, D.; Fesenko, I. DRAWID: User-friendly java software for chromosome measurements and idiogram drawing. Comp. Cytogenet. 2017, 11, 747. [Google Scholar] [CrossRef] [PubMed]
- De Vries, J. Onion chromosome nomenclature and homoeology relationships workshop report. Euphytica 1990, 49, 1–3. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Marker/Gene | Chromosome | Expected Probe Length, bp | Observed Probe Length, bp | Identity, % |
|---|---|---|---|---|
| Unigene23526 | 2 | 1777 | 1772 | 99.6 |
| Unigene10061 | 2 | 1517 | 1650 | 99.7 |
| CL5148.Contig1 | 2 | 1294 | 1294 | 100 |
| Unigene572 | 2 | 2777 | 2801 | 99.8 |
| Unigene28076 | 2 | 1464 | 1465 | 99.7 |
| CL4449.Contig1 | 2 | 1231 | 1231 | 100 |
| Unigene25645 | 2 | 1188 | 1188 | 100 |
| Unigene27326 | 2 | 1008 | 1008 | 99.7 |
| Unigene10683 | 2 | 1160 | 1160 | 100 |
| mlh1 | 2 | 1347 | 1349 | 99.9 |
| Unigene23418 | 2 | 1368 | 1368 | 100 |
| Unigene28713 | 2 | 1323 | 1323 | 100 |
| Unigene5305 | 2 | 1588 | 1627 | 99.0 |
| Unigene7941 | 6 | 2283 | 2286 | 99.0 |
| Unigene13863 | 6 | 1084 | 1084 | 100 |
| CL4877.Contig2 | 6 | 1180 | 1180 | 100 |
| Unigene49 | 6 | 1110 | 1110 | 100 |
| Unigene10558 | 6 | 1143 | 1143 | 100 |
| Unigene22659 | 6 | 1457 | 1457 | 100 |
| CL39.Contig3 | 6 | 1044 | 1044 | 100 |
| Unigene28149 | 6 | 1236 | 1236 | 100 |
| Unigene8201 | 6 | 3866 | 3866 | 100 |
| Unigene13813 | 6 | 3809 | 3809 | 100 |
| CL6133.Contig1 | 6 | 1018 | 1018 | 100 |
| Probe | Relative Position 1 | Detection Frequency, % | n 3 | ||
|---|---|---|---|---|---|
| Mean ± SD | Arm | n 2 | |||
| Unigene23526 | Short | 7 | 57.2 | 24 | |
| Unigene10061 | Short | 12 | 49.3 | 25 | |
| CL5148.Contig1 | Short | 9 | 45.7 | 26 | |
| Unigene572 | Short | 17 | 77.4 | 48 | |
| Unigene28076 | Short | 7 | 47.3 | 17 | |
| CL4449.Contig1 | Short | 6 | 42.8 | 23 | |
| Unigene25645 | Short | 6 | 52.6 | 26 | |
| Unigene27326 | Short | 14 | 45.1 | 19 | |
| Unigene28713 | Long | 6 | 52.4 | 16 | |
| Unigene5305 | Long | 14 | 51.6 | 32 | |
| mlh1 gene | Long | 8 | 48.1 | 13 | |
| Unigene10683 | Long | 8 | 47.8 | 23 | |
| Unigene23418 | Long | 5 | 46.3 | 16 | |
| Probe | Relative Position 1 | Detection Frequency, % | n 3 | ||
|---|---|---|---|---|---|
| Mean ± SD | Arm | n 2 | |||
| CL6133.Contig1 | Short | 5 | 42.1 | 17 | |
| Unigene13813 | Short | 9 | 77.8 | 20 | |
| Unigene7941 | Short | 10 | 73.4 | 14 | |
| Unigene13863 | Long | 7 | 43.6 | 21 | |
| CL4877.Contig2 | Long | 7 | 46.8 | 21 | |
| Unigene49 | Long | 6 | 43.6 | 22 | |
| Unigene10558 | Long | 6 | 49.5 | 22 | |
| Unigene22659 | Long | 5 | 54.8 | 21 | |
| CL39.Contig3 | Long | 7 | 42.1 | 18 | |
| Unigene28149 | Long | 6 | 46.7 | 19 | |
| Unigene8201 | Long | 16 | 86.7 | 15 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ermolaev, A.; Kudryavtseva, N.; Pivovarov, A.; Kirov, I.; Karlov, G.; Khrustaleva, L. Integrating Genetic and Chromosome Maps of Allium cepa: From Markers Visualization to Genome Assembly Verification. Int. J. Mol. Sci. 2022, 23, 10486. https://doi.org/10.3390/ijms231810486
Ermolaev A, Kudryavtseva N, Pivovarov A, Kirov I, Karlov G, Khrustaleva L. Integrating Genetic and Chromosome Maps of Allium cepa: From Markers Visualization to Genome Assembly Verification. International Journal of Molecular Sciences. 2022; 23(18):10486. https://doi.org/10.3390/ijms231810486
Chicago/Turabian StyleErmolaev, Aleksey, Natalia Kudryavtseva, Anton Pivovarov, Ilya Kirov, Gennady Karlov, and Ludmila Khrustaleva. 2022. "Integrating Genetic and Chromosome Maps of Allium cepa: From Markers Visualization to Genome Assembly Verification" International Journal of Molecular Sciences 23, no. 18: 10486. https://doi.org/10.3390/ijms231810486
APA StyleErmolaev, A., Kudryavtseva, N., Pivovarov, A., Kirov, I., Karlov, G., & Khrustaleva, L. (2022). Integrating Genetic and Chromosome Maps of Allium cepa: From Markers Visualization to Genome Assembly Verification. International Journal of Molecular Sciences, 23(18), 10486. https://doi.org/10.3390/ijms231810486