
LPS-Induced Coagulation and Neuronal Damage in a Mice Model Is Attenuated by Enoxaparin

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
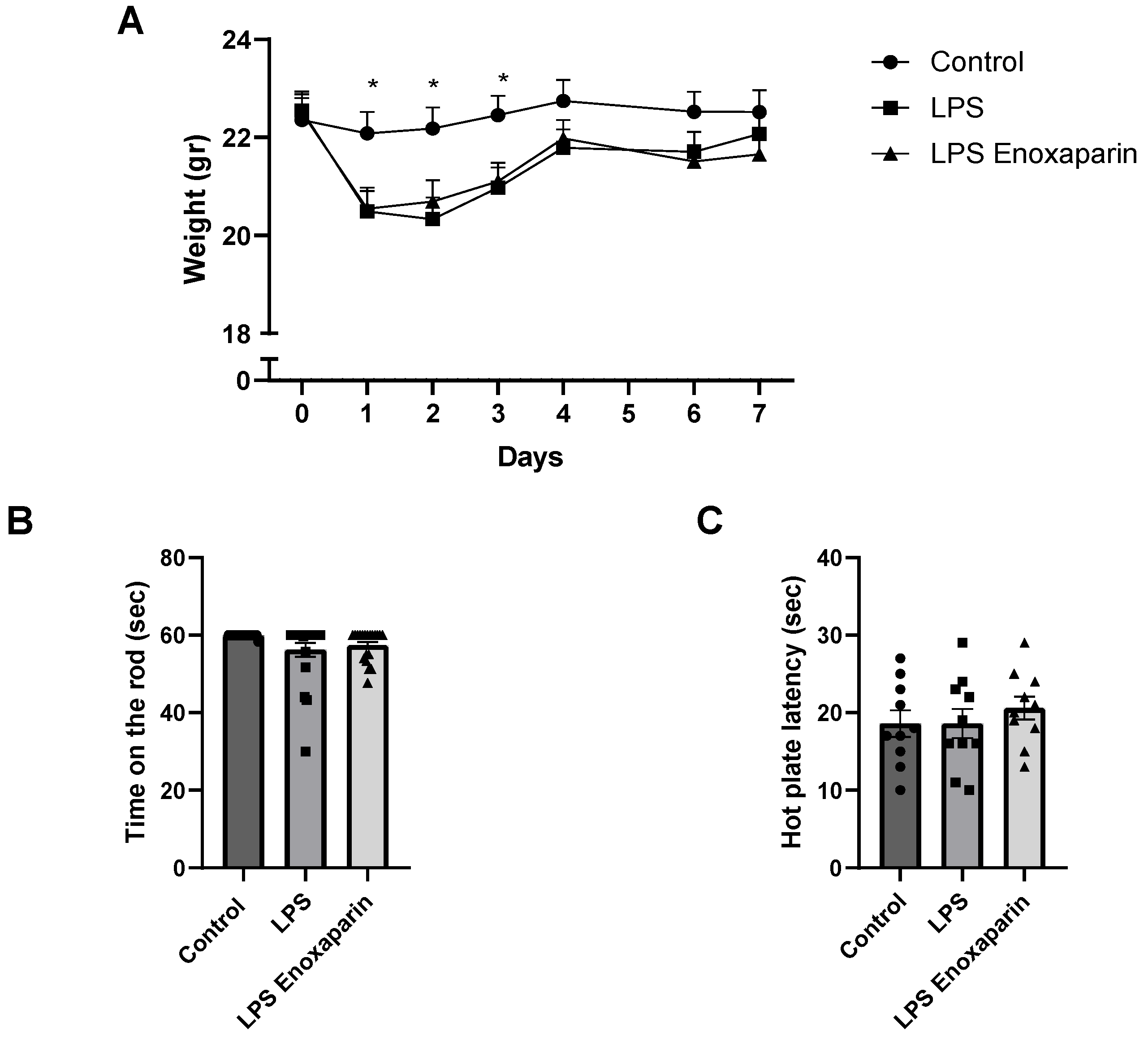
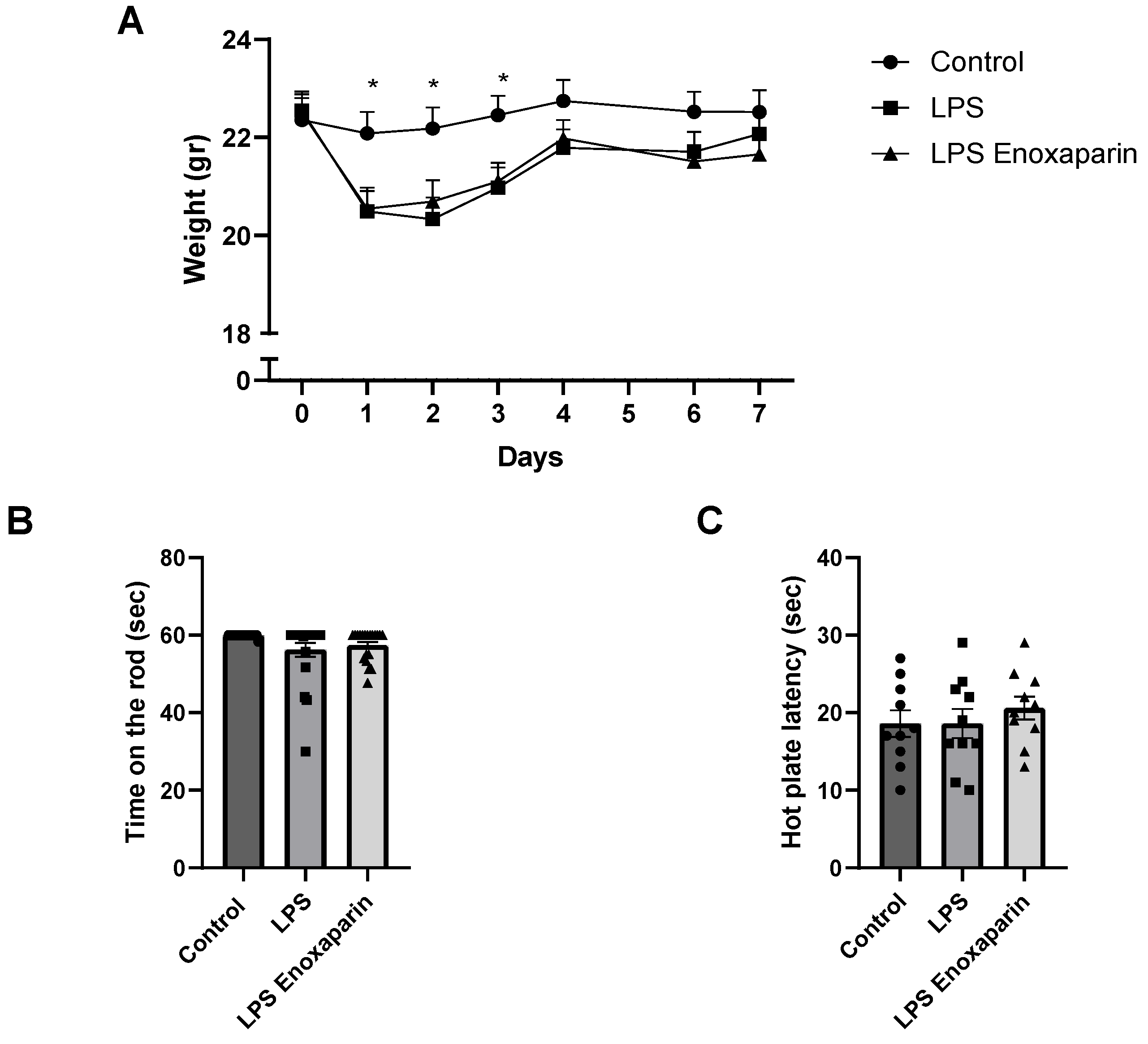
2.1. General Health of the LPS Mice
2.2. Motor Functions
2.3. Heat Sensitivity Function
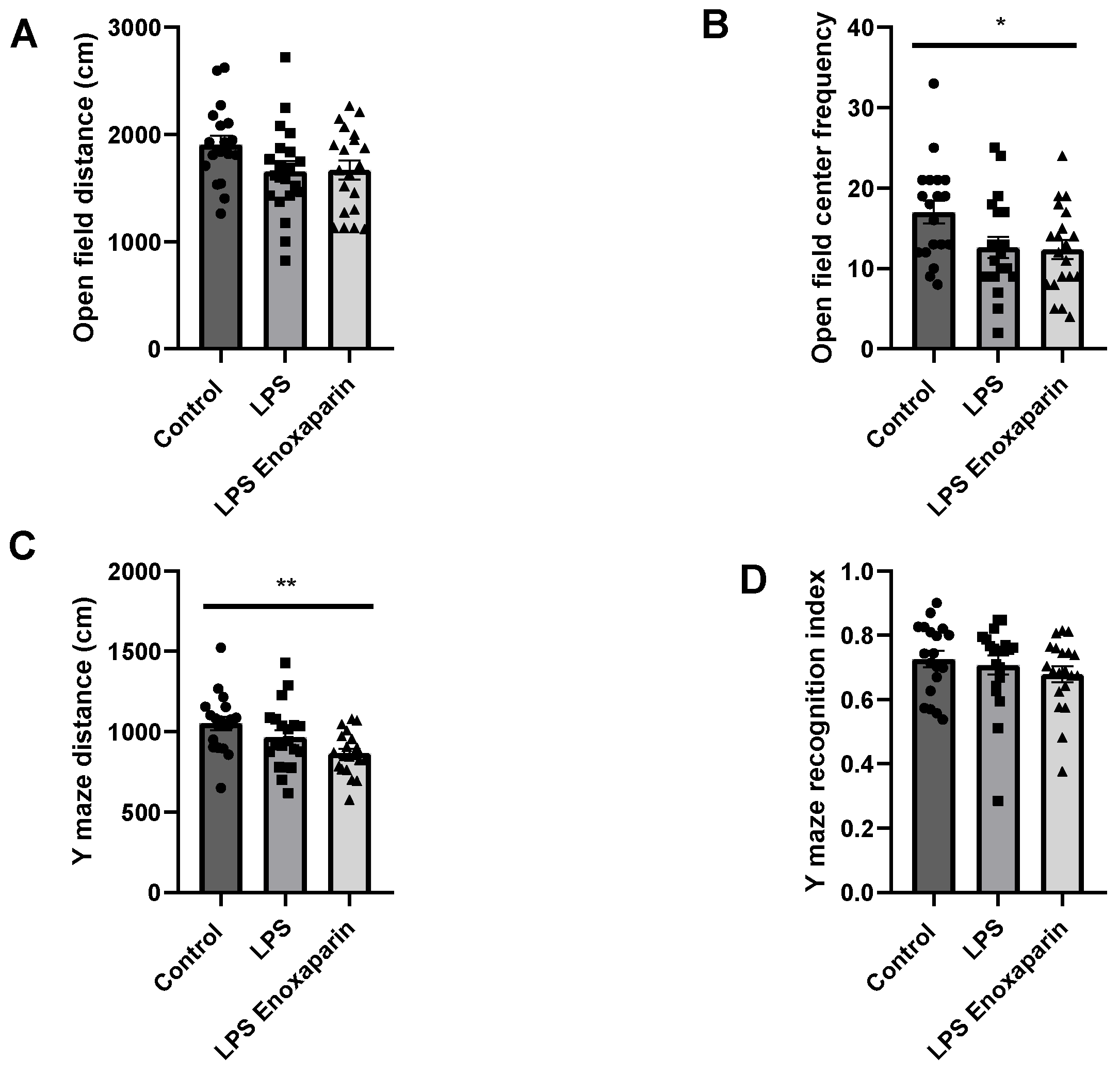
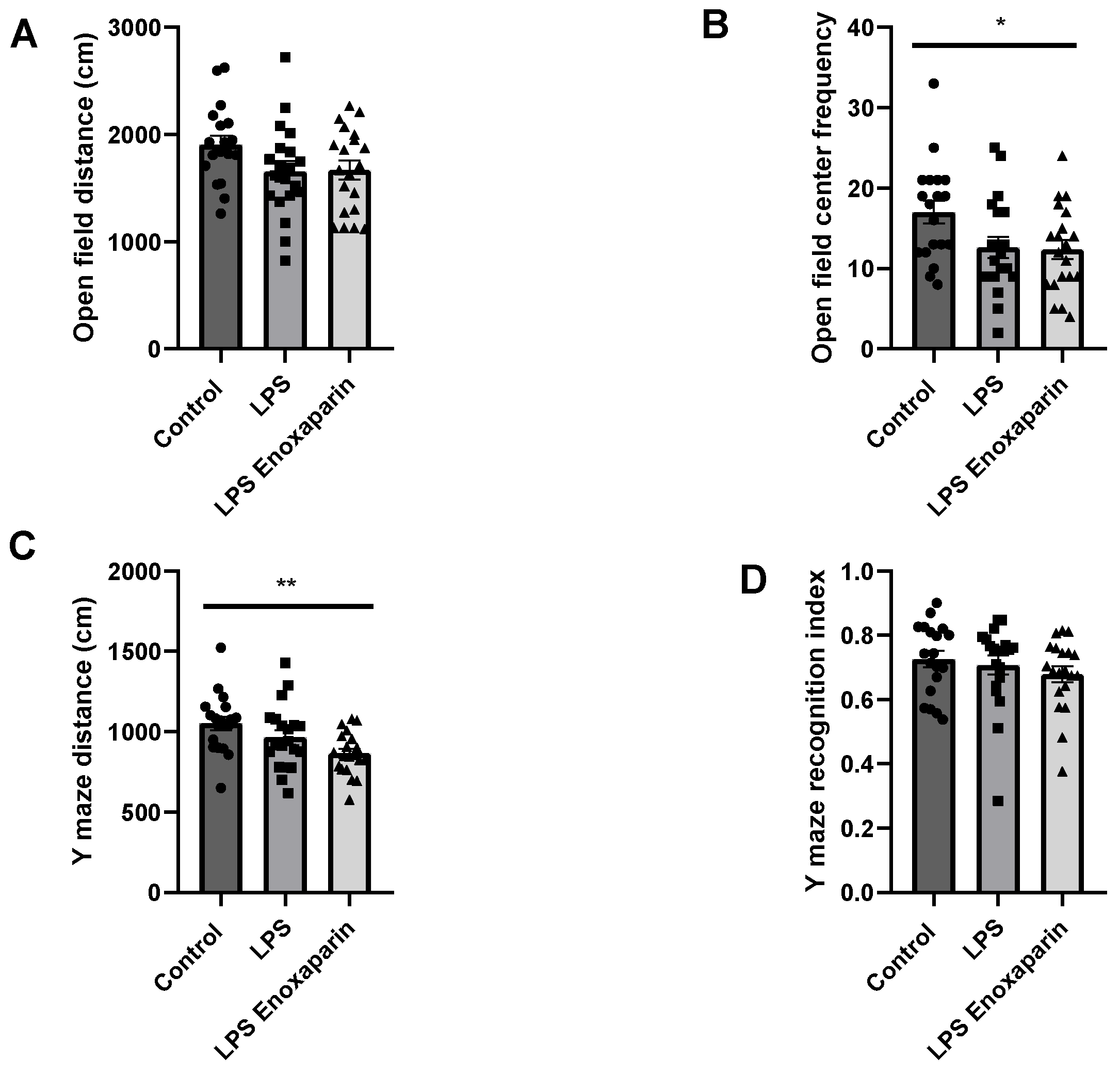
2.4. Open Field Test
2.5. Y-Maze
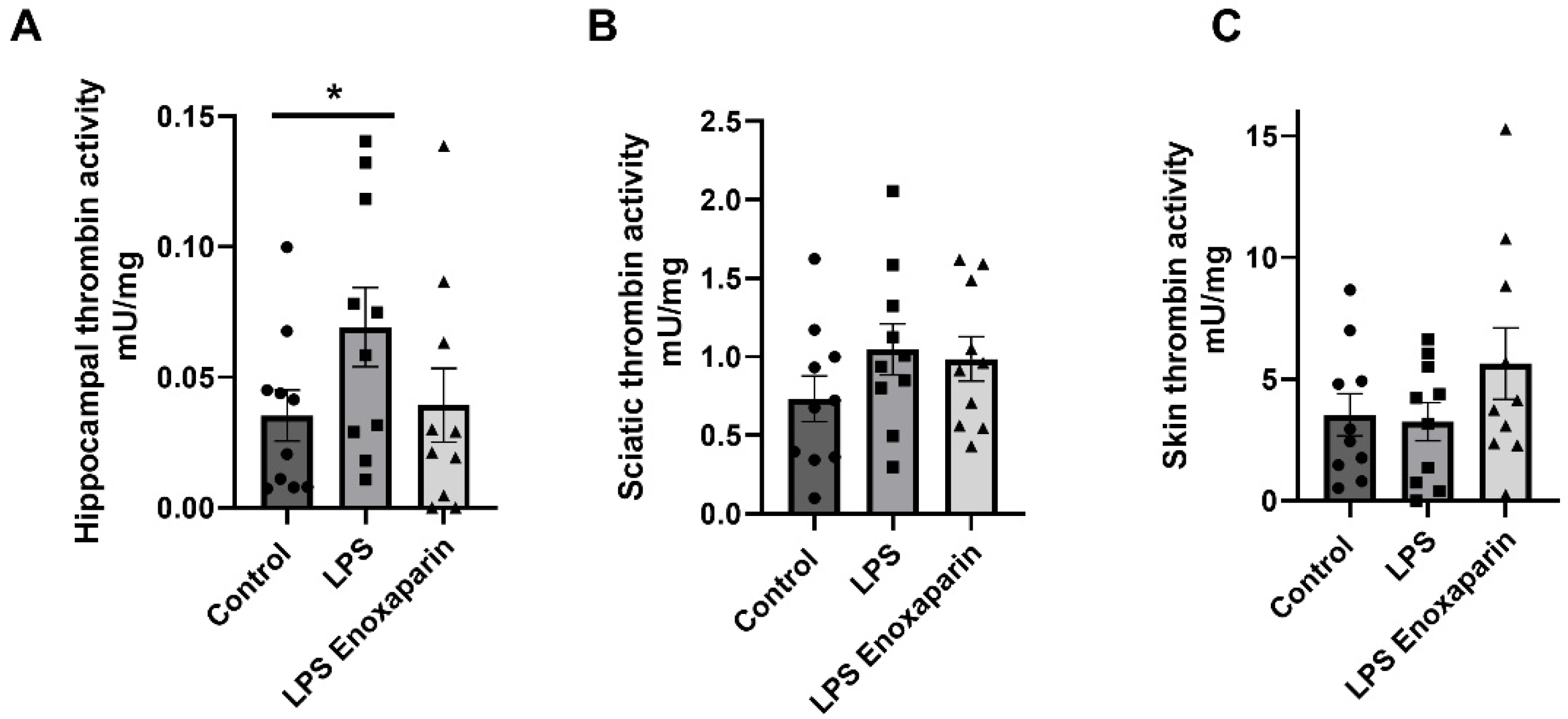
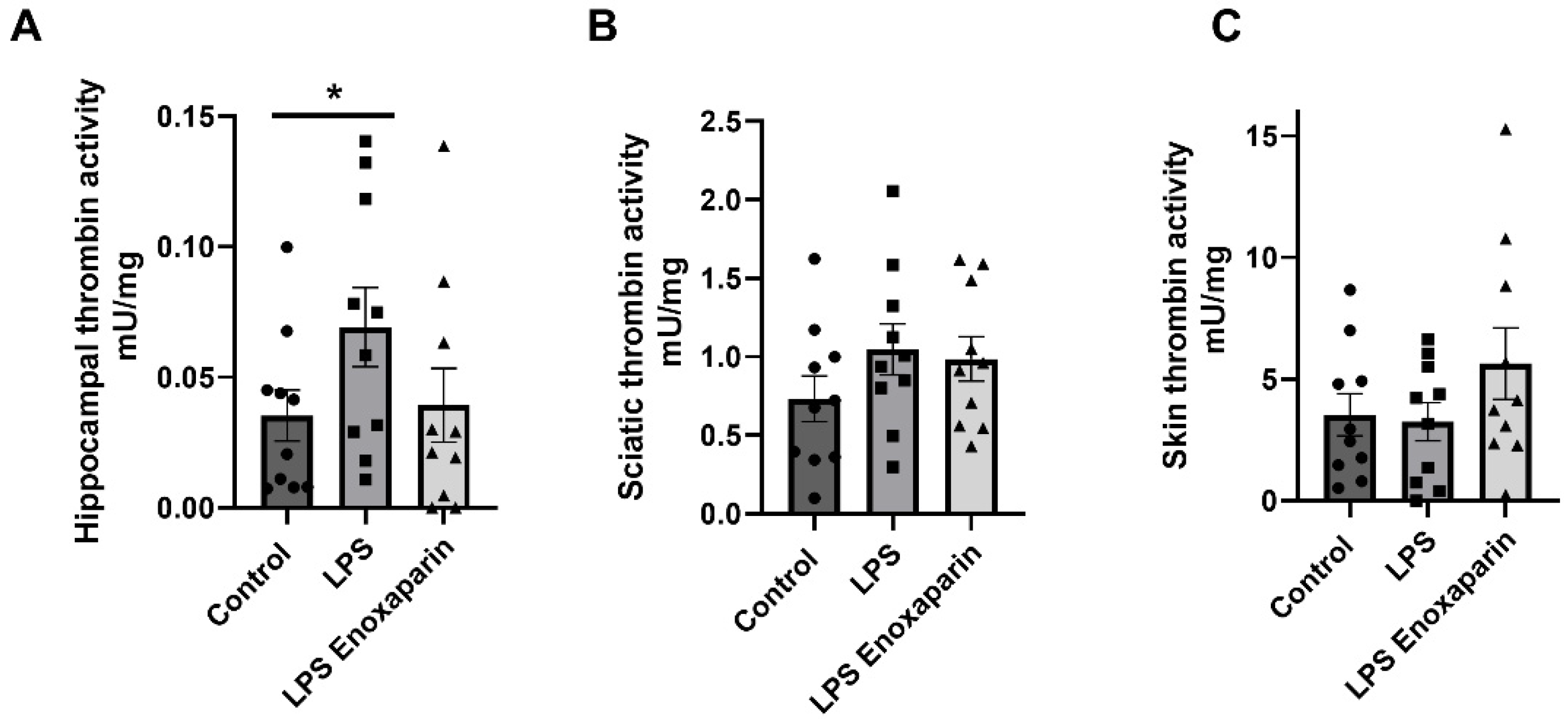
2.6. Thrombin Activity
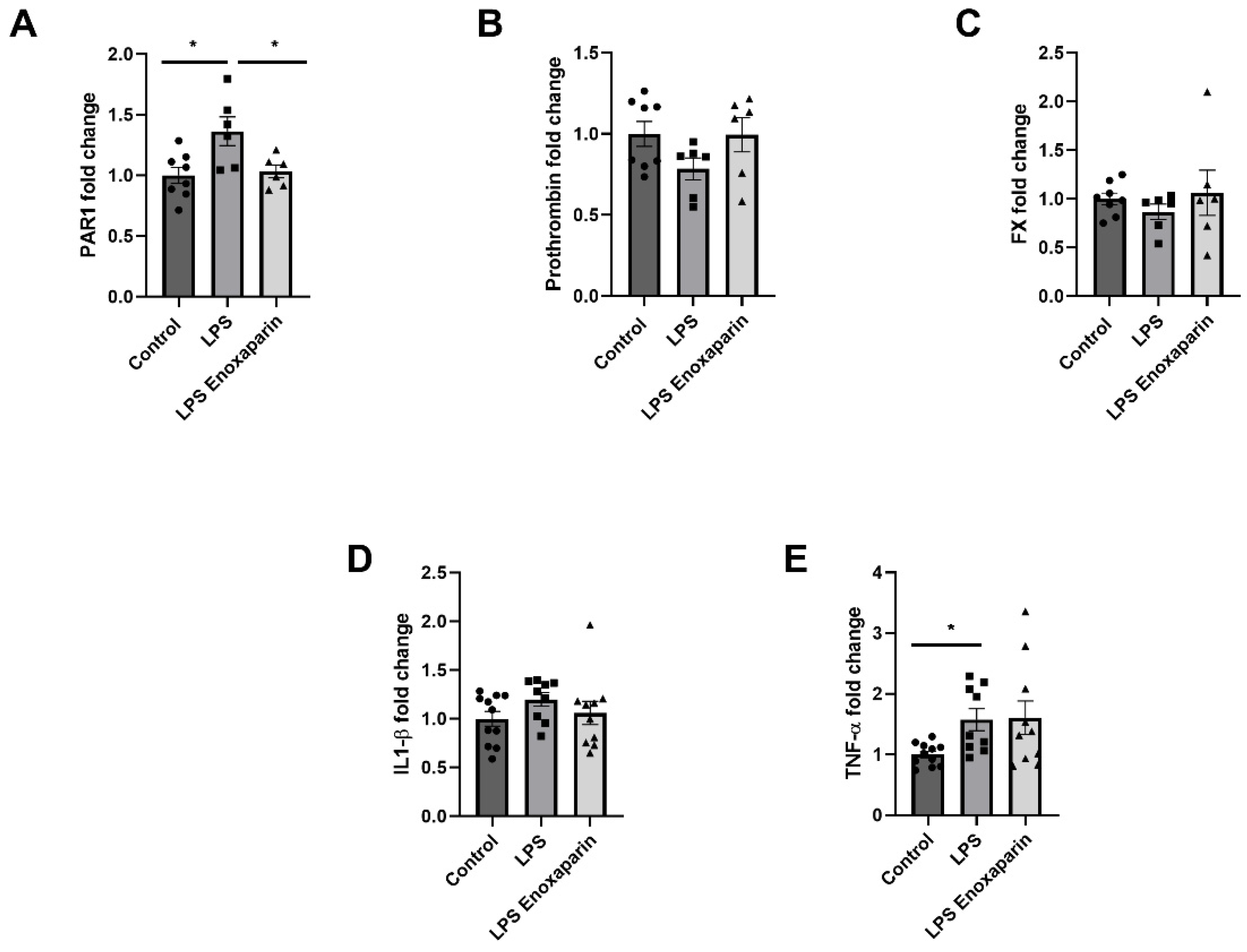
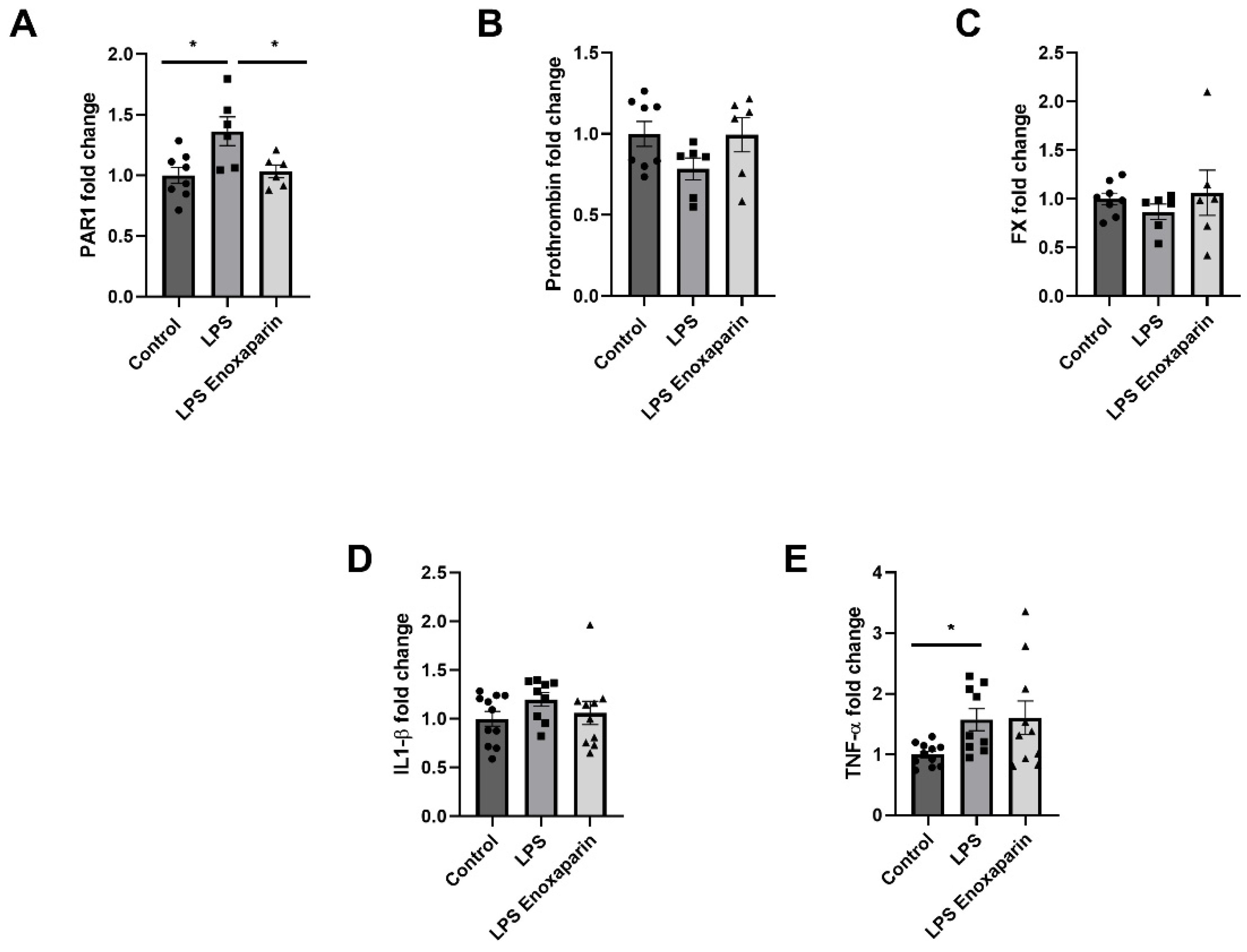
2.7. Hippocampal Gene Expression of Coagulation and Inflammatory Factors
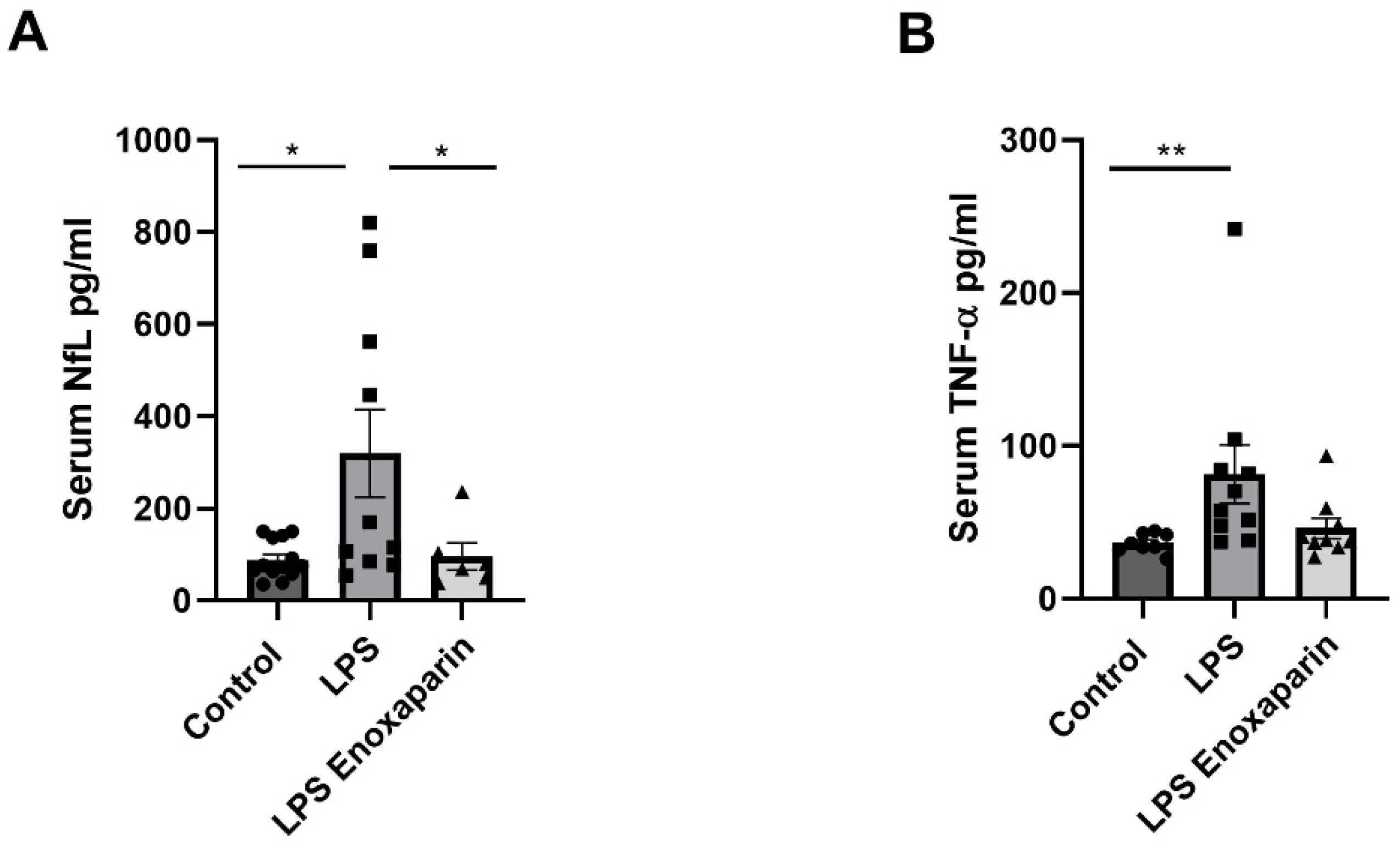
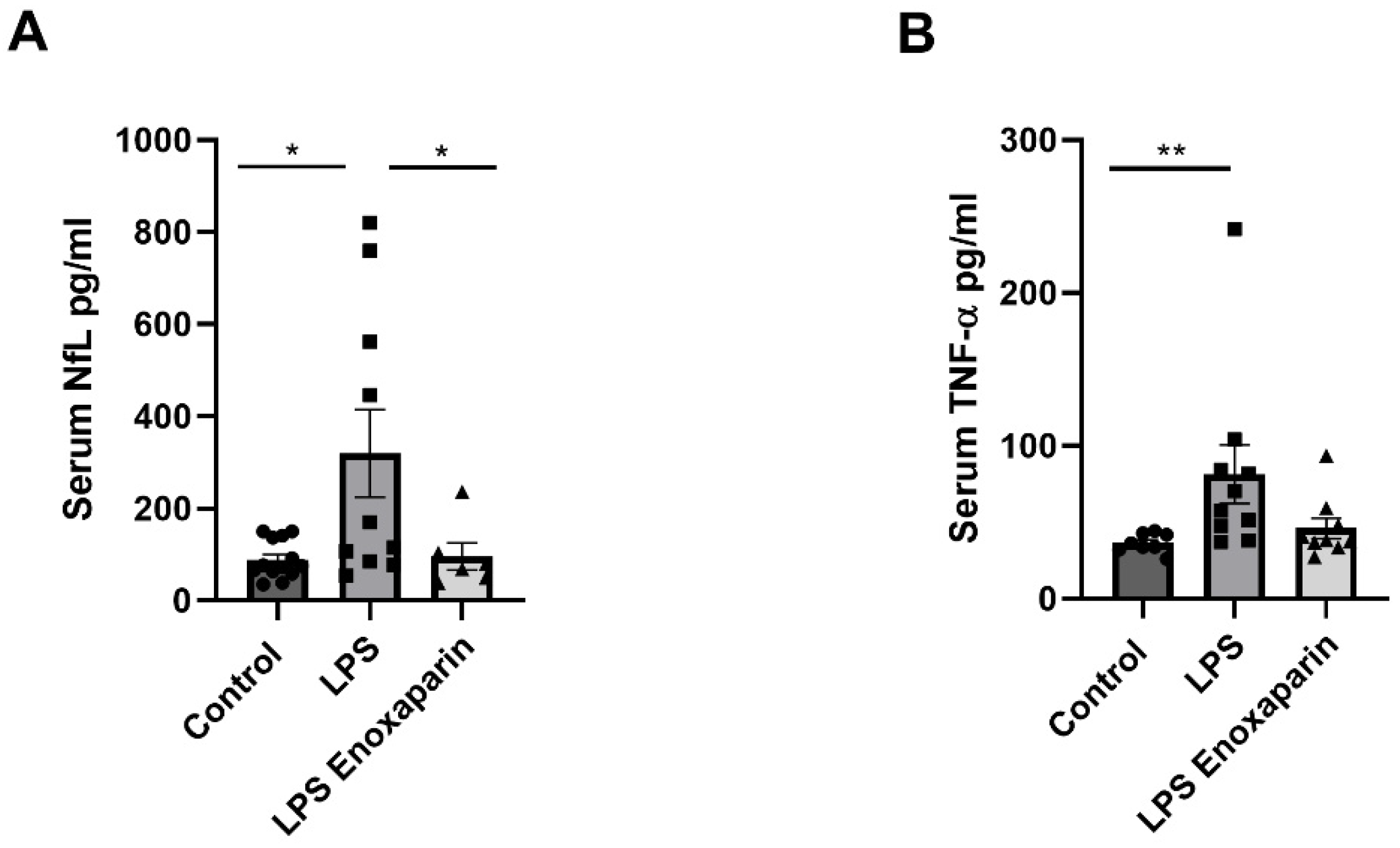
2.8. Serum Markers for Axonal Damage and Inflammation
3. Discussion
4. Materials and Methods
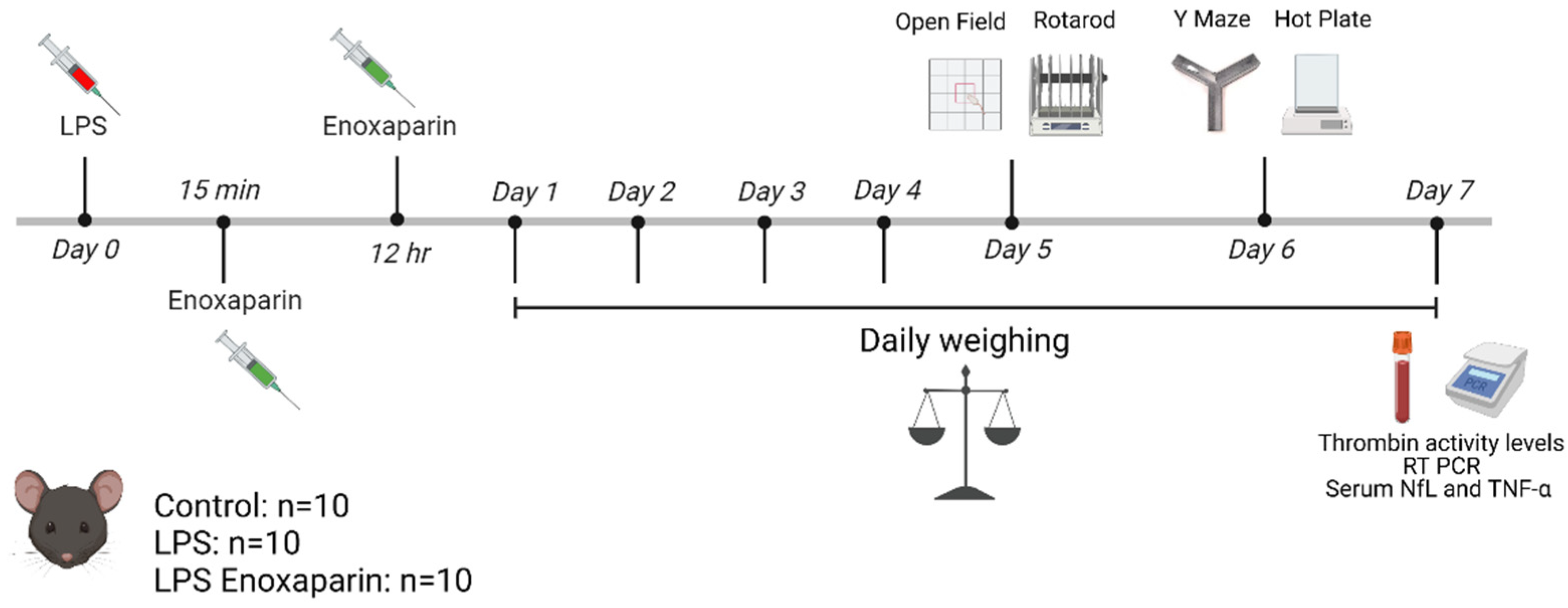
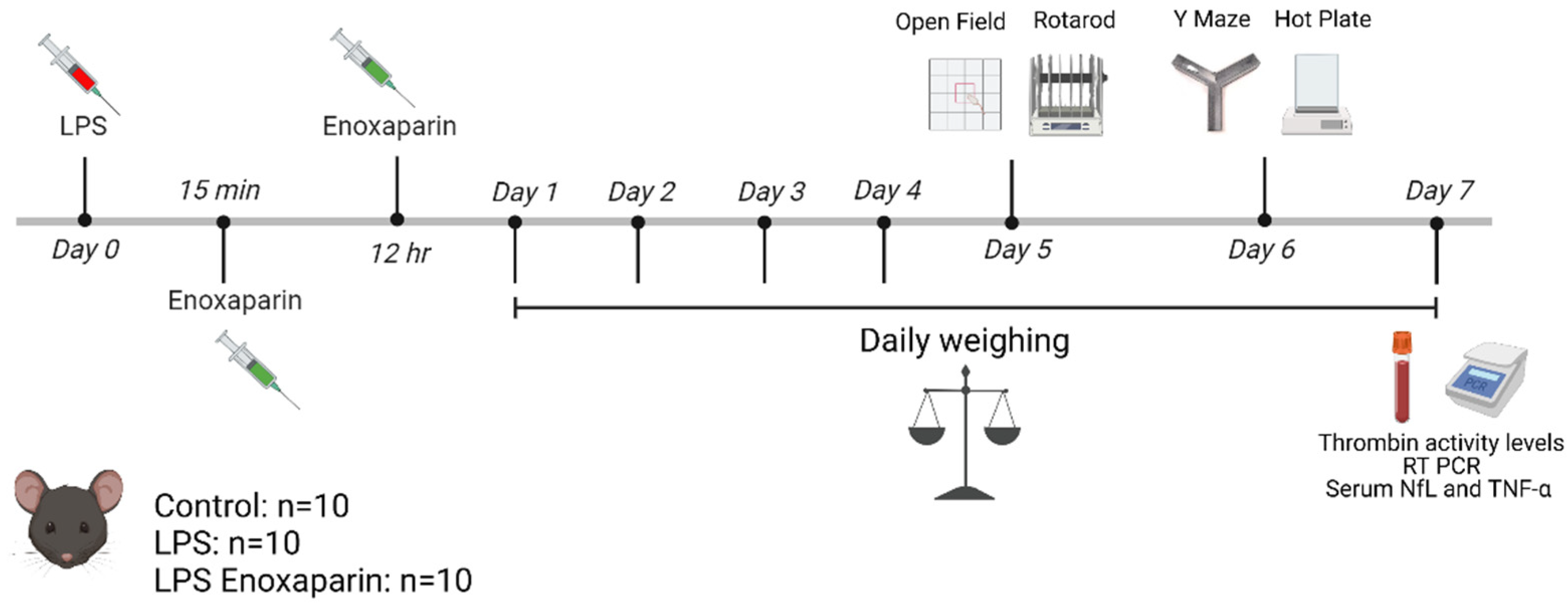
4.1. Model Establishment and Treatment Protocol
4.2. Rotarod
4.3. Hot Plate
4.4. Behavioral Tests
4.5. Open Field
4.6. Y-Maze
4.7. Thrombin Activity Assay
4.8. Coagulation and Inflammation Gene Expression in the Hippocampus
4.9. Neurofilament Light Chain and Tumor Necrosis Factor-α Measurement in the Serum
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, R.; Pan, M.-X.; Tang, J.-C.; Zhang, Y.; Liao, H.-B.; Zhuang, Y.; Zhao, D.; Wan, Q. Role of Neuroinflammation in Ischemic Stroke. Neuroimmunol. Neuroinflammation 2017, 4, 158. [Google Scholar] [CrossRef]
- Lozano, D.; Gonzales-Portillo, G.S.; Acosta, S.; de la Pena, I.; Tajiri, N.; Kaneko, Y.; Borlongan, C.V. Neuroinflammatory Responses to Traumatic Brain Injury: Etiology, Clinical Consequences, And Therapeutic Opportunities. Neuropsychiatr. Dis. Treat. 2015, 11, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Philips, T.; Robberecht, W. Neuroinflammation in Amyotrophic Lateral Sclerosis: Role of Glial Activation in Motor Neuron Disease. Lancet Neurol. 2011, 10, 253–263. [Google Scholar] [CrossRef]
- Rom, S.; Zuluaga-Ramirez, V.; Gajghate, S.; Seliga, A.; Winfield, M.; Heldt, N.A.; Kolpakov, M.A.; Bashkirova, Y.V.; Sabri, A.K.; Persidsky, Y. Hyperglycemia-Driven Neuroinflammation Compromises BBB Leading to Memory Loss in Both Diabetes Mellitus (DM) Type 1 and Type 2 Mouse Models. Mol. Neurobiol. 2019, 56, 1883–1896. [Google Scholar] [CrossRef]
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of Multiple Sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Shi, K.; Tian, D.C.; Li, Z.G.; Ducruet, A.F.; Lawton, M.T.; Shi, F.D. Global Brain Inflammation in Stroke. Lancet Neurol. 2019, 18, 1058–1066. [Google Scholar] [CrossRef]
- Witcher, K.G.; Bray, C.E.; Chunchai, T.; Zhao, F.; O’Neil, S.M.; Gordillo, A.J.; Campbell, W.A.; McKim, D.B.; Liu, X.; Dziabis, J.E.; et al. Traumatic Brain Injury Causes Chronic Cortical Inflammation and Neuronal Dysfunction Mediated by Microglia. J. Neurosci. 2021, 41, 1597–1616. [Google Scholar] [CrossRef] [PubMed]
- Bendorius, M.; Po, C.; Muller, S.; Jeltsch-David, H. From Systemic Inflammation to Neuroinflammation: The Case of Neurolupus. Int. J. Mol. Sci. 2018, 19, 3588. [Google Scholar] [CrossRef]
- Pikman, R.; Kivity, S.; Levy, Y.; Arango, M.-T.; Chapman, J.; Yonath, H.; Shoenfeld, Y.; Gofrit, S.G. Neuropsychiatric SLE: From Animal Model to Human. Lupus 2017, 26, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Meneses, G.; Cárdenas, G.; Espinosa, A.; Rassy, D.; Pérez-Osorio, I.N.; Bárcena, B.; Fleury, A.; Besedovsky, H.; Fragoso, G.; Sciutto, E. Sepsis: Developing New Alternatives to Reduce Neuroinflammation and Attenuate Brain Injury. Ann. N. Y. Acad. Sci. 2019, 1437, 43–56. [Google Scholar] [CrossRef]
- Oronsky, B.; Larson, C.; Hammond, T.C.; Oronsky, A.; Kesari, S.; Lybeck, M.; Reid, T.R. A Review of Persistent Post-COVID Syndrome (PPCS). Clin. Rev. Allergy Immunol. 2021, 1, 3. [Google Scholar] [CrossRef] [PubMed]
- Frantzeskaki, F.; Armaganidis, A.; Orfanos, S.E. Immunothrombosis in Acute Respiratory Distress Syndrome: Cross Talks between Inflammation and Coagulation. Respiration 2017, 93, 212–225. [Google Scholar] [CrossRef] [PubMed]
- Festoff, B.W.; Citron, B.A. Thrombin and the Coag-Inflammatory Nexus in Neurotrauma, ALS, and Other Neurodegenerative Disorders. Front. Neurol. 2019, 10, 59. [Google Scholar] [CrossRef] [PubMed]
- Lorenzano, S.; Inglese, M.; Koudriavtseva, T. Editorial: Role of Coagulation Pathways in Neurological Diseases. Front. Neurol. 2019, 10, 791. [Google Scholar] [CrossRef]
- Esmon, C.T. The Interactions between Inflammation and Coagulation. Br. J. Haematol. 2005, 131, 417–430. [Google Scholar] [CrossRef]
- O’Brien, M. The Reciprocal Relationship Between Inflammation and Coagulation. Top. Companion Anim. Med. 2012, 27, 46–52. [Google Scholar] [CrossRef]
- Levi, M.; Van Der Poll, T. Inflammation and Coagulation. Crit. Care Med. 2010, 38, S26–S34. [Google Scholar] [CrossRef]
- Coughlin, S. Thrombin Signalling and Protease-Activated Receptors. Nature 2000, 407, 258–264. [Google Scholar] [CrossRef]
- Sokolova, E.; Reiser, G. Prothrombin/Thrombin and the Thrombin Receptors PAR-1 and PAR-4 in the Brain: Localization, Expression and Participation in Neurodegenerative Diseases. Thromb. Haemost. 2008, 100, 576–581. [Google Scholar] [CrossRef]
- Heuberger, D.M.; Schuepbach, R.A. Protease-Activated Receptors (PARs): Mechanisms of Action and Potential Therapeutic Modulators in PAR-Driven Inflammatory Diseases. Thromb. J. 2019, 17, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shavit Stein, E.; Ben Shimon, M.; Artan Furman, A.; Golderman, V.; Chapman, J.; Maggio, N. Thrombin Inhibition Reduces the Expression of Brain Inflammation Markers upon Systemic LPS Treatment. Neural Plast. 2018, 2018, 7692182. [Google Scholar] [CrossRef] [PubMed]
- Shavit-Stein, E.; Aronovich, R.; Sylantiev, C.; Gera, O.; Gofrit, S.G.; Chapman, J.; Dori, A. Blocking Thrombin Significantly Ameliorates Experimental Autoimmune Neuritis. Front. Neurol. 2019, 9, 1139. [Google Scholar] [CrossRef]
- Radulovic, M.; Yoon, H.; Wu, J.; Mustafa, K.; Scarisbrick, I.A. Targeting the Thrombin Receptor Modulates Inflammation and Astrogliosis to Improve Recovery after Spinal Cord Injury. Neurobiol. Dis. 2016, 93, 226–242. [Google Scholar] [CrossRef] [PubMed]
- Noble, S.; Peters, D.H.; Goa, K.L. Enoxaparin: A Reappraisal of Its Pharmacology and Clinical Applications in the Prevention and Treatment of Thromboembolic Disease. Drugs 1995, 49, 388–410. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, I.S.; Rao, J.; Izadi, D.; Butler, P.E. Historical Article: Hirudo Medicinalis: Ancient Origins of, and Trends in the Use of Medicinal Leeches throughout History. Br. J. Oral Maxillofac. Surg. 2004, 42, 133–137. [Google Scholar] [CrossRef]
- Bergamaschini, L.; Rossi, E.; Storini, C.; Pizzimenti, S.; Distaso, M.; Perego, C.; De Luigi, A.; Vergani, C.; De Simoni, M.G. Peripheral Treatment with Enoxaparin, a Low Molecular Weight Heparin, Reduces Plaques and β-Amyloid Accumulation in a Mouse Model of Alzheimer’s Disease. J. Neurosci. 2004, 24, 4181–4186. [Google Scholar] [CrossRef]
- Wahl, F.; Grosjean-Piot, O.; Bareyre, F.; Uzan, A.; Stutzmann, J. marie Enoxaparin Reduces Brain Edema, Cerebral Lesions, and Improves Motor and Cognitive Impairments Induced by a Traumatic Brain Injury in Rats. J. Neurotrauma 2000, 17, 1055–1065. [Google Scholar] [CrossRef]
- Li, S.; Marks, J.A.; Eisenstadt, R.; Kumasaka, K.; Samadi, D.; Johnson, V.E.; Holena, D.N.; Allen, S.R.; Browne, K.D.; Smith, D.H.; et al. Enoxaparin Ameliorates Post-Traumatic Brain Injury Edema and Neurologic Recovery, Reducing Cerebral Leukocyte Endothelial Interactions and Vessel Permeability in Vivo. J. Trauma Acute Care Surg. 2015, 79, 78–84. [Google Scholar] [CrossRef]
- Li, S.; Eisenstadt, R.; Kumasaka, K.; Johnson, V.E.; Marks, J.; Nagata, K.; Browne, K.D.; Smith, D.H.; Pascual, J.L. Does Enoxaparin Interfere with HMGB1 Signaling after TBI? A Potential Mechanism for Reduced Cerebral Edema and Neurologic Recovery. J. Trauma Acute Care Surg. 2016, 80, 381–389. [Google Scholar] [CrossRef]
- Kerr, N.A.; de Rivero Vaccari, J.P.; Weaver, C.; Dietrich, W.D.; Ahmed, T.; Keane, R.W. Enoxaparin Attenuates Acute Lung Injury and Inflammasome Activation after Traumatic Brain Injury. J. Neurotrauma 2020, 38, 646–654. [Google Scholar] [CrossRef]
- Semmler, A.; Hermann, S.; Mormann, F.; Weberpals, M.; Paxian, S.A.; Okulla, T.; Schäfers, M.; Kummer, M.P.; Klockgether, T.; Heneka, M.T. Sepsis Causes Neuroinflammation and Concomitant Decrease of Cerebral Metabolism. J. Neuroinflammation 2008, 5, 38. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Wu, X.; Block, M.L.; Liu, Y.; Breese, G.R.; Hong, J.S.; Knapp, D.J.; Crews, F.T. Systemic LPS Causes Chronic Neuroinflammation and Progressive Neurodegeneration. Glia 2007, 55, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Skelly, D.T.; Hennessy, E.; Dansereau, M.A.; Cunningham, C. A Systematic Analysis of the Peripheral and CNS Effects of Systemic LPS, IL-1Β, TNF-α and IL-6 Challenges in C57BL/6 Mice. PLoS ONE 2013, 8, e69123. [Google Scholar] [CrossRef]
- Valero, J.; Mastrella, G.; Neiva, I.; Sánchez, S.; Malva, J.O. Long-Term Effects of an Acute and Systemic Administration of LPS on Adult Neurogenesis and Spatial Memory. Front. Neurosci. 2014, 8, 83. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Bi, W.; Xiao, S.; Lan, X.; Cheng, X.; Zhang, J.; Lu, D.; Wei, W.; Wang, Y.; Li, H.; et al. Neuroinflammation Induced by Lipopolysaccharide Causes Cognitive Impairment in Mice. Sci. Rep. 2019, 9, 5790. [Google Scholar] [CrossRef]
- Jiang, M.J.; Chen, Y.H.; Li, L.; Xu, L.; Liu, H.; Qu, X.L.; Xu, J.J.; Ge, B.B.; Qu, H.D. Protective Effects of DL-3-n-Butylphthalide in the Lipopolysaccharide-Induced Mouse Model of Parkinson’s Disease. Mol. Med. Rep. 2017, 16, 6184–6189. [Google Scholar] [CrossRef]
- Marchand, F.; Perretti, M.; McMahon, S.B. Role of the Immune System in Chronic Pain. Nat. Rev. Neurosci. 2005, 6, 521–532. [Google Scholar] [CrossRef]
- Acosta, C.; Davies, A. Bacterial Lipopolysaccharide Regulates Nociceptin Expression in Sensory Neurons. J. Neurosci. Res. 2008, 86, 1077–1086. [Google Scholar] [CrossRef]
- Seibenhener, M.L.; Wooten, M.C. Use of the Open Field Maze to Measure Locomotor and Anxiety-like Behavior in Mice. J. Vis. Exp. 2015, 96, 52434. [Google Scholar] [CrossRef]
- Golderman, V.; Berkowitz, S.; Gofrit, S.G.; Gera, O.; Aharoni, S.A.; Zohar, D.N.; Keren, D.; Dori, A.; Chapman, J.; Shavit-Stein, E. Thrombin Activity in Rodent and Human Skin: Modified by Inflammation and Correlates with Innervation. Biomedicines 2022, 10, 1461. [Google Scholar] [CrossRef]
- Dantzer, R.; Kelley, K. Twenty Years of Research on Cytokine-Induced Sickness Behavior. Brain Behav. Immun. 2007, 21, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Frenois, F.; Moreau, M.; Connor, J.O.; Lawson, M.; Micon, C.; Lestage, J.; Kelley, K.W.; Dantzer, R.; Castanon, N. Lipopolysaccharide Induces Delayed FosB/DeltaFosB Immunostaining within the Mouse Extended Amygdala, Hippocampus and Hypothalamus, That Parallel the Expression of Depressive-like Behavior. Psychoneuroendocrinology 2007, 32, 516. [Google Scholar] [CrossRef] [PubMed]
- Mastinu, A.; Bonini, S.A.; Premoli, M.; Maccarinelli, G.; Sweeney, E.M.; Zhang, L.; Lucini, L.; Memo, M. Protective Effects of Gynostemma Pentaphyllum (Var. Ginpent) against Lipopolysaccharide-Induced Inflammation and Motor Alteration in Mice. Molecules 2021, 26, 570. [Google Scholar] [CrossRef]
- Deacon, R.M.J. Measuring Motor Coordination in Mice. J. Vis. Exp. 2013, 75, 2609. [Google Scholar] [CrossRef] [PubMed]
- Perez-Dominguez, M.; Ávila-Muñoz, E.; Domínguez-Rivas, E.; Zepeda, A. The Detrimental Effects of Lipopolysaccharide-Induced Neuroinflammation on Adult Hippocampal Neurogenesis Depend on the Duration of the pro-Inflammatory Response. Neural Regen. Res. 2019, 14, 817. [Google Scholar] [CrossRef]
- Weiss, R.; Bushi, D.; Mindel, E.; Bitton, A.; Diesendruck, Y.; Gera, O.; Drori, T.; Zmira, O.; Aharoni, S.A.; Agmon-Levin, N.; et al. Autoantibodies to Annexin A2 and Cerebral Thrombosis: Insights from a Mouse Model. Lupus 2021, 30, 775–784. [Google Scholar] [CrossRef]
- Shrot, S.; Katzav, A.; Korczyn, A.D.; Litvinju, Y.; Hershenson, R.; Pick, C.G.; Blank, M.; Zaech, J.; Shoenfeld, Y.; Sirota, P.; et al. Behavioral and Cognitive Deficits Occur Only after Prolonged Exposure of Mice to Antiphospholipid Antibodies. Lupus 2002, 11, 736–743. [Google Scholar] [CrossRef]
- Katzav, A.; Pick, C.G.; Korczyn, A.D.; Oest, E.; Blank, M.; Shoenfeld, Y.; Chapman, J. Hyperactivity in a Mouse Model of the Antiphospholipid Syndrome. Lupus 2001, 10, 496–499. [Google Scholar] [CrossRef]
- Banks, W.A.; Gray, A.M.; Erickson, M.A.; Salameh, T.S.; Damodarasamy, M.; Sheibani, N.; Meabon, J.S.; Wing, E.E.; Morofuji, Y.; Cook, D.G.; et al. Lipopolysaccharide-Induced Blood-Brain Barrier Disruption: Roles of Cyclooxygenase, Oxidative Stress, Neuroinflammation, and Elements of the Neurovascular Unit. J. Neuroinflammation 2015, 12, 223. [Google Scholar] [CrossRef]
- Yan, Y.; Ji, Y.; Su, N.; Mei, X.; Wang, Y.; Du, S.; Zhu, W.; Zhang, C.; Lu, Y.; Xing, X.H. Non-Anticoagulant Effects of Low Molecular Weight Heparins in Inflammatory Disorders: A Review. Carbohydr. Polym. 2017, 160, 71–81. [Google Scholar] [CrossRef]
- Khalil, M.; Teunissen, C.E.; Otto, M.; Piehl, F.; Sormani, M.P.; Gattringer, T.; Barro, C.; Kappos, L.; Comabella, M.; Fazekas, F.; et al. Neurofilaments as Biomarkers in Neurological Disorders. Nat. Rev. Neurol. 2018, 14, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Gisslén, M.; Price, R.W.; Andreasson, U.; Norgren, N.; Nilsson, S.; Hagberg, L.; Fuchs, D.; Spudich, S.; Blennow, K.; Zetterberg, H. Plasma Concentration of the Neurofilament Light Protein (NFL) Is a Biomarker of CNS Injury in HIV Infection: A Cross-Sectional Study. EBioMedicine 2016, 3, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Varhaug, K.N.; Torkildsen, Ø.; Myhr, K.-M.; Vedeler, C.A. Neurofilament Light Chain as a Biomarker in Multiple Sclerosis. Front. Neurol. 2019, 10, 338. [Google Scholar] [CrossRef] [PubMed]
- Stutzmann, J.M.; Mary, V.; Wahl, F.; Grosjean-Piot, O.; Uzan, A.; Pratt, J. Neuroprotective Profile of Enoxaparin, a Low Molecular Weight Heparin, in in Vivo Models of Cerebral Ischemia or Traumatic Brain Injury in Rats: A Review. CNS Drug Rev. 2002, 8, 1–30. [Google Scholar] [CrossRef]
- Shastri, M.D.; Stewart, N.; Eapen, M.; Peterson, G.M.; Zaidi, S.T.R.; Gueven, N.; Sohal, S.S.; Patel, R.P. Opposing Effects of Low Molecular Weight Heparins on the Release of Inflammatory Cytokines from Peripheral Blood Mononuclear Cells of Asthmatics. PLoS ONE 2015, 10, e0118798. [Google Scholar] [CrossRef]
- Nutescu, E.A.; Burnett, A.; Fanikos, J.; Spinler, S.; Wittkowsky, A. Pharmacology of Anticoagulants Used in the Treatment of Venous Thromboembolism. J. Thromb. Thrombolysis 2016, 41, 15–31. [Google Scholar] [CrossRef]
- Gerotziafas, G.; Petropoulou, A.; Verdy, E.; Samama, M.; Elalamy, I. Effect of the Anti-Factor Xa and Anti-Factor IIa Activities of Low-Molecular-Weight Heparins upon the Phases of Thrombin Generation. J. Thromb. Haemost. 2007, 5, 955–962. [Google Scholar] [CrossRef]
- Gofrit, S.; Shavit-Stein, E. The Neuro-Glial Coagulonome: The Thrombin Receptor and Coagulation Pathways as Major Players in Neurological Diseases. Neural Regen. Res. 2019, 14, 2043–2053. [Google Scholar] [PubMed]
- Shavit-Stein, E.; Gofrit, S.G.; Gayster, A.; Teldan, Y.; Ron, A.; Bandora, E.A.; Golderman, V.; Gera, O.; Harnof, S.; Chapman, J.; et al. Treatment of Diabetic Neuropathy with a Novel PAR1-Targeting Molecule. Biomolecules 2020, 10, 1552. [Google Scholar] [CrossRef]
- Tanne, D.; Katzav, A.; Beilin, O.; Grigoriadis, N.C.; Blank, M.; Pick, C.G.; von Landenberg, P.; Shoenfeld, Y.; Chapman, J. Interaction of Inflammation, Thrombosis, Aspirin and Enoxaparin in CNS Experimental Antiphospholipid Syndrome. Neurobiol. Dis. 2008, 30, 56–64. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, M.; Yang, N.; Gao, S.; Li, S.; Zhang, J.; Bi, Y.; Ren, S.; Hou, Y.; Jiang, M.; et al. A Thrombin-Responsive Nanoprobe for In Vivo Visualization of Thrombus Formation through Three-Dimensional Optical/Computed Tomography Hybrid Imaging. ACS Appl. Mater. Interfaces 2021, 13, 27814–27824. [Google Scholar] [CrossRef]
- Chen, B.; Friedman, B.; Whitney, M.A.; van Winkle, J.A.; Le, I.F.; Olson, E.S.; Cheng, Q.; Pereira, B.; Zhao, L.; Tsien, R.Y.; et al. Thrombin Activity Associated with Neuronal Damage during Acute Focal Ischemia. J. Neurosci. 2012, 32, 7622–7631. [Google Scholar] [CrossRef]
- Conrad, C.D.; Galea, L.A.M.; Kuroda, Y.; McEwen, B.S. Chronic Stress Impairs Rat Spatial Memory on the Y Maze, and This Effect Is Blocked by Tianeptine Pretreatment. Behav. Neurosci. 1996, 110, 1321–1334. [Google Scholar] [CrossRef]
- Stein, E.S.; Itsekson-Hayosh, Z.; Aronovich, A.; Reisner, Y.; Bushi, D.; Pick, C.G.; Tanne, D.; Chapman, J.; Vlachos, A.; Maggio, N. Thrombin Induces Ischemic LTP (ILTP): Implications for Synaptic Plasticity in the Acute Phase of Ischemic Stroke. Sci. Rep. 2015, 5, 7912. [Google Scholar] [CrossRef]
- Ben Shimon, M.; Zeimer, T.; Shavit Stein, E.; Artan-Furman, A.; Harnof, S.; Chapman, J.; Eisenkraft, A.; Pick, C.G.; Maggio, N. Recovery from Trauma Induced Amnesia Correlates with Normalization of Thrombin Activity in the Mouse Hippocampus. PLoS ONE 2017, 12, e0188524. [Google Scholar] [CrossRef]
- Bushi, D.; Stein, E.S.; Golderman, V.; Feingold, E.; Gera, O.; Chapman, J.; Tanne, D. A Linear Temporal Increase in Thrombin Activity and Loss of Its Receptor in Mouse Brain Following Ischemic Stroke. Front. Neurol. 2017, 8, 138. [Google Scholar] [CrossRef]
- Bushi, D.; Ben Shimon, M.; Shavit Stein, E.; Chapman, J.; Maggio, N.; Tanne, D. Increased Thrombin Activity Following Reperfusion after Ischemic Stroke Alters Synaptic Transmission in the Hippocampus. J. Neurochem. 2015, 135, 1140–1148. [Google Scholar] [CrossRef]
- Bushi, D.; Chapman, J.; Katzav, A.; Shavit-Stein, E.; Molshatzki, N.; Maggio, N.; Tanne, D. Quantitative Detection of Thrombin Activity in an Ischemic Stroke Model. J. Mol. Neurosci. 2013, 51, 844–850. [Google Scholar] [CrossRef]
- Beilin, O.; Karussis, D.M.; Korczyn, A.D.; Gurwitz, D.; Aronovich, R.; Hantai, D.; Grigoriadis, N.; Mizrachi-Kol, R.; Chapman, J. Increased Thrombin Inhibition in Experimental Autoimmune Encephalomyelitis. J. Neurosci. Res. 2005, 79, 351–359. [Google Scholar] [CrossRef]
- Beilin, O.; Gurwitz, D.; Korczyn, A.D.; Chapman, J. Quantitative Measurements of Mouse Brain Thrombin-like and Thrombin Inhibition Activities. Neuroreport 2001, 12, 2347–2351. [Google Scholar] [CrossRef]
- Itsekson-Hayosh, Z.; Shavit-Stein, E.; Last, D.; Goez, D.; Daniels, D.; Bushi, D.; Gera, O.; Zibly, Z.; Mardor, Y.; Chapman, J.; et al. Thrombin Activity and Thrombin Receptor in Rat Glioblastoma Model: Possible Markers and Targets for Intervention? J. Mol. Neurosci. 2015, 56, 644–651. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward | Reverse |
|---|---|---|
| PAR1 | GCCTCCATCATGCTCATGAC | AAAGCAGACGATGAAGATGCA |
| PT | CCGAAAGGGCAACCTAGAGC | GGCCCAGAACACGTCTGTG |
| FX | GTGGCCGGGAATGCAA | AACCCTTCATTGTCTTCGTTAATGA |
| TNF-α | GACCCTCACACTCAGATCATCTTCT | CCTCCACTTGGTGGTTTGCT |
| IL1-β | CTGGTGTGTGACGTTCCCATTA | CCGACAGCACGAGGCTTT |
| HPRT | GATTAGCGATGATGAACCAGGTT | CCTCCCATCTCCTTCATGA CA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berkowitz, S.; Gofrit, S.G.; Aharoni, S.A.; Golderman, V.; Qassim, L.; Goldberg, Z.; Dori, A.; Maggio, N.; Chapman, J.; Shavit-Stein, E. LPS-Induced Coagulation and Neuronal Damage in a Mice Model Is Attenuated by Enoxaparin. Int. J. Mol. Sci. 2022, 23, 10472. https://doi.org/10.3390/ijms231810472
Berkowitz S, Gofrit SG, Aharoni SA, Golderman V, Qassim L, Goldberg Z, Dori A, Maggio N, Chapman J, Shavit-Stein E. LPS-Induced Coagulation and Neuronal Damage in a Mice Model Is Attenuated by Enoxaparin. International Journal of Molecular Sciences. 2022; 23(18):10472. https://doi.org/10.3390/ijms231810472
Chicago/Turabian StyleBerkowitz, Shani, Shany Guly Gofrit, Shay Anat Aharoni, Valery Golderman, Lamis Qassim, Zehavit Goldberg, Amir Dori, Nicola Maggio, Joab Chapman, and Efrat Shavit-Stein. 2022. "LPS-Induced Coagulation and Neuronal Damage in a Mice Model Is Attenuated by Enoxaparin" International Journal of Molecular Sciences 23, no. 18: 10472. https://doi.org/10.3390/ijms231810472
APA StyleBerkowitz, S., Gofrit, S. G., Aharoni, S. A., Golderman, V., Qassim, L., Goldberg, Z., Dori, A., Maggio, N., Chapman, J., & Shavit-Stein, E. (2022). LPS-Induced Coagulation and Neuronal Damage in a Mice Model Is Attenuated by Enoxaparin. International Journal of Molecular Sciences, 23(18), 10472. https://doi.org/10.3390/ijms231810472