
The Nursehound Scyliorhinus stellaris Mitochondrial Genome—Phylogeny, Relationships among Scyliorhinidae and Variability in Waters of the Balearic Islands



Abstract
:1. Introduction
2. Results and Discussion
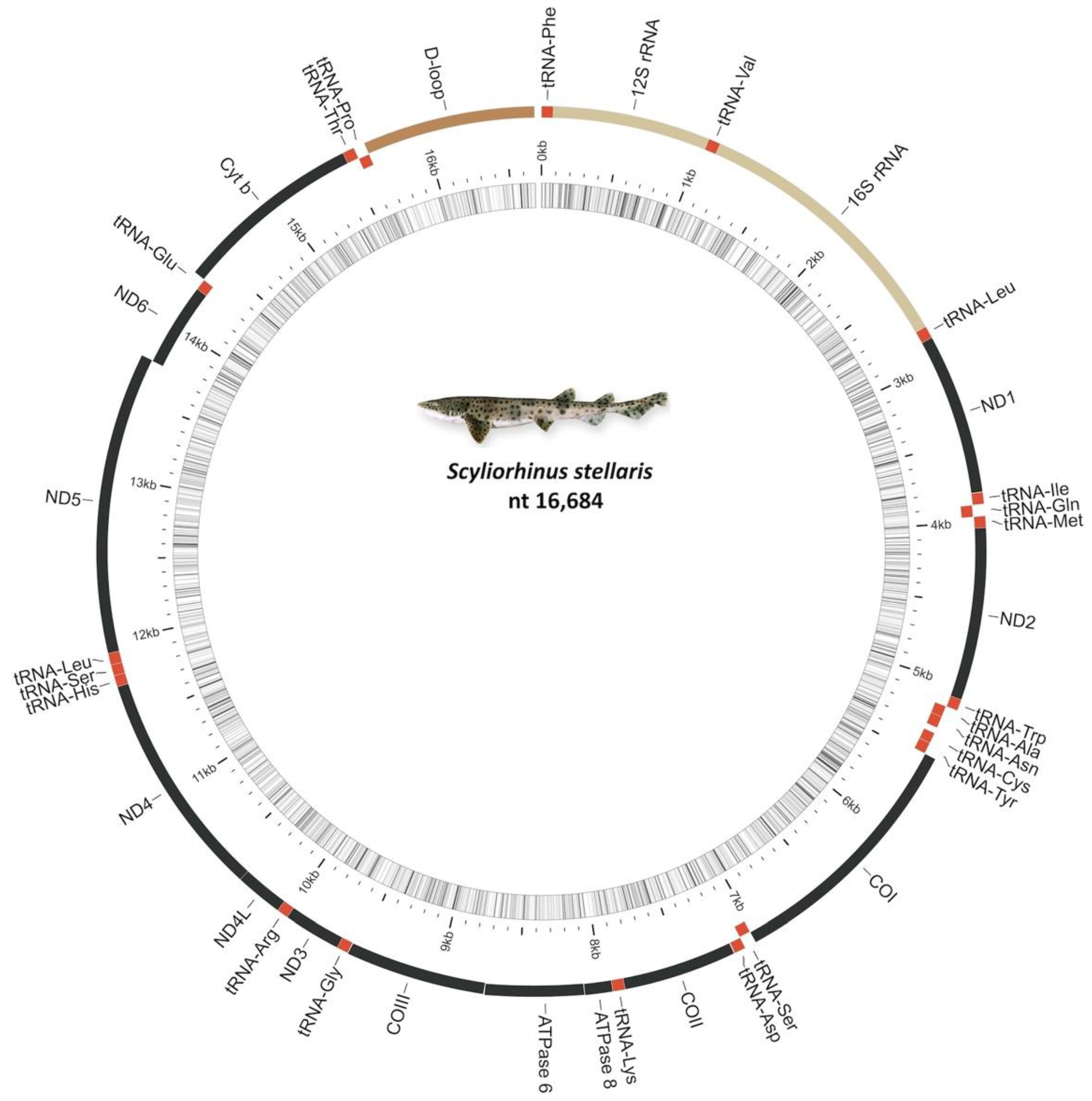
2.1. Mitochondrial Genome Organization
2.2. Ribosomal and Transfer RNA Genes
2.3. Noncoding Regions
Variability in Balearic Sea
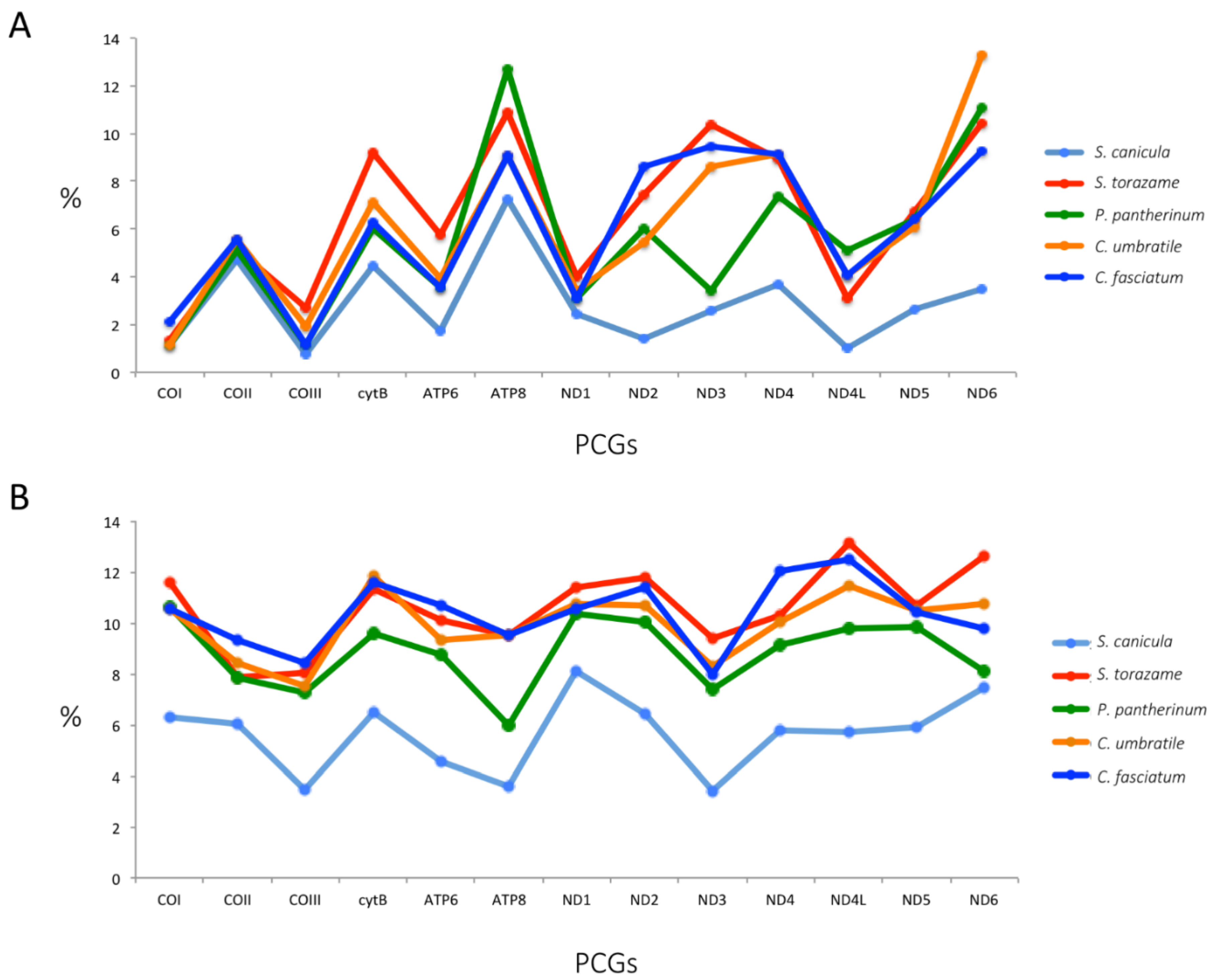
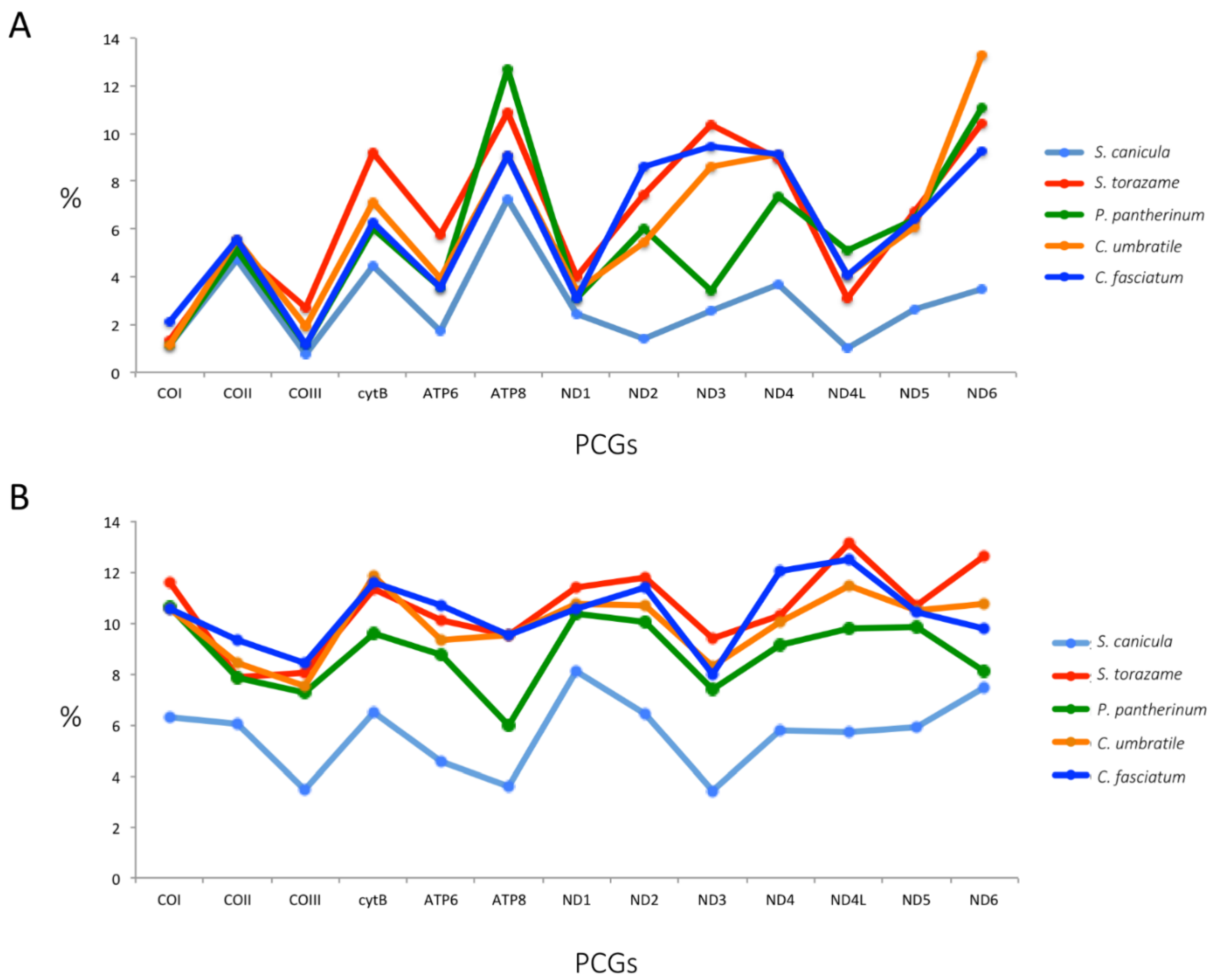
2.4. Protein-Coding Genes
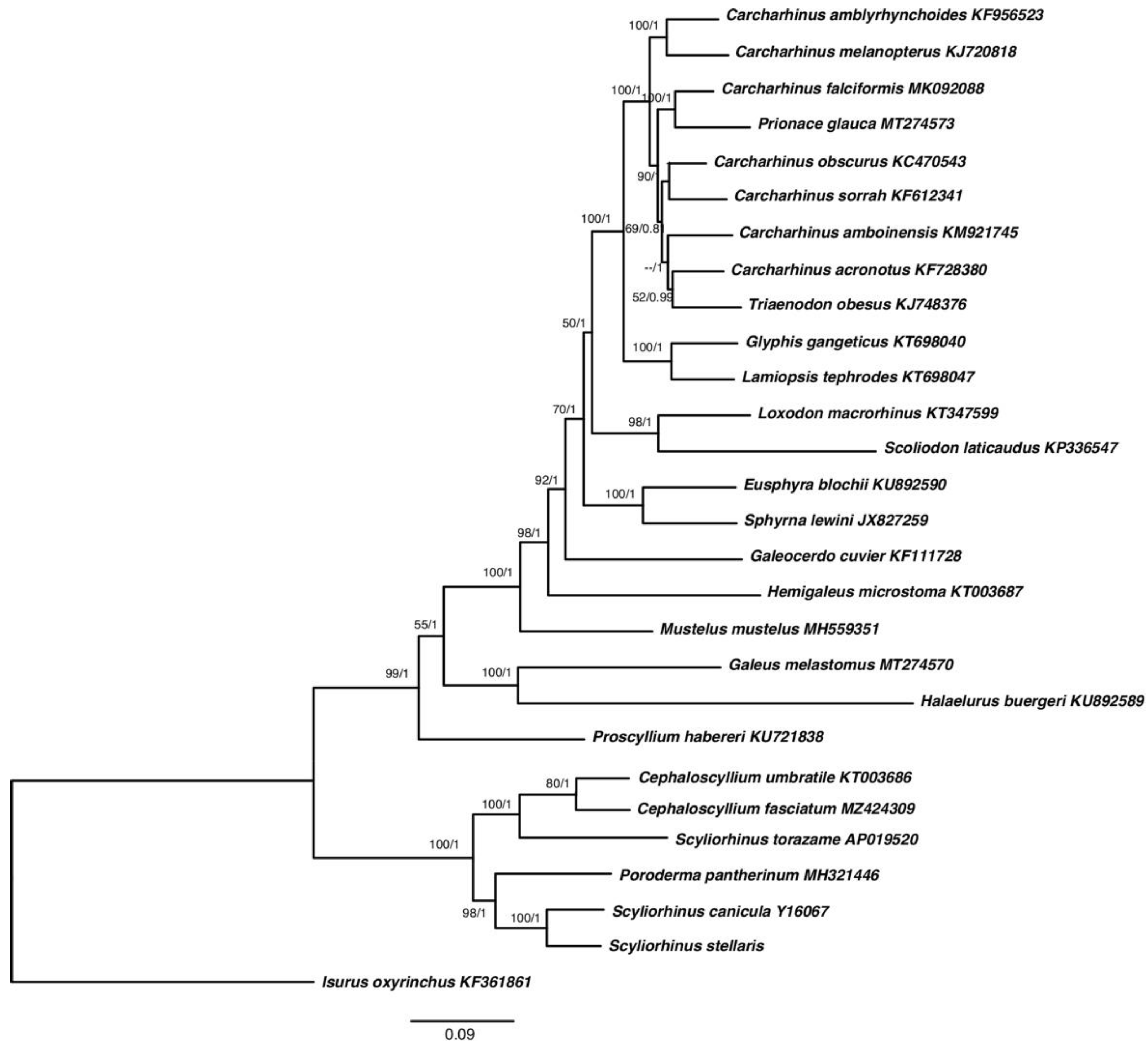
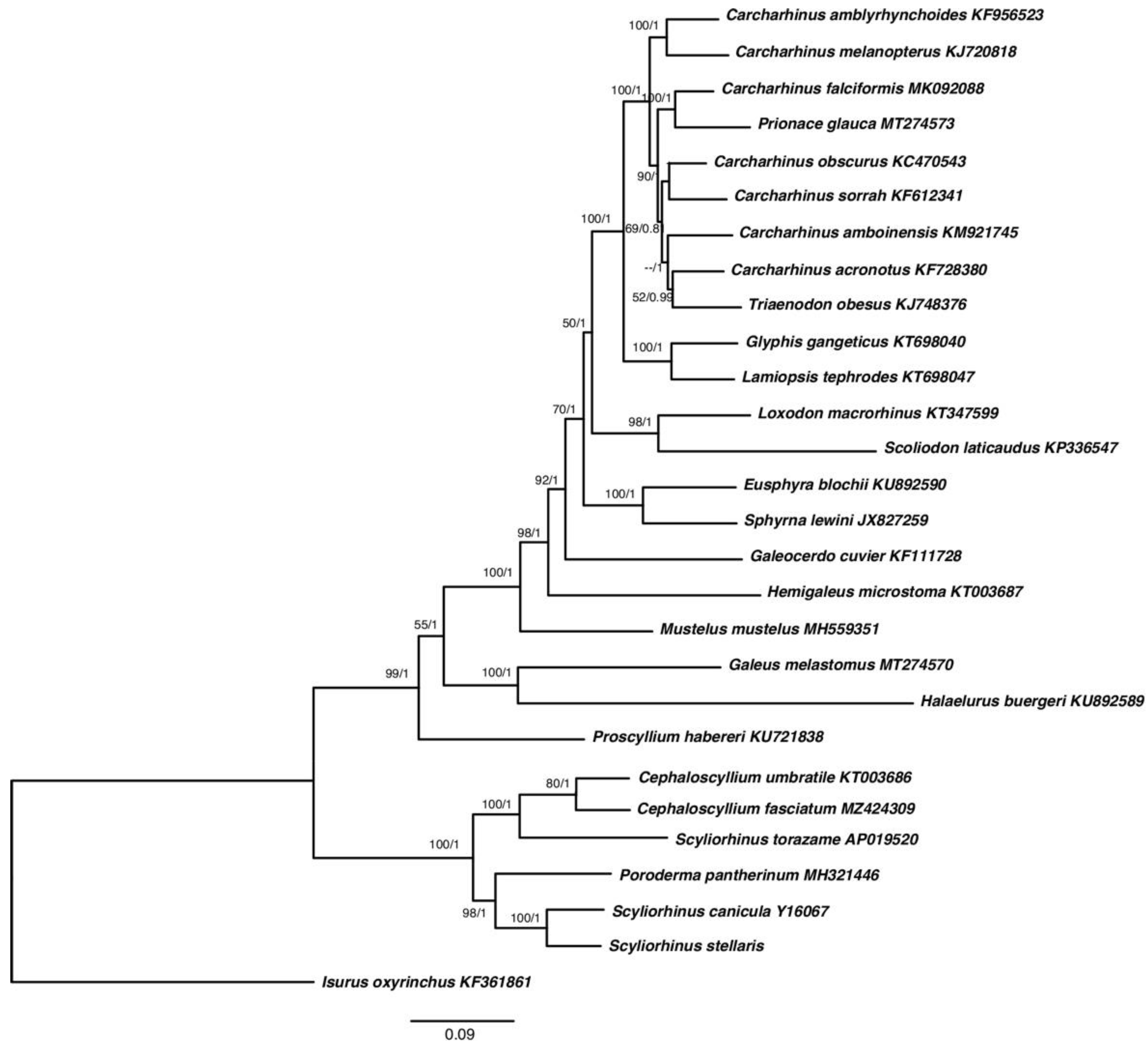
2.5. Multigenes Phylogenies and Genetic Distance
3. Materials and Methods
3.1. Fish Sampling
3.2. DNA Isolation, Amplification and Purification
3.3. Sequence Analysis
3.4. Mitogenomes Comparison, Genetic Distance and Phylogenetic Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barausse, A.; Correale, V.; Curkovic, A.; Finotto, L.; Riginella, E.; Visentin, E.; Mazzoldi, C. The role of fisheries and the environment in driving the decline of elasmobranchs in the northern Adriatic Sea. ICES J. Mar. Sci. 2014, 71, 1593–1603. [Google Scholar] [CrossRef]
- Koehler, L.; Smith, L.E.; Nowell, G. Recovered and released-A novel approach to oviparous shark conservation. Ocean Coast. Manag. 2018, 154, 178–185. [Google Scholar] [CrossRef]
- Silva, J.F.; Ellis, J.R. Bycatch and discarding patterns of dogfish and sharks taken in English and Welsh commercial fisheries. J. Fish Biol. 2019, 94, 966–980. [Google Scholar] [CrossRef] [PubMed]
- Ebert, D.A.; Dando, M.; Fowler, S. Sharks of the World: A Complete Guide; Princeton University Press: Princeton, NJ, USA, 2021. [Google Scholar] [CrossRef]
- Finucci, B.; Derrick, D.; Pacoureau, N. Scyliorhinus Stellaris. The IUCN Red List of Threatened Species 2021; e.T161484A124493465; IUCN: Gland, Switzerland, 2021. [Google Scholar]
- Ellis, J.; Serena, F.; Mancusi, C.; Haka, F.; Morey, G.; Guallart, J.; Schembri, T. Scyliorhinus Stellaris. The IUCN Red List of Threatened Species 2015; e.T161484A48923567; IUCN: Gland, Switzerland, 2015. [Google Scholar]
- Ellis, J.R.; Serena, F.; Mancusi, C.; Haka, F.; Morey, G.; Guallart, J.; Schembri, T. Scyliorhinus Stellaris. The IUCN Red List of Threatened Species 2016; e.T161484A16527873; IUCN: Gland, Switzerland, 2016. [Google Scholar]
- Grau, A.M.; Mayol, J.; Oliver, J.; Riera, F.; Riera, M.I. Llibre vermell dels peixos de les Illes Balears; Conselleria de Medi Ambient, Agricultura i Pesca: Palma, Spain, 2015; 150p. [Google Scholar]
- Aldebert, Y. Demersal Resources of the Gulf of Lions (NW Mediterranean). Impact of exploitation of fish diversity. Milieu 1997, 47, 275–284. [Google Scholar]
- Ragonese, S.; Vitale, S.; Dimech, M.; Mazzola, S. Abundances of Demersal Sharks and Chimaera from 1994–2009 Scientific Surveys in the Central Mediterranean Sea. PLoS ONE 2013, 8, e74865. [Google Scholar] [CrossRef]
- Ramírez-Amaro, S.; Ordines, F.; Esteban, A.; García, C.; Guijarro, B.; Salmerón, F.; Terrrasa, B.; Massutí, E. The diversity of recent trends for chondrichthyans in the Mediterranean reflects fishing exploitation and a potential evolutionary pressure towards early maturation. Sci. Rep. 2020, 10, 1–18. [Google Scholar] [CrossRef]
- Maynou, F.; Sbrana, M.; Sartor, P.; Maravelias, C.; Kavadas, S.; Damalas, D.; Cartes, J.E.; Osio, G. Estimating Trends of Population Decline in Long-Lived Marine Species in the Mediterranean Sea Based on Fishers’ Perceptions. PLoS ONE 2011, 6, e21818. [Google Scholar] [CrossRef]
- Colloca, F.; Scarcella, G.; Libralato, S. Recent Trends and Impacts of Fisheries Exploitation on Mediterranean Stocks and Ecosystems. Front. Mar. Sci. 2017, 4, 244. [Google Scholar] [CrossRef]
- Ellis, J.; Serena, F.; Mancusi, C.; Haka, F.; Morey, G.; Guallart, J.; Schembri, T. Scyliorhinus Stellaris. The IUCN Red List of Threatened Species 2009; e.T161484A5434281; IUCN: Gland, Switzerland, 2009. [Google Scholar]
- Compagno, L.J.V. Sharks of the Order Carcharhiniformes; First Reprint, 2003; The Blackburn Press: Caldwell, NJ, USA, 1988; pp. 1–572. [Google Scholar]
- Iglésias, S.P.; Lecointre, G.; Sellos, D.Y. Extensive paraphylies within sharks of the order Carcharhiniformes inferred from nuclear and mitochondrial genes. Mol. Phylogenet. Evol. 2005, 34, 569–583. [Google Scholar] [CrossRef]
- Human, B.A.; Owen, E.P.; Compagno, L.J.; Harley, E.H. Testing morphologically based phylogenetic theories within the cartilaginous fishes with molecular data, with special reference to the catshark family (Chondrichthyes; Scyliorhinidae) and the interrelationships within them. Mol. Phylogenetics Evol. 2006, 39, 384–391. [Google Scholar] [CrossRef]
- Schaaf-da Silva, J.A.; Ebert, D.A. A revision of the western North Pacific swellsharks, genus Cephaloscyllium Gill 1862 (Chondrichthys: Carcharhiniformes: Scyliorhinidae), including descriptions of two new species. Zootaxa 2008, 1872, 1–8. [Google Scholar] [CrossRef]
- Kousteni, V.; Kasapidis, P.; Kotoulas, G.; Megalofonou, P. Evidence of high genetic connectivity for the longnose spurdog Squalus blainville in the Mediterranean Sea. Med. Mar. Sci. 2016, 17, 371–383. [Google Scholar] [CrossRef]
- Soares, K.D.A.; de Carvalho, M.R. Phylogenetic relationship of catshark species of the genus Scyliorhinus (Chondrichthyes, Carcharhiniformes, Scyliorhinidae) based on comparative morphology. Zoosys. Evol. 2020, 96, 345–395. [Google Scholar] [CrossRef]
- Naylor, G.J.P.; Caira, J.N.; Jensen, K.; Rosana, K.A.M.; Straube, N.; Lakner, C. Elasmobranch Phylogeny: A Mitochondrial Estimate Based on 595 Species. In Biology of Sharks and Their Relatives, 2nd ed.; Carrier, J.C., Musick, J.A., Heithaus, M.R., Eds.; CRC Press: Boca Raton, FL, USA, 2012; pp. 31–56. [Google Scholar]
- Chen, H.; Lin, L.; Chen, X.; Ai, W.; Chen, S. (Complete mitochondrial genome and the phylogenetic position of the Blotchy swell shark Cephaloscyllium umbratile. Mitochondrial DNA Part A 2016, 27, 3045–3047. [Google Scholar] [CrossRef]
- van Staden, M.; Gledhill, K.S.; Rhode, C.; Bester-van der Merwe, A.E. The complete mitochondrial genome and phylogenetic position of the leopard catshark, Poroderma pantherinum. Mitochondrial DNA Part B 2018, 3, 750–752. [Google Scholar] [CrossRef]
- Curole, J.P.; Kocher, T.D. Mitogenomics: Digging deeper with complete mitochondrial genomes. Trends Ecol. Evol. 1999, 14, 394–398. [Google Scholar] [CrossRef]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef]
- Miya, M.; Nishida, M. The mitogenomic contributions to molecular phylogenetics and evolution of fishes: A 15-year retrospect. Ichthyol. Res. 2015, 62, 29–71. [Google Scholar] [CrossRef]
- Kousteni, V.; Mazzoleni, S.; Vasileiadou, K.; Rovatsos, M. Complete mitochondrial DNA genome of nine species of Sharks and Rays and their phylogenetic placement among modern Elasmobranchs. Genes 2021, 12, 324. [Google Scholar] [CrossRef] [PubMed]
- Wolstenholme, D.R. Animal mitochondrial DNA: Structure and evolution. Int. Rev. Cytol. 1992, 141, 173–216. [Google Scholar]
- Manchado, M.; Catanese, G.; Ponce, M.; Funes, V.; Infante, C. The complete mitochondrial genome of the Senegal sole, Solea senegalensis Kaup. Comparative analysis of tandem repeats in the control region among soles. DNA Seq. 2007, 18, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Catanese, G.; Coupé, S.; Bunet, R. Mitogenome sequence comparison in the endangered congeneric Pinna nobilis and Pinna rudis bivalves. Mol. Biol. Rep. 2022, 49, 3627–3635. [Google Scholar] [CrossRef] [PubMed]
- Clayton, D.A. Replication and transcription of vertebrate mitochondrial DNA. Annu. Rev. Cell Biol. 1991, 7, 453–478. [Google Scholar] [CrossRef]
- Shadel, G.S.; Clayton, D.A. Mitochondrial DNA maintenance in vertebrates. Annu. Rev. Biochem. 1997, 66, 409–435. [Google Scholar] [CrossRef]
- Kousteni, V.; Kasapidis, P.; Kotoulas, G.; Megalofonou, P. Strong population genetic structure and contrasting demographic histories for the small-spotted catshark (Scyliorhinus canicula) in the Mediterranean Sea. Heredity 2015, 114, 333–343. [Google Scholar] [CrossRef]
- Broughton, R.E.; Milam, J.E.; Roe, B.A. The complete mitochondrial sequence of the zebrafish (Danio rerio) mitochondrial genome and evolutionary patterns in vertebrate mitochondrial DNA. Genome Res. 2001, 11, 1958–1967. [Google Scholar] [CrossRef]
- Inoue, J.G.; Miya, M.; Tsukamoto, K.; Nishida, M. A mitogenomic perspective on the basal teleostean phylogeny: Resolving higher-level relationships with longer DNA sequences. Mol. Phylogenet Evol. 2001, 20, 275–285. [Google Scholar] [CrossRef]
- Manchado, M.; Catanese, G.; Infante, C. Complete mitochondrial DNA sequence of the Atlantic bluefin tuna (Thunnus thynnus). Fish. Sci. 2004, 70, 68–73. [Google Scholar] [CrossRef]
- Catanese, G.; Infante, C.; Manchado, M. Complete mitochondrial DNA sequences of the frigate tuna Auxis thazard and the bullet tuna Auxis rochei. Mitochondrial DNA 2008, 19, 159–166. [Google Scholar] [CrossRef]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef]
- Sebastian, W.; Sukumaran, S.; Zacharia, P.U.; Muraleedharan, K.R.; Kumar, P.K.D.; Gopalakrishnan, A. Signals of selection in the mitogenome provide insights into adaptation mechanisms in heterogeneous habitats in a widely distributed pelagic fish. Sci. Rep. 2020, 10, 9081. [Google Scholar] [CrossRef] [PubMed]
- Vélez-Zuazo, X.; Agnarsson, I. Shark tales: A molecular species-level phylogeny of sharks (Selachimorpha, Chondrichthyes). Mol. Phylogenet Evol. 2011, 58, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Froese, R.; Pauly, D. FishBase. World Wide Web Electronic Publication. version (02/2022). 2022. Available online: www.fishbase.org (accessed on 22 July 2022).
- Gabora, L. Convergent Evolution. In Brenner’s Encyclopedia of Genetics, 2nd ed.; Maloy, S., Hughes, K., Eds.; Academic Press: Cambridge, MA, USA, 2013; pp. 178–180. [Google Scholar]
- Shen, X.; Pu, Z.; Chen, X.; Murphy, R.W.; Shen, Y. Convergent Evolution of Mitochondrial Genes in Deep-Sea Fishes. Front. Genet. 2019, 10, 925. [Google Scholar] [CrossRef] [PubMed]
- Greenway, R.; Barts, N.; Henpita, C.; Brown, A.P.; Arias Rodriguez, L.; Rodríguez Peña, C.M.; Arndt, S.; Lau, G.Y.; Murphy, M.P.; Wu, L.; et al. Convergent evolution of conserved mitochondrial pathways underlies repeated adaptation to extreme environments. Proc. Natl. Acad. Sci. USA 2020, 117, 16424–16430. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, W.; Fukunaga, T.; Isagozawa, R.; Yamada, K.; Maeda, Y.; Satoh, T.P.; Sado, T.; Mabuchi, K.; Takeshima, H.; Miya, M.; et al. MitoFish and MitoAnnotator: A Mitochondrial Genome Database of Fish with an Accurate and Automatic Annotation Pipeline. Mol. Biol. Evol. 2013, 30, 2531–2540. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Datasets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA 11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 7, 3022–3027. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Posada, D.; Buckley, T.R. Model selection and model averaging in phylogenetics: Advantages of Akaike information criterion and Bayesian approaches over likelihood ratio tests. Syst. Biol. 2004, 53, 793–808. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | Strand | Length | Codons | Spacer | ||||
|---|---|---|---|---|---|---|---|---|
| Gene Description | Start | Stop | nt | AA | Start | Stop | nt | |
| tRNA-F PHE (TTC) | 1 | 70 | H | 70 | 0 | |||
| rRNA12S | 71 | 1028 | H | 958 | 0 | |||
| tRNA-V VAL (GTA) | 1029 | 1100 | H | 72 | 0 | |||
| rRNA16S | 1101 | 2772 | H | 1672 | 0 | |||
| tRNA-L2 LEU (TTA) | 2773 | 2847 | H | 75 | 0 | |||
| ND1 | 2848 | 3822 | H | 975 | 324 | ATG | TAA | 0 |
| tRNA-I ILE (ATC) | 3826 | 3895 | H | 70 | 3 | |||
| tRNA-Q GLN (CAA) | 3897 | 3968 | L | 72 | 1 | |||
| tRNA-M MET (ATG) | 3969 | 4038 | H | 70 | 0 | |||
| ND2 | 4039 | 5085 | H | 1047 | 348 | ATG | TAA | 0 |
| tRNA-W TRP (TGA) | 5085 | 5153 | H | 69 | −1 | |||
| tRNA-A ALA (GCA) | 5155 | 5223 | L | 69 | 1 | |||
| tRNA-N ASN (AAC) | 5224 | 5296 | L | 73 | 0 | |||
| OriL | 5294 | 5344 | - | 51 | −3 | |||
| tRNA-C CYS (TGC) | 5334 | 5400 | L | 67 | −11 | |||
| tRNA-Y TYR (TAC) | 5402 | 5471 | L | 70 | 1 | |||
| COI | 5473 | 7026 | H | 1554 | 517 | GTG | TAA | 1 |
| tRNA-S2 SER (TCA) | 7027 | 7097 | L | 71 | 0 | |||
| tRNA-D ASP (GAC) | 7098 | 7166 | H | 69 | 0 | |||
| COII | 7172 | 7867 | H | 696 | 231 | ATG | AGA | 5 |
| tRNA-K LYS (AAA) | 7860 | 7933 | H | 74 | −8 | |||
| ATP8 | 7935 | 8102 | H | 168 | 55 | ATG | TAA | 1 |
| ATP6 | 8093 | 8776 | H | 684 | 227 | ATG | TAA | −10 |
| COIII | 8776 | 9561 | H | 786 | 261 | ATG | TAA | −1 |
| tRNA-G GLY (GGA) | 9564 | 9633 | H | 70 | 3 | |||
| ND3 | 9634 | 9984 | H | 351 | 116 | ATG | TAA | 0 |
| tRNA-R ARG (CGA) | 9983 | 10,052 | H | 70 | −2 | |||
| ND4L | 10,053 | 10,349 | H | 297 | 98 | ATG | TAA | 0 |
| ND4 | 10,343 | 11,723 | H | 1381 | 460 | ATG | T-- | −7 |
| tRNA-H HIS (CAC) | 11,724 | 11,792 | H | 69 | 0 | |||
| tRNA-S1 SER (AGC) | 11,793 | 11,859 | H | 67 | 0 | |||
| tRNA-L1 LEU (CTA) | 11,860 | 11,931 | H | 72 | 0 | |||
| ND5 | 11,932 | 13,761 | H | 1830 | 609 | ATG | TAG | 0 |
| ND6 | 13,758 | 14,279 | L | 522 | 173 | ATG | TAA | −4 |
| tRNA-E GLU (GAA) | 14,280 | 14,349 | L | 70 | 0 | |||
| CYTB | 14,352 | 15,495 | H | 1144 | 381 | ATG | T-- | 2 |
| tRNA-T THR (ACA) | 15,496 | 15,567 | H | 72 | 0 | |||
| tRNA-P PRO (CCA) | 15,568 | 15,636 | L | 69 | 0 | |||
| DLOOP | 15,637 | 16,684 | - | 1048 | 0 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Catanese, G.; Morey, G.; Verger, F.; Grau, A.M. The Nursehound Scyliorhinus stellaris Mitochondrial Genome—Phylogeny, Relationships among Scyliorhinidae and Variability in Waters of the Balearic Islands. Int. J. Mol. Sci. 2022, 23, 10355. https://doi.org/10.3390/ijms231810355
Catanese G, Morey G, Verger F, Grau AM. The Nursehound Scyliorhinus stellaris Mitochondrial Genome—Phylogeny, Relationships among Scyliorhinidae and Variability in Waters of the Balearic Islands. International Journal of Molecular Sciences. 2022; 23(18):10355. https://doi.org/10.3390/ijms231810355
Chicago/Turabian StyleCatanese, Gaetano, Gabriel Morey, Francesc Verger, and Antonio Maria Grau. 2022. "The Nursehound Scyliorhinus stellaris Mitochondrial Genome—Phylogeny, Relationships among Scyliorhinidae and Variability in Waters of the Balearic Islands" International Journal of Molecular Sciences 23, no. 18: 10355. https://doi.org/10.3390/ijms231810355
APA StyleCatanese, G., Morey, G., Verger, F., & Grau, A. M. (2022). The Nursehound Scyliorhinus stellaris Mitochondrial Genome—Phylogeny, Relationships among Scyliorhinidae and Variability in Waters of the Balearic Islands. International Journal of Molecular Sciences, 23(18), 10355. https://doi.org/10.3390/ijms231810355