

Assemblages of Plasmodium and Related Parasites in Birds with Different Migration Statuses


Abstract
:1. Introduction
2. Results
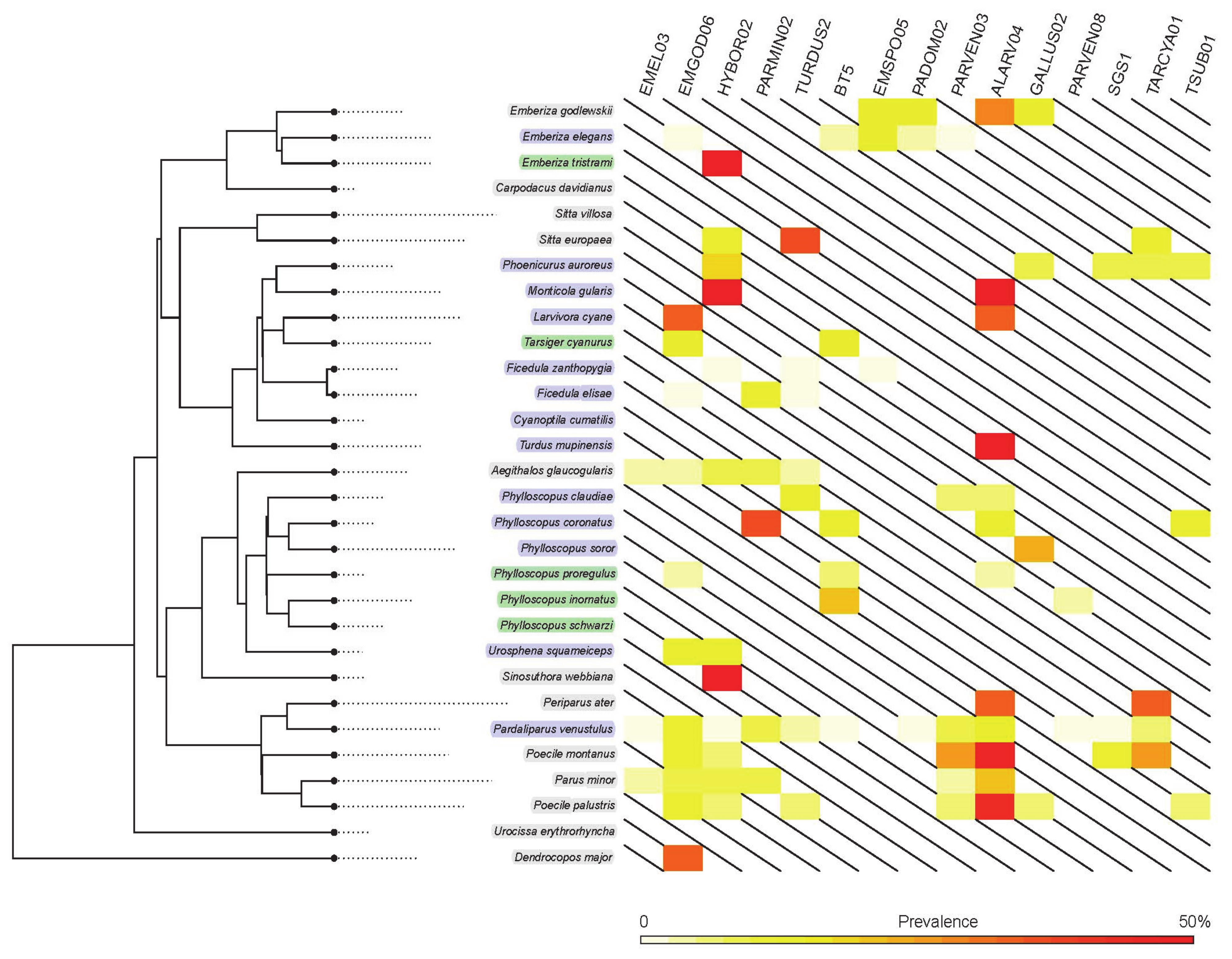
2.1. Prevalence and Lineage Diversity of the Haemosporidian Parasites
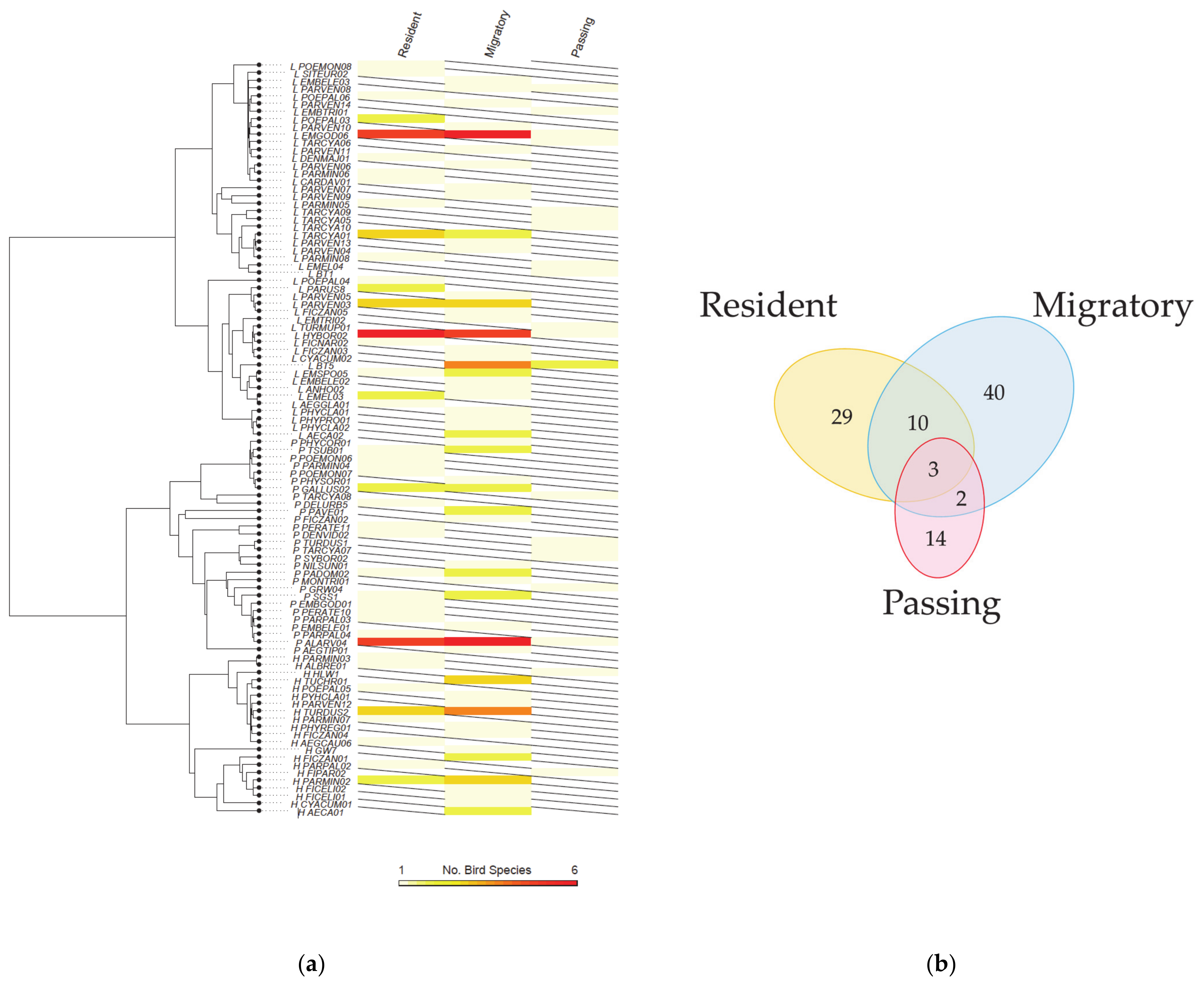
2.2. Shared Lineages among the Host Groups
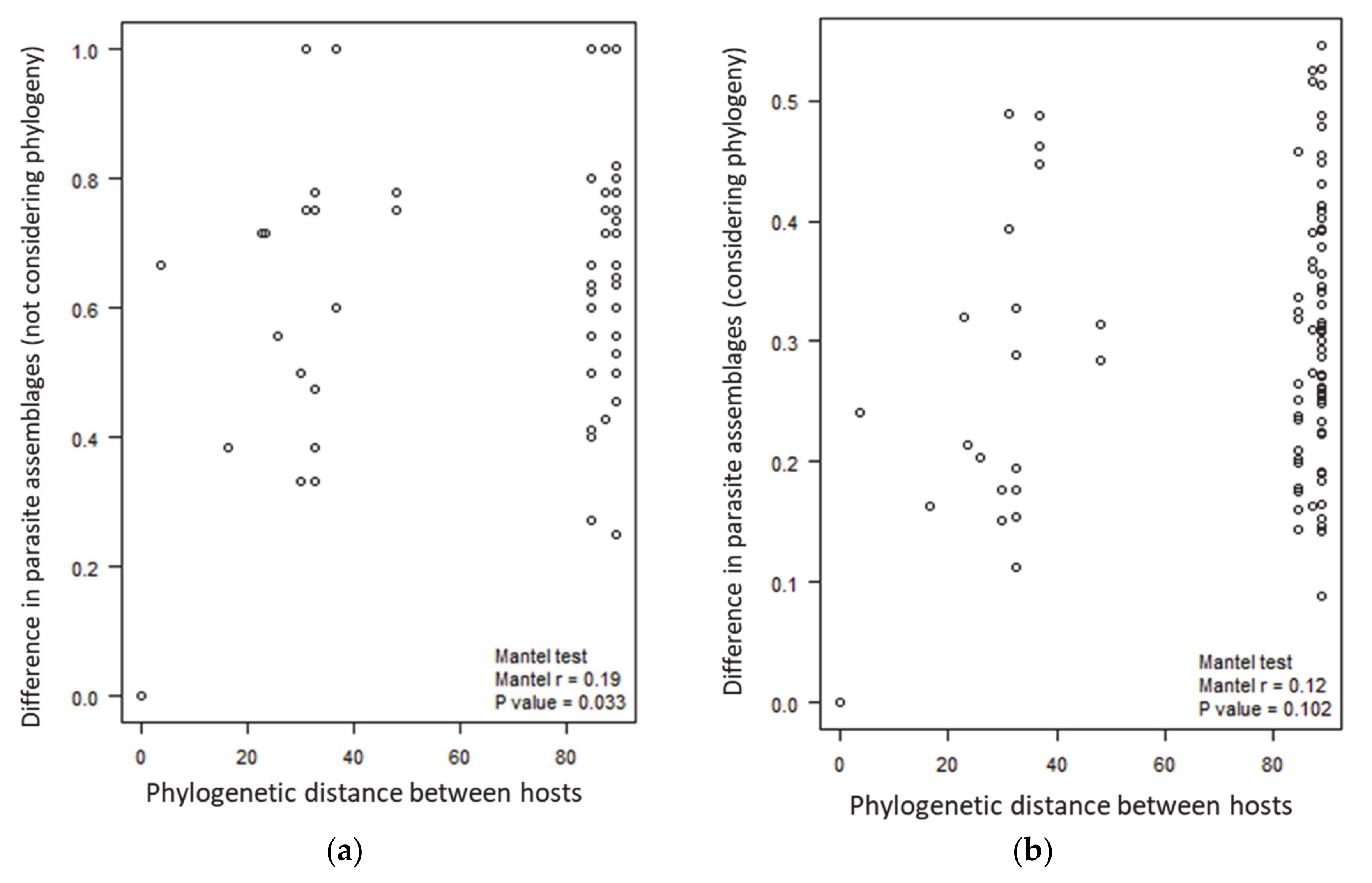
2.3. Parasite Assemblages in the Different Host Groups
3. Discussion
4. Materials and Methods
4.1. Sample Collection and Haemosporidian Identification
4.2. Phylogenetic Analysis
4.3. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Jones, K.E.; Patel, N.G.; Levy, M.A.; Storeygard, A.; Balk, D.; Gittleman, J.L.; Daszak, P. Global trends in emerging infectious diseases. Nature 2008, 451, 990. [Google Scholar] [CrossRef]
- Woodworth, B.L.; Atkinson, C.T.; LaPointe, D.A.; Hart, P.J.; Spiegel, C.S.; Tweed, E.J.; Henneman, C.; LeBrun, J.; Denette, T.; DeMots, R.; et al. Host population persistence in the face of introduced vector-borne diseases: Hawaii amakihi and avian malaria. Proc. Natl. Acad. Sci. USA 2005, 102, 1531–1536. [Google Scholar] [CrossRef] [PubMed]
- Valkiūnas, G. Avian Malaria Parasites and Other Haemosporidia; CRC Press: Boca Raton, FL, USA, 2005. [Google Scholar]
- Rivero, A.; Gandon, S. Evolutionary Ecology of Avian Malaria: Past to Present. Trends Parasitol. 2018, 34, 712–726. [Google Scholar] [CrossRef] [PubMed]
- Bukauskaitė, D.; Žiegytė, R.; Palinauskas, V.; Iezhova, T.A.; Dimitrov, D.; Ilgūnas, M.; Bernotienė, R.; Markovets, M.Y.; Valkiūnas, G. Biting midges (Culicoides, Diptera) transmit Haemoproteus parasites of owls: Evidence from sporogony and molecular phylogeny. Parasites Vectors 2015, 8, 303. [Google Scholar] [CrossRef] [PubMed]
- Bensch, S.; Hellgren, O.; Pérez-Tris, J. MalAvi: A public database of malaria parasites and related haemosporidians in avian hosts based on mitochondrial cytochrome b lineages. Mol. Ecol. Resour. 2009, 9, 1353–1358. [Google Scholar] [CrossRef] [PubMed]
- Ellis, V.A.; Sari, E.H.; Rubenstein, D.R.; Dickerson, R.C.; Bensch, S.; Ricklefs, R.E. The global biogeography of avian haemosporidian parasites is characterized by local diversification and intercontinental dispersal. Parasitology 2018, 146, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Altizer, S.; Bartel, R.; Han, B.A. Animal Migration and Infectious Disease Risk. Science 2011, 331, 296–302. [Google Scholar] [CrossRef]
- Rolland, J.; Jiguet, F.; Jønsson, K.A.; Condamine, F.L.; Morlon, H. Settling down of seasonal migrants promotes bird diversification. Proc. R. Soc. B Biol. Sci. 2014, 281, 20140473. [Google Scholar] [CrossRef]
- Ricklefs, R.E.; Medeiros, M.; Ellis, V.A.; Svensson-Coelho, M.; Blake, J.G.; Loiselle, B.A.; Soares, L.; Fecchio, A.; Outlaw, D.; Marra, P.P. Avian migration and the distribution of malaria parasites in New World passerine birds. J. Biogeogr. 2017, 44, 1113–1123. [Google Scholar] [CrossRef]
- Teitelbaum, C.S.; Huang, S.; Hall, R.J.; Altizer, S. Migratory behaviour predicts greater parasite diversity in ungulates. Proc. R. Soc. B Biol. Sci. 2018, 285, 20180089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, A.K.; Binning, S.A. Recovery from infection is more likely to favour the evolution of migration than social escape from infection. J. Anim. Ecol. 2020, 89, 1448–1457. [Google Scholar] [CrossRef] [PubMed]
- Peacock, S.J.; Krkošek, M.; Lewis, M.A.; Molnár, P.K. A unifying framework for the transient parasite dynamics of migratory hosts. Proc. Natl. Acad. Sci. USA 2020, 117, 10897–10903. [Google Scholar] [CrossRef] [PubMed]
- Vienne, D.d.; Refrégier, G.; López-Villavicencio, M.; Tellier, A.; Hood, M.; Giraud, T. Cospeciation vs host-shift speciation: Methods for testing, evidence from natural associations and relation to coevolution. New Phytol. 2013, 198, 347–385. [Google Scholar] [CrossRef] [PubMed]
- Alcala, N.; Jenkins, T.; Christe, P.; Vuilleumier, S. Host shift and cospeciation rate estimation from co-phylogenies. Ecol. Lett. 2017, 20, 1014–1024. [Google Scholar] [CrossRef]
- Huang, X.; Jönsson, J.; Bensch, S. Persistence of avian haemosporidians in the wild: A case study to illustrate seasonal infection patterns in relation to host life stages. Int. J. Parasitol. 2020, 50, 611–619. [Google Scholar] [CrossRef]
- Pulgarín-R, P.C.; Gómez, C.; Bayly, N.J.; Bensch, S.; FitzGerald, A.M.; Starkloff, N.; Kirchman, J.J.; González-Prieto, A.M.; Hobson, K.A.; Ungvari-Martin, J.; et al. Migratory birds as vehicles for parasite dispersal? Infection by avian haemosporidians over the year and throughout the range of a long-distance migrant. J. Biogeogr. 2019, 46, 83–96. [Google Scholar] [CrossRef]
- Soares, L.; Latta, S.C.; Ricklefs, R.E. Neotropical migratory and resident birds occurring in sympatry during winter have distinct haemosporidian parasite assemblages. J. Biogeogr. 2020, 47, 748–759. [Google Scholar] [CrossRef]
- Davies, T.J.; Pedersen, A.B. Phylogeny and geography predict pathogen community similarity in wild primates and humans. Proc. R. Soc. B 2008, 275, 1695–1701. [Google Scholar] [CrossRef]
- Clark, N.J.; Clegg, S.M. Integrating phylogenetic and ecological distances reveals new insights into parasite host specificity. Mol. Ecol. 2017, 26, 3074–3086. [Google Scholar] [CrossRef]
- Antonovics, J.; Boots, M.; Ebert, D.; Koskella, B.; Poss, M.; Sadd, B.M. The origin of specificity by means of natural selection: Evolved and nonhost resistance in host–pathogen interactions. Evolution 2013, 67, 1–9. [Google Scholar] [CrossRef]
- Bensch, S.; Waldenström, J.; Jonzén, N.; Westerdahl, H.; Hansson, B.; Sejberg, D.; Hasselquist, D. Temporal dynamics and diversity of avian malaria parasites in a single host species. J. Anim. Ecol. 2007, 76, 112–122. [Google Scholar] [CrossRef]
- Becker, K.; Tilley, L.; Vennerstrom, J.L.; Roberts, D.; Rogerson, S.; Ginsburg, H. Oxidative stress in malaria parasite-infected erythrocytes: Host–parasite interactions. Int. J. Parasitol. 2004, 34, 163–189. [Google Scholar] [CrossRef] [PubMed]
- Huang, X. Assessment of Associations between Malaria Parasites and Avian Hosts—A Combination of Classic System and Modern Molecular Approach. Biology 2021, 10, 636. [Google Scholar] [CrossRef] [PubMed]
- Shurulinkov, P.; Chakarov, N.; Daskalova, G. Blood parasites, body condition, and wing length in two subspecies of yellow wagtail (Motacilla flava) during migration. Parasitol. Res. 2012, 110, 2043–2051. [Google Scholar] [CrossRef]
- Combes, C. Parasitism: The Ecology and Evolution of Intimate Interactions; University of Chicago Press: Chicago, IL, USA, 2001. [Google Scholar]
- Valkiūnas, G.; Iezhova, T.A. Exo-erythrocytic development of avian malaria and related haemosporidian parasites. Malar. J. 2017, 16, 101. [Google Scholar] [CrossRef]
- Santiago-Alarcon, D.; Palinauskas, V.; Schaefer, H.M. Diptera vectors of avian Haemosporidian parasites: Untangling parasite life cycles and their taxonomy. Biol. Rev. 2012, 87, 928–964. [Google Scholar] [CrossRef]
- Hellgren, O.; Wood, M.J.; Waldenstrom, J.; Hasselquist, D.; Ottosson, U.; Stervander, M.; Bensch, S. Circannual variation in blood parasitism in a sub-Saharan migrant passerine bird, the garden warbler. J. Evol. Biol. 2013, 26, 1047–1059. [Google Scholar] [CrossRef]
- Knowles, S.C.L.; Palinauskas, V.; Sheldon, B.C. Chronic malaria infections increase family inequalities and reduce parental fitness: Experimental evidence from a wild bird population. J. Evol. Biol. 2010, 23, 557–569. [Google Scholar] [CrossRef] [PubMed]
- O'Connor, E.A.; Strandh, M.; Hasselquist, D.; Nilsson, J.-Å.; Westerdahl, H. The evolution of highly variable immunity genes across a passerine bird radiation. Mol. Ecol. 2016, 25, 977–989. [Google Scholar] [CrossRef] [PubMed]
- Dufour, P.; Descamps, S.; Chantepie, S.; Renaud, J.; Guéguen, M.; Schiffers, K.; Thuiller, W.; Lavergne, S. Reconstructing the geographic and climatic origins of long-distance bird migrations. J. Biogeogr. 2020, 47, 155–166. [Google Scholar] [CrossRef] [Green Version]
- Hellgren, O.; Waldenström, J.; Bensch, S. A new PCR assay for simultaneous studies of Leucocytozoon, Plasmodium, and Haemoproteus from avian blood. J. Parasitol. 2004, 90, 797–802. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772–772. [Google Scholar] [CrossRef] [PubMed]
- Revell, L.J. phytools: An R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 2012, 3, 217–223. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Pagel, M. Inferring the historical patterns of biological evolution. Nature 1999, 401, 877. [Google Scholar] [CrossRef] [PubMed]
- Jetz, W.; Thomas, G.H.; Joy, J.B.; Hartmann, K.; Mooers, A.O. The global diversity of birds in space and time. Nature 2012, 491, 444. [Google Scholar] [CrossRef]
- Bryant, J.A.; Lamanna, C.; Morlon, H.; Kerkhoff, A.J.; Enquist, B.J.; Green, J.L. Microbes on mountainsides: Contrasting elevational patterns of bacterial and plant diversity. Proc. Natl. Acad. Sci. USA 2008, 105, 11505–11511. [Google Scholar] [CrossRef]
- Baselga, A.; Orme, C.D.L. betapart: An R package for the study of beta diversity. Methods Ecol. Evol. 2012, 3, 808–812. [Google Scholar] [CrossRef]
- Paradis, E.; Claude, J.; Strimmer, K. APE: Analyses of phylogenetics and evolution in R language. Bioinformatics 2004, 20, 289–290. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Lineage | Genus | Host Group | Pagel’s λ | p Value |
|---|---|---|---|---|
| PARMIN02 | Haemoproteus | Res + Mig | 6.7 × 10−5 | 1 |
| TURDUS2 | Haemoproteus | Res + Mig | 6.7 × 10−5 | 1 |
| BT5 | Leucocytozoon | Mig + Pass | 6.7 × 10−5 | 1 |
| PARVEN08 | Leucocytozoon | Mig + Pass | 6.7 × 10−5 | 1 |
| EMEL03 | Leucocytozoon | Res + Mig | 6.7 × 10−5 | 1 |
| EMSPO05 | Leucocytozoon | Res + Mig | 0.63 | 0.13 |
| PARVEN03 | Leucocytozoon | Res + Mig | 0.57 | 0.09 |
| TARCYA01 | Leucocytozoon | Res + Mig | 0.64 | 0.45 |
| EMGOD06 | Leucocytozoon | Res + Mig + Pass | 1 | 0.05 |
| HYBOR02 | Leucocytozoon | Res + Mig + Pass | 6.7 × 10−5 | 1 |
| GALLUS02 | Plasmodium | Res + Mig | 6.7 × 10−5 | 1 |
| PADOM02 | Plasmodium | Res + Mig | 0.43 | 0.61 |
| SGS1 | Plasmodium | Res + Mig | 6.7 × 10−5 | 1 |
| TSUB01 | Plasmodium | Res + Mig | 6.7 × 10−5 | 1 |
| ALARV04 | Plasmodium | Res + Mig + Pass | 0.45 | 0.47 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, X.; Chen, Z.; Yang, G.; Xia, C.; Luo, Q.; Gao, X.; Dong, L. Assemblages of Plasmodium and Related Parasites in Birds with Different Migration Statuses. Int. J. Mol. Sci. 2022, 23, 10277. https://doi.org/10.3390/ijms231810277
Huang X, Chen Z, Yang G, Xia C, Luo Q, Gao X, Dong L. Assemblages of Plasmodium and Related Parasites in Birds with Different Migration Statuses. International Journal of Molecular Sciences. 2022; 23(18):10277. https://doi.org/10.3390/ijms231810277
Chicago/Turabian StyleHuang, Xi, Zelin Chen, Guocheng Yang, Canwei Xia, Qiujin Luo, Xiang Gao, and Lu Dong. 2022. "Assemblages of Plasmodium and Related Parasites in Birds with Different Migration Statuses" International Journal of Molecular Sciences 23, no. 18: 10277. https://doi.org/10.3390/ijms231810277
APA StyleHuang, X., Chen, Z., Yang, G., Xia, C., Luo, Q., Gao, X., & Dong, L. (2022). Assemblages of Plasmodium and Related Parasites in Birds with Different Migration Statuses. International Journal of Molecular Sciences, 23(18), 10277. https://doi.org/10.3390/ijms231810277