


Idiopathic Plasmacytic Lymphadenopathy Forms an Independent Subtype of Idiopathic Multicentric Castleman Disease

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
2.1. Clinical Findings
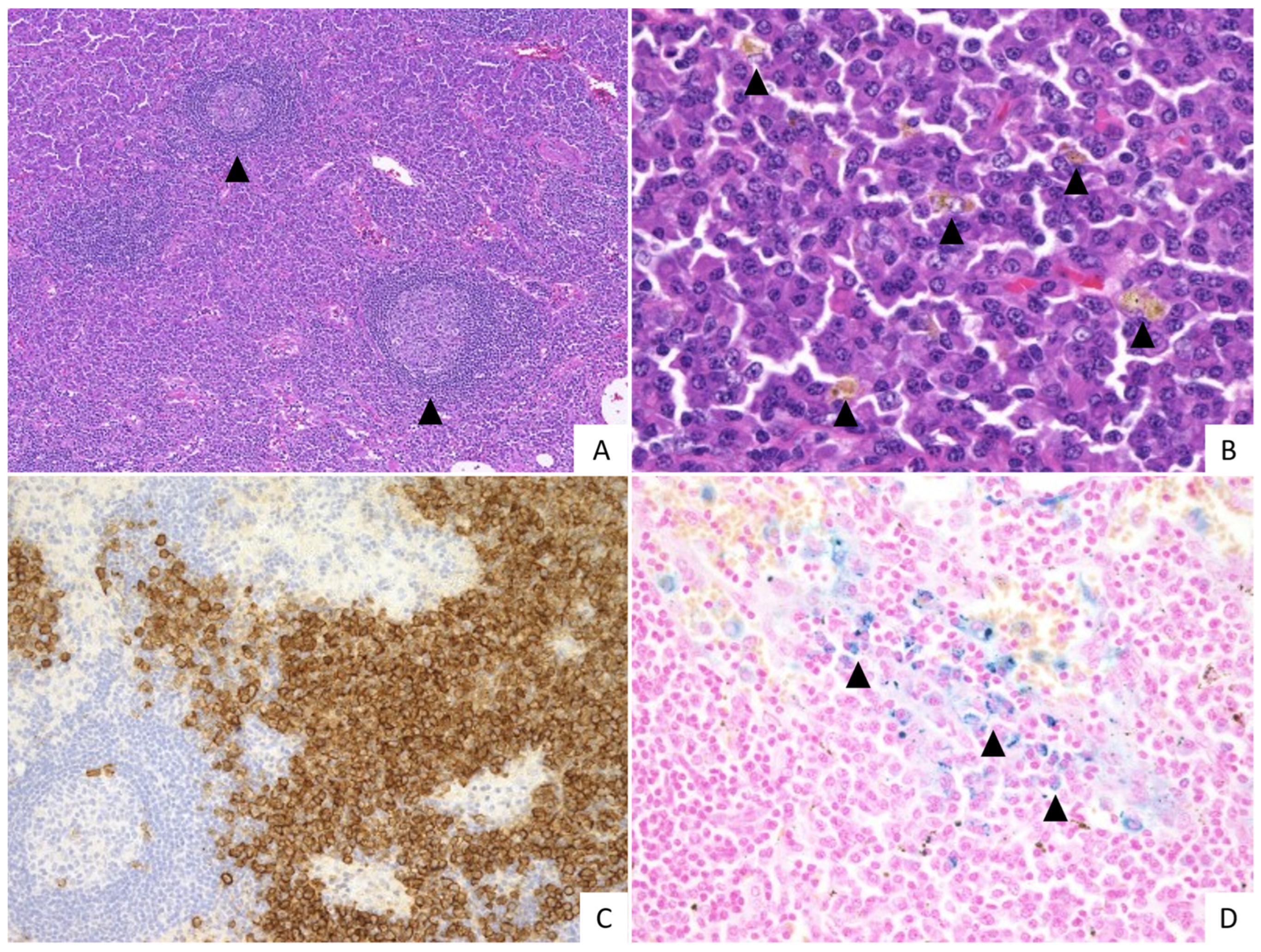
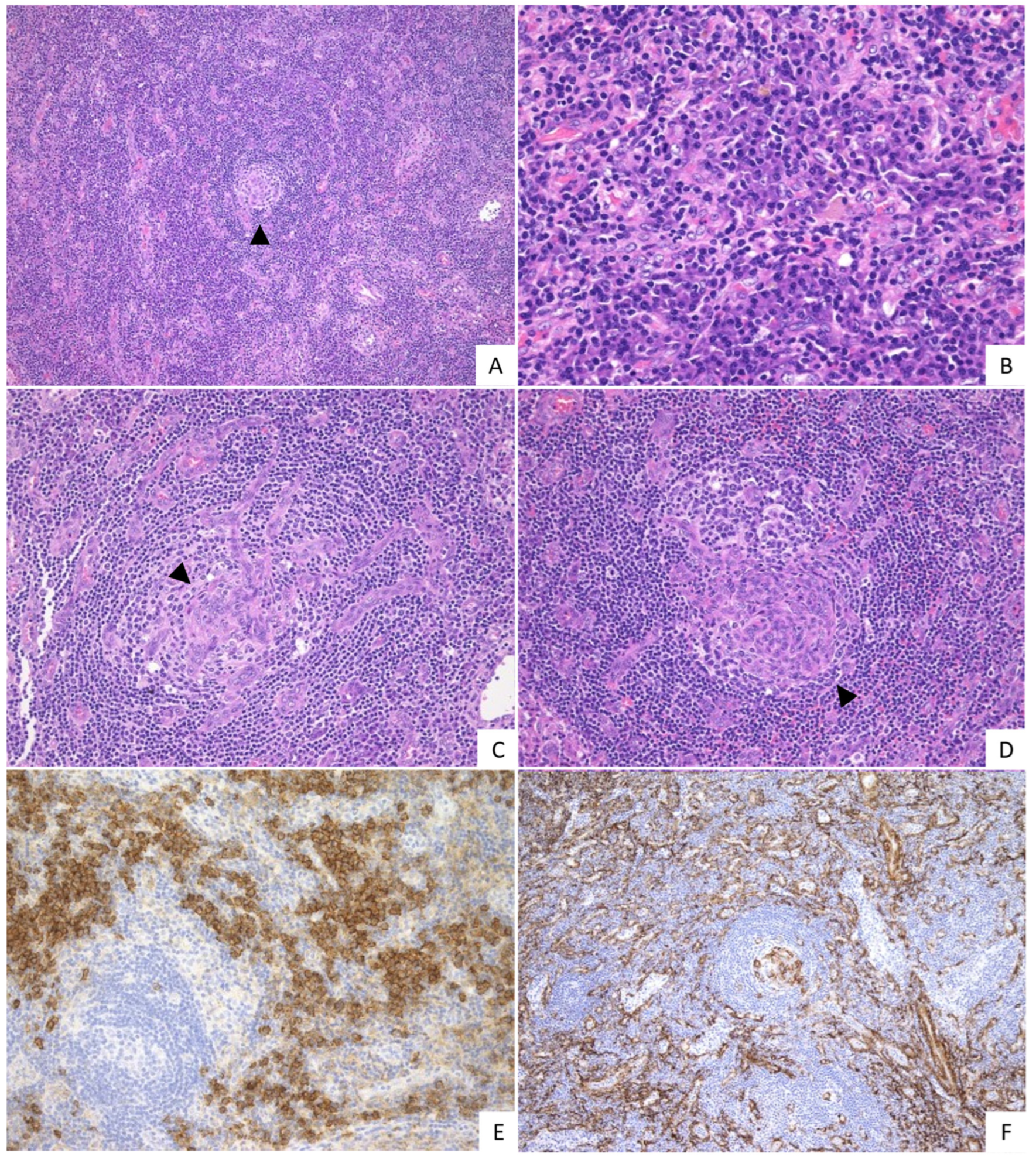
2.2. Pathological Findings
2.3. Treatment and Clinical Course
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Histological Evaluation
4.3. Classification of iMCD-NOS
4.4. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Castleman, B.; Iverson, L.; Menendez, V.P. Localized mediastinal lymphnode hyperplasia resembling thymoma. Cancer 1956, 9, 822–830. [Google Scholar] [CrossRef]
- Nishimura, M.F.; Nishimura, Y.; Nishikori, A.; Maekawa, Y.; Maehama, K.; Yoshino, T.; Sato, Y. Clinical and Pathological Characteristics of Hyaline-Vascular Type Unicentric Castleman Disease: A 20-Year Retrospective Analysis. Diagnostics 2021, 11, 2008. [Google Scholar] [CrossRef] [PubMed]
- Frizzera, G.; Banks, P.M.; Massarelli, G.; Rosai, J. A systemic lymphoproliferative disorder with morphologic features of Castleman’s disease. Pathological findings in 15 patients. Am. J. Surg. Pathol. 1983, 7, 211–231. [Google Scholar] [CrossRef]
- Soulier, J.; Grollet, L.; Oksenhendler, E.; Cacoub, P.; Cazals-Hatem, D.; Babinet, P.; d’Agay, M.F.; Clauvel, J.P.; Raphael, M.; Degos, L.; et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood 1995, 86, 1276–1280. [Google Scholar] [CrossRef]
- Fajgenbaum, D.C.; Uldrick, T.S.; Bagg, A.; Frank, D.; Wu, D.; Srkalovic, G.; Simpson, D.; Liu, A.Y.; Menke, D.; Chandrakasan, S.; et al. International, evidence-based consensus diagnostic criteria for HHV-8-negative/idiopathic multicentric Castleman disease. Blood 2017, 129, 1646–1657. [Google Scholar] [CrossRef] [PubMed]
- Iwaki, N.; Fajgenbaum, D.C.; Nabel, C.S.; Gion, Y.; Kondo, E.; Kawano, M.; Masunari, T.; Yoshida, I.; Moro, H.; Nikkuni, K.; et al. Clinicopathologic analysis of TAFRO syndrome demonstrates a distinct subtype of HHV-8-negative multicentric Castleman disease. Am. J. Hematol. 2016, 91, 220–226. [Google Scholar] [CrossRef]
- Nishimura, Y.; Fajgenbaum, D.C.; Pierson, S.K.; Iwaki, N.; Nishikori, A.; Kawano, M.; Nakamura, N.; Izutsu, K.; Takeuchi, K.; Nishimura, M.F.; et al. Validated international definition of the thrombocytopenia, anasarca, fever, reticulin fibrosis, renal insufficiency, and organomegaly clinical subtype (TAFRO) of idiopathic multicentric Castleman disease. Am. J. Hematol. 2021, 96, 1241–1252. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, Y.; Nishimura, M.F.; Sato, Y. International definition of iMCD-TAFRO: Future perspectives. J. Clin. Exp. Hematop. 2022, 62, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Keller, A.R.; Hochholzer, L.; Castleman, B. Hyaline-vascular and plasma-cell types of giant lymph node hyperplasia of the mediastinum and other locations. Cancer 1972, 29, 670–683. [Google Scholar] [CrossRef]
- Frizzera, G. Castleman’s disease and related disorders. Semin. Diagn. Pathol. 1988, 5, 346–364. [Google Scholar]
- Wang, H.W.; Pittaluga, S.; Jaffe, E.S. Multicentric Castleman disease: Where are we now? Semin Diagn. Pathol. 2016, 33, 294–306. [Google Scholar] [CrossRef] [Green Version]
- Frizzera, G.; Peterson, B.A.; Bayrd, E.D.; Goldman, A. A systemic lymphoproliferative disorder with morphologic features of Castleman’s disease: Clinical findings and clinicopathologic correlations in 15 patients. J. Clin. Oncol. 1985, 3, 1202–1216. [Google Scholar] [CrossRef]
- Kojima, M.; Nakamura, N.; Tsukamoto, N.; Otuski, Y.; Shimizu, K.; Itoh, H.; Kobayashi, S.; Kobayashi, H.; Murase, T.; Masawa, N.; et al. Clinical implications of idiopathic multicentric castleman disease among Japanese: A report of 28 cases. Int. J. Surg. Pathol. 2008, 16, 391–398. [Google Scholar] [CrossRef]
- Pierson, S.K.; Shenoy, S.; Oromendia, A.B.; Gorzewski, A.M.; Langan Pai, R.A.; Nabel, C.S.; Ruth, J.R.; Parente, S.A.T.; Arenas, D.J.; Guilfoyle, M.; et al. Discovery and validation of a novel subgroup and therapeutic target in idiopathic multicentric Castleman disease. Blood Adv. 2021, 5, 3445–3456. [Google Scholar] [CrossRef]
- Endo, Y.; Koga, T.; Ubara, Y.; Sumiyoshi, R.; Furukawa, K.; Kawakami, A. Mediterranean fever gene variants modify clinical phenotypes of idiopathic multi-centric Castleman disease. Clin. Exp. Immunol. 2021, 206, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Mohri, N. Clinicopathological analysis of systemic nodal plasmacytosis with severe polyclonal hyperimmunoglobulinemia. Proc. Jpn. Soc. Pathol. 1978, 67, 252–253. [Google Scholar]
- Han, E.J.; O, J.H.; Jung, S.E.; Park, G.; Choi, B.O.; Jeon, Y.W.; Min, G.J.; Cho, S.G. FDG PET/CT Findings of Castleman Disease Assessed by Histologic Subtypes and Compared with Laboratory Findings. Diagnostics 2020, 10, 998. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, M.F.; Nishimura, Y.; Nishikori, A.; Yoshino, T.; Sato, Y. Historical and pathological overview of Castleman disease. J. Clin. Exp. Hematop. 2022, 62, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Igawa, T.; Ogino, K.; Nishikori, A.; Gion, Y.; Yoshino, T.; Sato, Y. Hemosiderin deposition in lymph nodes of patients with plasma cell-type Castleman disease. J. Clin. Exp. Hematop. 2020, 60, 1–6. [Google Scholar] [CrossRef]
- Nishikori, A.; Nishimura, M.F.; Nishimura, Y.; Notohara, K.; Satou, A.; Moriyama, M.; Nakamura, S.; Sato, Y. Investigation of IgG4-positive cells in idiopathic multicentric Castleman disease and validation of the 2020 exclusion criteria for IgG4-related disease. Pathol. Int. 2022, 72, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, M.F.; Igawa, T.; Gion, Y.; Tomita, S.; Inoue, D.; Izumozaki, A.; Ubara, Y.; Nishimura, Y.; Yoshino, T.; Sato, Y. Pulmonary Manifestations of Plasma Cell Type Idiopathic Multicentric Castleman Disease: A Clinicopathological Study in Comparison with IgG4-Related Disease. J. Pers. Med. 2020, 10, 269. [Google Scholar] [CrossRef]
- Takeuchi, K. Idiopathic plasmacytic lymphadenopathy: A conceptual history along with a translation of the original Japanese article published in 1980. J. Clin. Exp. Hematop. 2022, 62, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, Y.; Nishikori, A.; Sawada, H.; Czech, T.; Otsuka, Y.; Nishimura, M.F.; Mizuno, H.; Sawa, N.; Momose, S.; Ohsawa, K.; et al. Idiopathic multicentric Castleman disease with positive antiphospholipid antibody: Atypical and undiagnosed autoimmune disease? J. Clin. Exp. Hematop. 2022, 62, 99–105. [Google Scholar] [CrossRef]
- Kojima, M.; Nakamura, S.; Itoh, H.; Yoshida, K.; Asano, S.; Yamane, N.; Komatsumoto, S.; Ban, S.; Joshita, T.; Suchi, T. Systemic lupus erythematosus (SLE) lymphadenopathy presenting with histopathologic features of Castleman’ disease: A clinicopathologic study of five cases. Pathol. Res. Pract. 1997, 193, 565–571. [Google Scholar] [CrossRef]
- Kojima, M.; Nakamura, N.; Tsukamoto, N.; Yokohama, A.; Itoh, H.; Kobayashi, S.; Kashimura, M.; Masawa, N.; Nakamura, S. Multicentric Castleman’s disease representing effusion at initial clinical presentation: Clinicopathological study of seven cases. Lupus 2011, 20, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Kojima, M.; Motoori, T.; Asano, S.; Nakamura, S. Histological diversity of reactive and atypical proliferative lymph node lesions in systemic lupus erythematosus patients. Pathol. Res. Pract. 2007, 203, 423–431. [Google Scholar] [CrossRef]
- Narazaki, M.; Kishimoto, T. The Two-Faced Cytokine IL-6 in Host Defense and Diseases. Int. J. Mol. Sci. 2018, 19, 3528. [Google Scholar] [CrossRef] [PubMed]
- Fajgenbaum, D.C.; Langan, R.A.; Japp, A.S.; Partridge, H.L.; Pierson, S.K.; Singh, A.; Arenas, D.J.; Ruth, J.R.; Nabel, C.S.; Stone, K.; et al. Identifying and targeting pathogenic PI3K/AKT/mTOR signaling in IL-6-blockade-refractory idiopathic multicentric Castleman disease. J. Clin. Investig. 2019, 129, 4451–4463. [Google Scholar] [CrossRef]
- Sumiyoshi, R.; Koga, T.; Kawakami, A. Candidate biomarkers for idiopathic multicentric Castleman disease. J. Clin. Exp. Hematop. 2022, 62, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Arenas, D.J.; Floess, K.; Kobrin, D.; Pai, R.L.; Srkalovic, M.B.; Tamakloe, M.A.; Rasheed, R.; Ziglar, J.; Khor, J.; Parente, S.A.T.; et al. Increased mTOR activation in idiopathic multicentric Castleman disease. Blood 2020, 135, 1673–1684. [Google Scholar] [CrossRef] [PubMed]
- Pai, R.L.; Japp, A.S.; Gonzalez, M.; Rasheed, R.F.; Okumura, M.; Arenas, D.; Pierson, S.K.; Powers, V.; Layman, A.A.K.; Kao, C.; et al. Type I IFN response associated with mTOR activation in the TAFRO subtype of idiopathic multicentric Castleman disease. JCI Insight 2020, 5, e135031. [Google Scholar] [CrossRef] [PubMed]

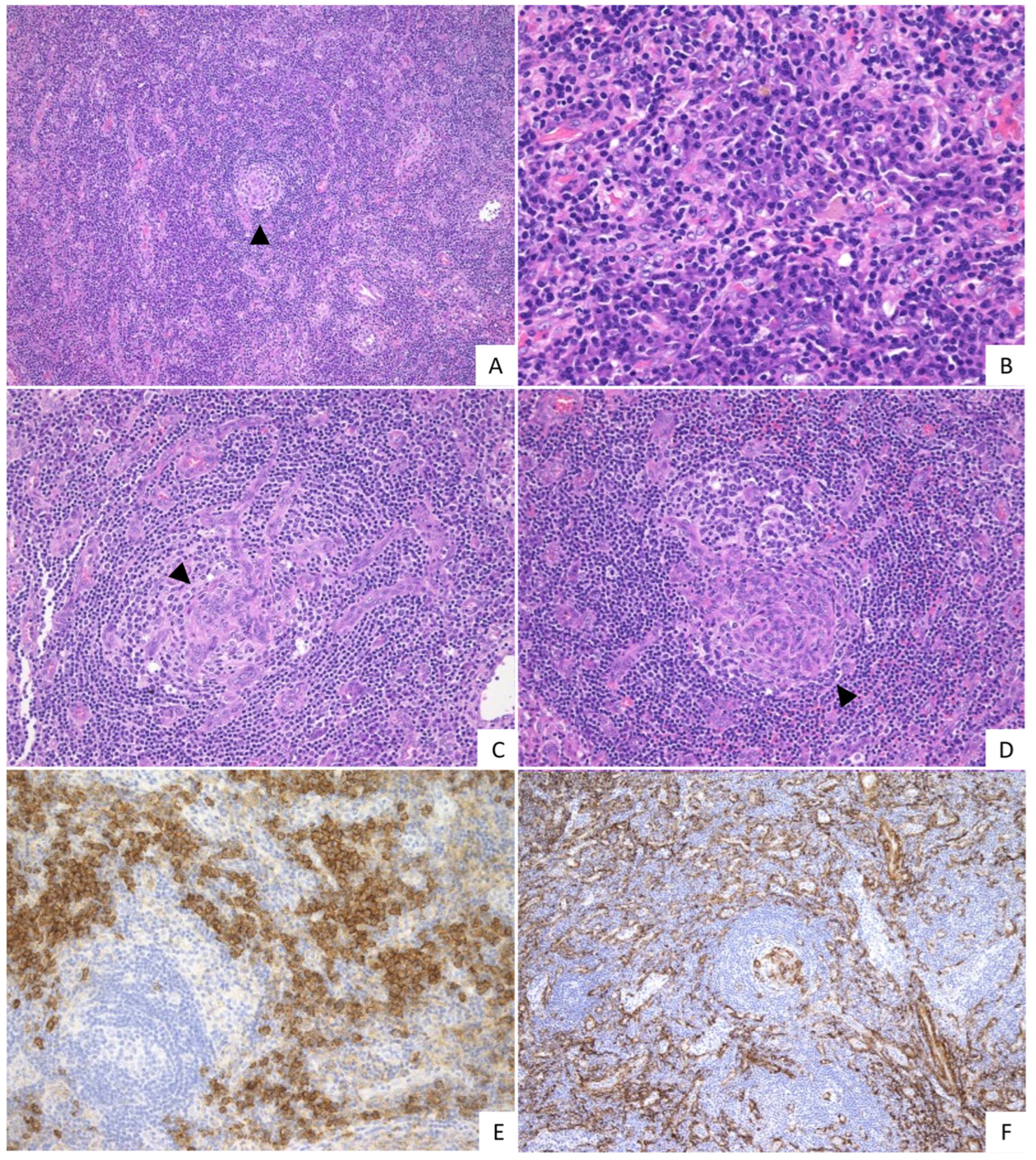

{kind=link}
{kind=link}
| IPL (n = 34) | Non-IPL (n = 8) | p-Value | |
|---|---|---|---|
| Age (median ± SD) | 54.8 ± 12.2 | 57.9 ± 16.9 | 0.471 |
| Sex (M/F) | 21/13 | 3/5 | |
| WBC (×10³/µL) | 7.7 ± 2.5 † | 14.1 ± 9.1 ‡ | 0.200 |
| CRP (mg/dL) | 6.5 ± 3.5 † | 13.2 ± 8.4 ‡ | 0.391 |
| Hb (g/dL) | 10.1 ± 2.1 † | 9.3 ± 1.6 | 0.080 |
| Plt (×10⁴/µL) | 36.9 ± 15.2 † | 23.8 ± 10.9 ‡ | 0.007 * |
| Serum IgG (mg/dL) | 5140.3 ± 1453.1 | 2502.0 ± 752.3 | <0.001 ** |
| Serum IL-6 (pg/mL) | 27.3 ± 16.8 † | 107.2 ± 94.2 ‡ | 0.149 |
| Pleural effusions or/and ascites (%) | 1 (2.9) | 5 (62.5) | <0.001 ** |
| Disease-specific autoantibody (%) | 6/26 (23.1) | 3/6 (50.0) | 0.420 |
| IPL (n = 34) | Non-IPL (n = 8) | p-Value | |
|---|---|---|---|
| Vascularity | |||
| Median | 0.7 | 2.0 | 0.003 * |
| Grade 0 (%) | 17 (50.0) | 1 (12.5) | |
| Grade 1 (%) | 11 (32.4) | 2 (25.0) | |
| Grade 2 (%) | 4 (11.8) | 1 (12.5) | |
| Grade 3 (%) | 2 (5.9) | 4 (50.0) | |
| Plasmacytosis | |||
| Median | 2.9 | 2.1 | 0.001 * |
| Grade 0 (%) | 0 (0.0) | 0 (0.0) | |
| Grade 1 (%) | 0 (0.0) | 1 (12.5) | |
| Grade 2 (%) | 3 (8.8) | 5 (62.5) | |
| Grade 3 (%) | 31 (91.2) | 2 (25.0) | |
| Regressed GCs | |||
| Median | 0.9 | 1.4 | 0.255 |
| Grade 0 (%) | 8 (23.5) | 2 (25.0) | |
| Grade 1 (%) | 20 (58.8) | 3 (37.5) | |
| Grade 2 (%) | 6 (17.6) | 1 (12.5) | |
| Grade 3 (%) | 0 (0.0) | 2 (25.0) | |
| Hyperplastic GCs | |||
| Median | 2.4 | 0.9 | 0.003 * |
| Grade 0 (%) | 1 (2.9) | 5 (62.5) | |
| Grade 1 (%) | 6 (17.6) | 0 (0.0) | |
| Grade 2 (%) | 7 (20.6) | 2 (25.0) | |
| Grade 3 (%) | 20 (58.8) | 1 (12.5) |
| Subtype | Case No. | Age/Sex | 1st Treatment | 2nd Treatment | 3rd Treatment | Outcome | Follow-Up Period (Month) |
|---|---|---|---|---|---|---|---|
| IPL | 1 | 35/F | PSL 20 mg | rituximab | tocilizumab | Improved | 122 |
| 2 | 62/M | PSL 30 mg | tocilizumab | Improved | 93 | ||
| 3 | 55/F | tocilizumab | Improved | 57 | |||
| 4 | 37/M | follow-up | - † | 4 | |||
| 5 | 39/M | tocilizumab | Improved | 39 | |||
| 6 | 49/F | tocilizumab | Improved | 38 | |||
| 7 | 59/M | PSL 50 mg | no response | 39 | |||
| 8 | 64/M | PSL 30 mg | tocilizumab | Improved | 130 | ||
| 9 | 70/M | PSL 30 mg | tocilizumab | Improved | 32 | ||
| 10 | 54/F | follow-up | no change | 161 | |||
| 11 | 65/M | PSL 25 mg | PSL 6 mg | Improved ‡ | 37 | ||
| 12 | 62/F | PSL 10 mg | PSL 5 mg | Improved | 175 | ||
| 13 | 55/F | tocilizumab | Improved | 13 | |||
| 14 | 74/F | tocilizumab | Improved | 10 | |||
| 15 | 52/F | tocilizumab | Improved ‡ | 202 | |||
| 16 | 70/M | follow-up | no change | 198 | |||
| 17 | 43/M | PSL 15 mg | tocilizumab | Improved | 131 | ||
| 18 | 41/M | PSL 5 mg | no response | 126 | |||
| 19 | 49/M | PSL 30 mg | Improved | 90 | |||
| 20 | 48/F | PSL 15 mg | tocilizumab | Improved | 77 | ||
| 21 | 76/M | PSL 40 mg | PSL 10 mg | Improved | 6 | ||
| 22 | 67/M | PSL 20 mg | tocilizumab | Improved | 4 | ||
| 23 | 72/M | tocilizumab | Improved | 4 | |||
| non-IPL | 1 | 52/F | PSL 50 mg + tocilizumab | rituximab | PR | 96 | |
| 2 | 89/M | PSL 60 mg | Repeatedly worsened during tapering | 10 | |||
| 3 | 73/F | PSL 35 mg | Improved | 74 | |||
| 4 | 49/F | follow-up | progression | 41 | |||
| 5 | 49/F | mPSL 500 mg | PSL 40 mg | no response | 143 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nishikori, A.; Nishimura, M.F.; Nishimura, Y.; Otsuka, F.; Maehama, K.; Ohsawa, K.; Momose, S.; Nakamura, N.; Sato, Y. Idiopathic Plasmacytic Lymphadenopathy Forms an Independent Subtype of Idiopathic Multicentric Castleman Disease. Int. J. Mol. Sci. 2022, 23, 10301. https://doi.org/10.3390/ijms231810301
Nishikori A, Nishimura MF, Nishimura Y, Otsuka F, Maehama K, Ohsawa K, Momose S, Nakamura N, Sato Y. Idiopathic Plasmacytic Lymphadenopathy Forms an Independent Subtype of Idiopathic Multicentric Castleman Disease. International Journal of Molecular Sciences. 2022; 23(18):10301. https://doi.org/10.3390/ijms231810301
Chicago/Turabian StyleNishikori, Asami, Midori Filiz Nishimura, Yoshito Nishimura, Fumio Otsuka, Kanna Maehama, Kumiko Ohsawa, Shuji Momose, Naoya Nakamura, and Yasuharu Sato. 2022. "Idiopathic Plasmacytic Lymphadenopathy Forms an Independent Subtype of Idiopathic Multicentric Castleman Disease" International Journal of Molecular Sciences 23, no. 18: 10301. https://doi.org/10.3390/ijms231810301
APA StyleNishikori, A., Nishimura, M. F., Nishimura, Y., Otsuka, F., Maehama, K., Ohsawa, K., Momose, S., Nakamura, N., & Sato, Y. (2022). Idiopathic Plasmacytic Lymphadenopathy Forms an Independent Subtype of Idiopathic Multicentric Castleman Disease. International Journal of Molecular Sciences, 23(18), 10301. https://doi.org/10.3390/ijms231810301