To optimize cell isolation and preservation, we compared different protocols for cell processing and fixation before cell sorting and scRNA-seq analysis. First, the isolation of cells from murine pancreatic tumors (Pan02) by mechanical dissociation was compared with enzymatic/mechanical digestion using a gentleMACS
TM Dissociator (Miltenyi Biotec). We also independently investigated whether the DSP fixation process affected FACS or scRNA-seq. Two independent experiments were performed, each with three samples.
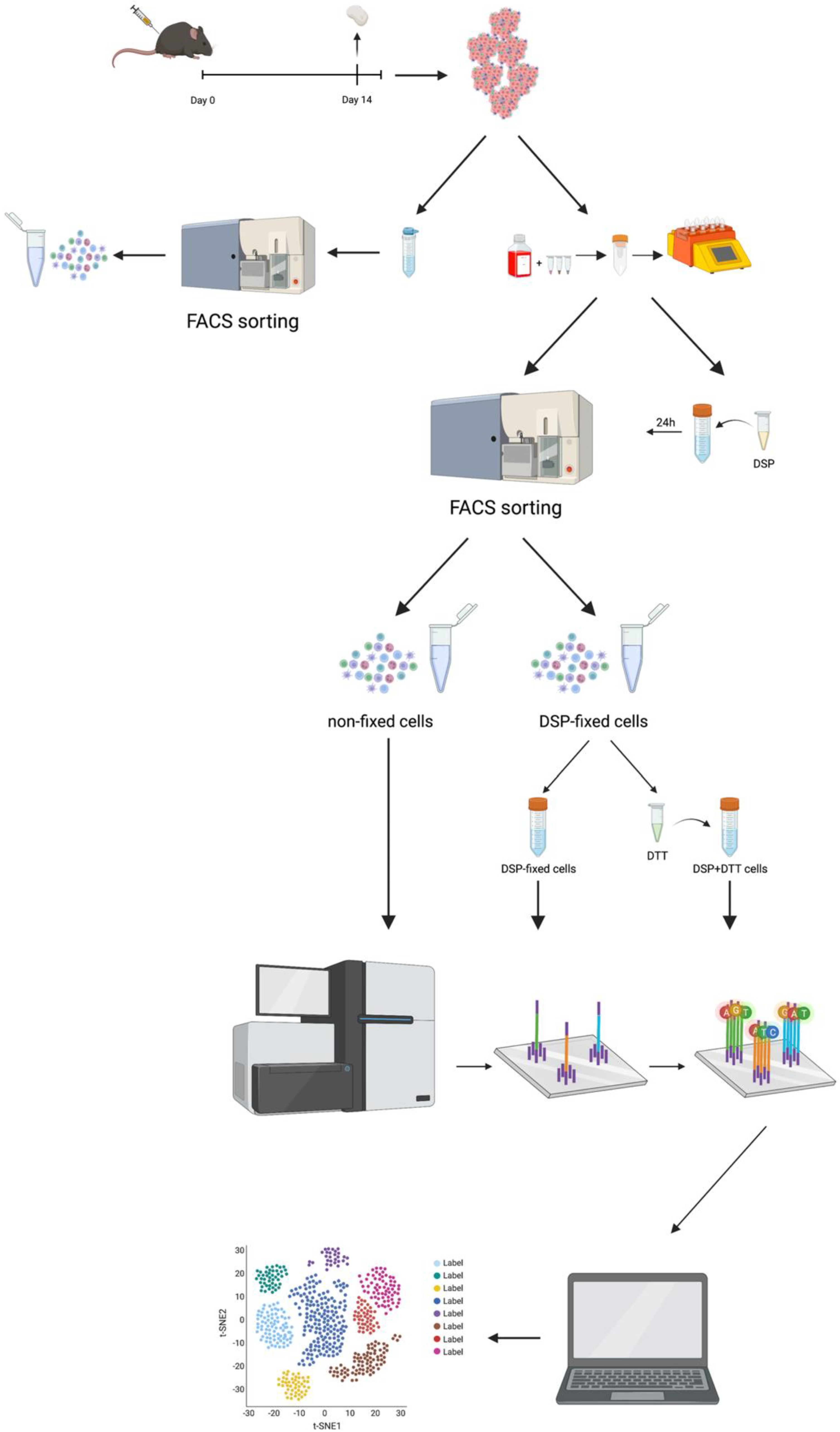
Figure 1 presents a graphical representation of the workflow.
Tumors were inoculated in mice and harvested on day 14 post-injection. Tumors were either dissociated mechanically or mechanically/enzymatically digested (using gentleMACSTM Dissociator, Miltenyi Biotec) prior to antibody labeling. One sample was DSP fixed and stored for at least 24 h prior to FACS, one sample was directly subjected to FACS for live CD45+ immune cells. Half of the DSP fixed samples were de-crosslinked (using DTT) prior to scRNA-seq. All three samples were sequenced on Illumina NovaSeq 6000 sequencer, and data were analyzed using the Seurat package in R.
2.1. Tumor Dissociation Protocol Optimization
As previously discussed, one of the constraints of studying primary dissociated pancreatic cancer tissue is the limited number of live cells, which we observed to be correlated with the number of live immune cells. Since our goal was to process clinical tissue samples obtained from surgeries for scRNA-seq, it was crucial to first optimize the dissociation and cell recovery steps using a more plentiful tissue source, such as murine Pan02 tumors. Tumors were isolated from the flank of mice on day 14 and divided in half for parallel processing to avoid intertumoral variability and to ensure a relatively similar distribution of cell types in each dissociation method. One half of each tumor was subjected to mechanical dissociation and strained through a filter to obtain a homogeneous cell suspension. The second half of each tumor was enzymatically dissociated using the gentleMACS
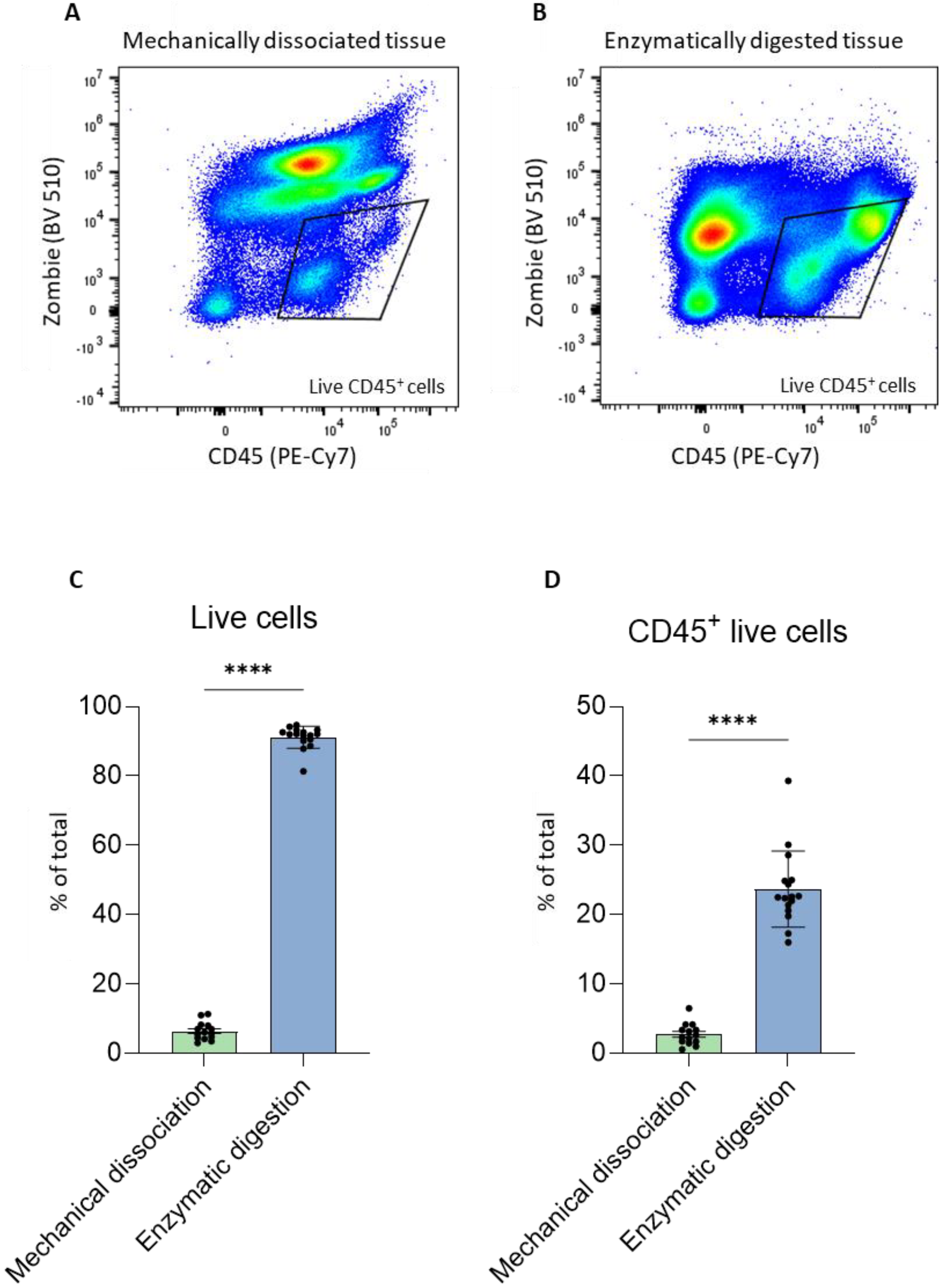
TM Dissociator according to the manufacturer’s protocol. Cells isolated from each dissociation protocol were subsequently incubated with the dye Zombie Aqua, which enables discrimination between dead and live cells, followed by a fluorochrome-labeled antibody specific for the immune cell marker CD45 to permit sorting of live immune cells from heterogeneous samples. An identical gating strategy to analyze the percentage of live CD45
+ cells was used for both dissociation protocols (
Figure 2A,B). In addition, several tumor cell suspensions from mechanical and mechanical/enzymatic digestion with gentleMACS
TM were acquired using FACS and the results were analyzed using FlowJo. Mechanical dissociation resulted in only approximately 10% live cells, whereas mechanical/enzymatic digestion with gentleMACS
TM resulted in 90% of the recovered cells being alive (
Figure 2C). Moreover, the higher percentage of live cells correlated with an increased percentage of live immune cells (
Figure 2D). Therefore, for the remaining experiments, we chose the enzymatic/mechanical method using gentleMACS
TM to dissociate tumors, as it resulted in a significantly higher percentage of live cells.
2.2. Sorting of DSP-Fixed Cells by FACS
After establishing a dissociation method that vastly improved the recovery of live cells, we next investigated whether DSP crosslinking affected cell viability, FACS staining, or sorting of live immune cells.
Tumor cell suspensions obtained after enzymatic digestion were labeled with Zombie dye and anti-CD45 antibody to distinguish live (zombie-negative) immune cells (CD45
+) from dead ones. The cell suspensions were divided into two identical samples. The fresh, unfixed cells were immediately subjected to FACS. The other sample was treated with DSP, as previously described [
7], stored at 4 °C for at least 24 h, and acquired by FACS the following day (a detailed fixation protocol is provided in the Materials and Methods section).
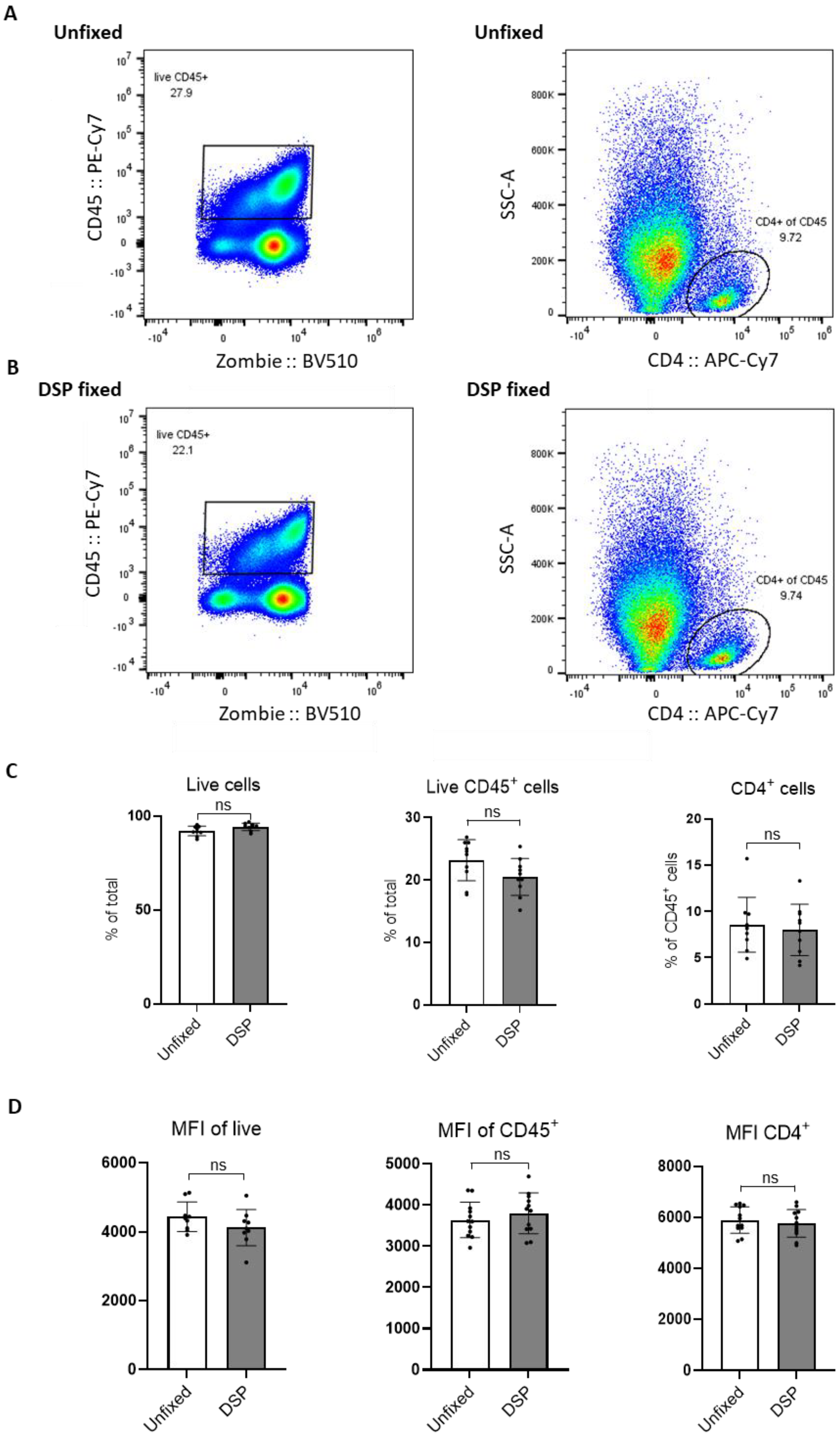
FACS analysis of multiple Pan02 tumors gated on live (Zombie
−), CD45
+, and CD4
+ cells in fresh (unfixed,
Figure 3A) and DSP-fixed (
Figure 3B) samples (for at least 24 h) showed no difference in the percentage of positive cells (
Figure 3C). In addition, there was no difference in the MFI of these markers with and without DSP fixation (
Figure 3D). Therefore, to proceed with scRNA-seq of fresh and DSP-fixed samples, live immune cells from these two sample groups were sorted using identical settings and gating strategies to avoid bias. Cells from multiple tumors (2–3) were pooled to obtain a sufficient number for scRNA-seq. Cell sorting reports revealed no obvious differences in cellular composition nor staining intensities between unfixed and DSP-fixed cells (
Supplemental Figure S1). These data show that DSP fixation did not alter cell morphology (size and granularity), antibody binding, or fluorochrome intensity. Thus, no deleterious effect of DSP crosslinking on FACS was observed after 24 h of fixation. Instead, we found that DSP fixation effectively preserved cell integrity and FACS staining to obtain pure, high-quality samples for subsequent scRNA-sequencing.
2.3. QC of Samples before Sequencing
After sorting for live immune cells, all samples were processed for scRNA-seq. Therefore, cells were resuspended according to the 10× Genomics protocol to achieve the maximum targeted cell recovery of 10,000 cells and used at a cell concentration of 1200 cells/µL. An optimal range of cell concentration before sequencing ensures that there are enough cells for sequencing yet prevents cell aggregation (multiplets) which occurs at an undesirably high concentration. Therefore, appropriate resuspension is important for ensuring high-quality sequencing data.
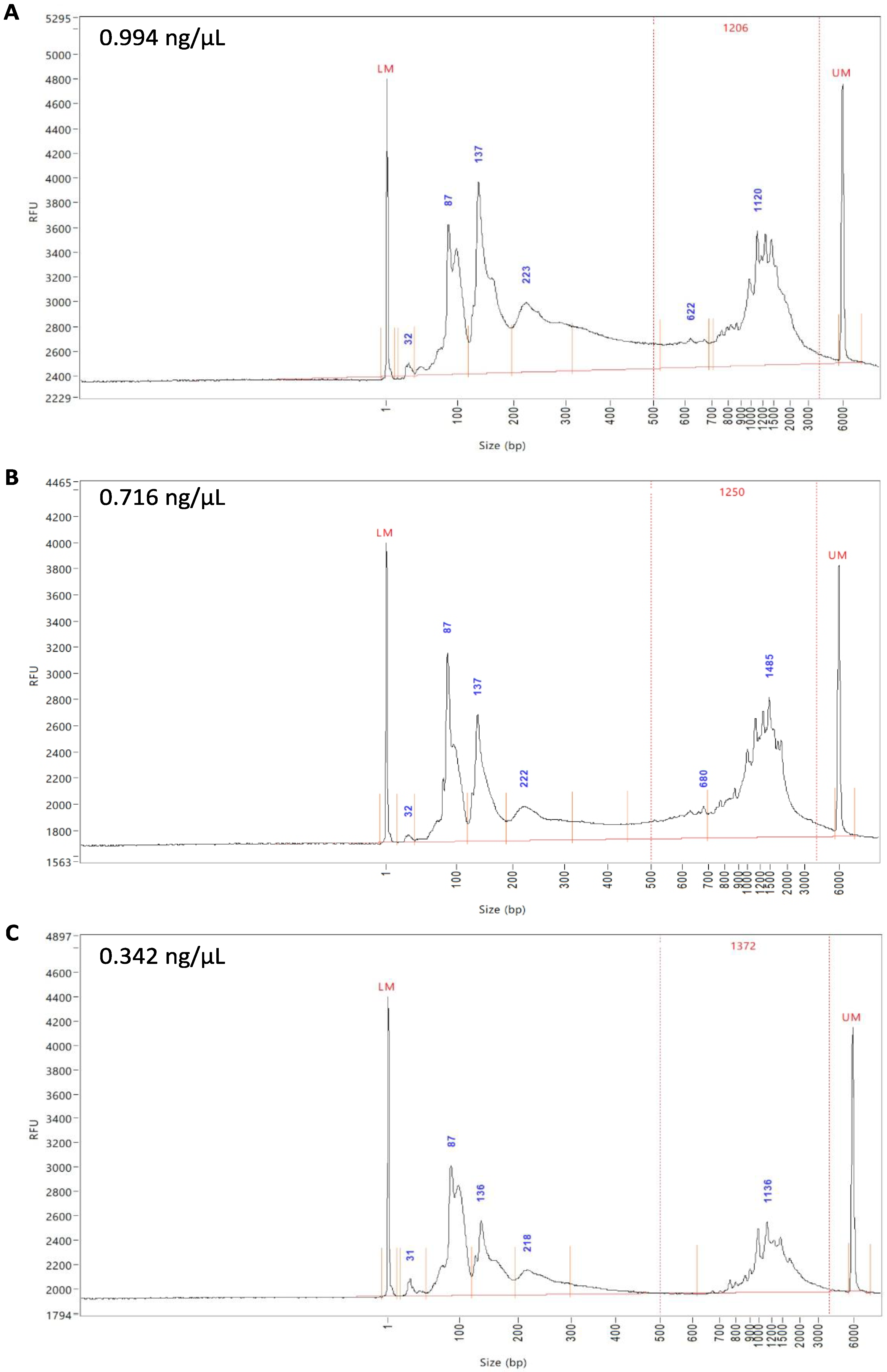
To assess whether DTT de-crosslinking affects the subsequent scRNA-seq of samples, half of the DSP-fixed, FACS-sorted samples were de-crosslinked with DTT. All three samples (fresh unfixed, DSP-fixed, and DSP-fixed/DTT de-crosslinked) were subjected to QC after cDNA generation on a Fragment Analyzer
TM (
Figure 4A–C). As the name suggests, fragment analysis represents the fragments of fluorescently labeled cDNA from the samples separated by automated capillary electrophoresis and then accurately sized by comparison with an internal standard. The expression peaks represent specific cDNA fragments based on their size and expression levels. To demonstrate an intact cDNA library, peaks are to be observed between 400 and 1000 base pairs (bp), which was the case for all three samples (
Figure 4A–C). The peaks at ~130 bp were indicative of the presence of empty adaptors, as well as low levels of primer carryover with peaks < 100 bp; therefore, a standard SPRIselect clean-up was performed to remove these primers during library preparation, which improves the signal and allows a better sequencing quality. cDNA quality of the unfixed and DSP-fixed samples was approximately 1 ng/µL (
Figure 4A,B) and that of the DSP-fixed/DTT de-crosslinked sample was around 0.34 ng/µL (
Figure 4C). These values are all in the optimal range for library preparation and sequencing.
2.4. QC of scRNA-Seq Data
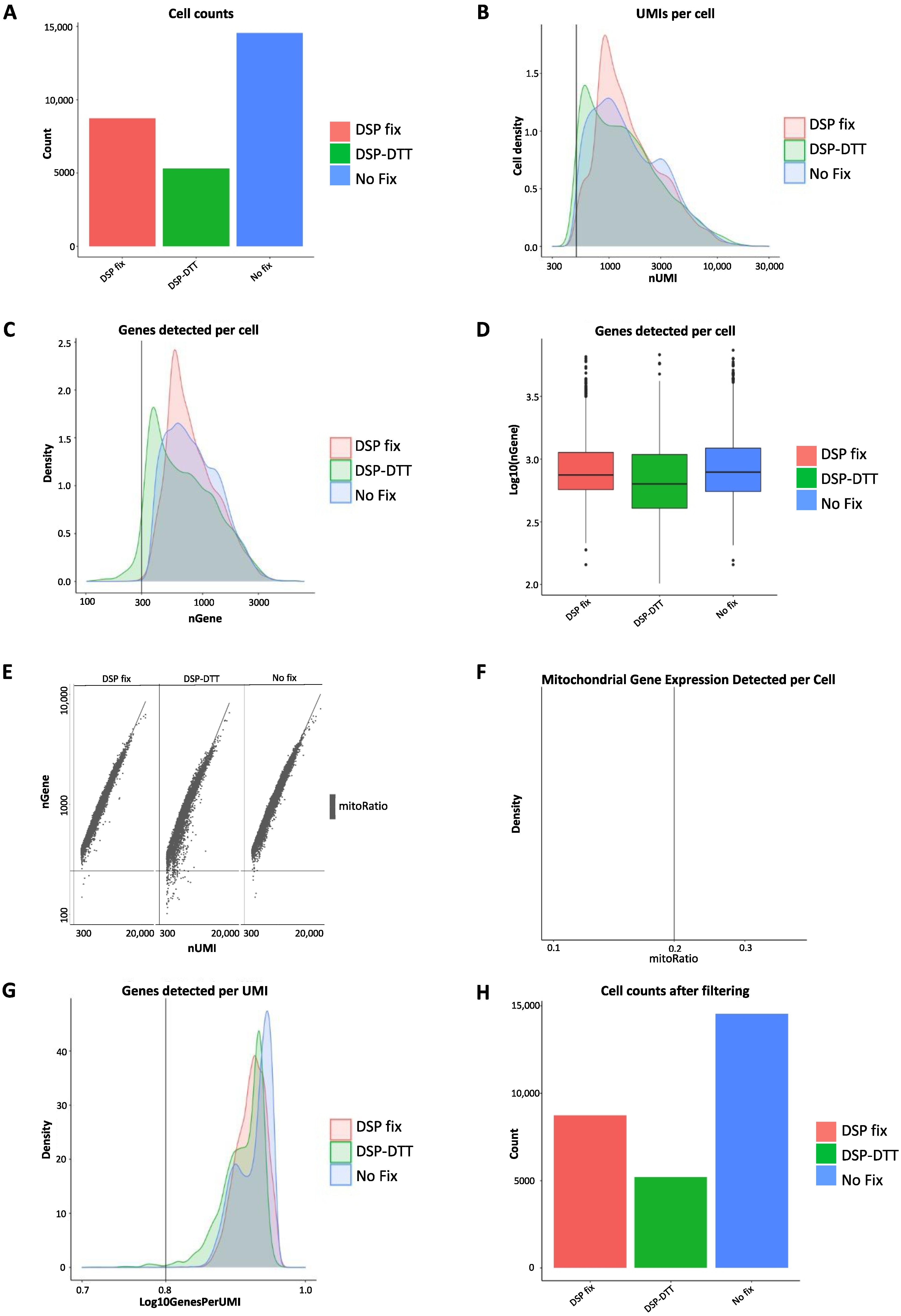
After confirming that each sample was sufficiently pure and with optimal fragment sizes and peaks, cellular RNA was sequenced using the 10× Genomics scRNA-seq platform on an Illumina sequencer, and the data were subjected to QC (
Figure 5). Data were extracted from Cell Ranger (10× Genomics Cell Ranger 3.1.0) and subsequent QC was performed in R using the Seurat package [
13]. For the analysis on the pooled datasets, the two experiments were merged using the Seurat package (
Supplemental Figure S2). Several metrics were assessed, such as cell counts, the number of unique molecular identifier (UMI) per cell, genes detected per cell, UMIs vs. genes detected, and mitochondrial count ratio based on the Seurat’s recommended default settings. The tests were performed to identify and remove cells of poor quality or insufficient complexity and to obtain only high-quality and biologically relevant cells for further analysis. Data analysis was performed as follows [
14]:
First, we determined the cell counts based on the number of unique cellular barcodes recovered. We were able to capture approximately 15,000 cells from the fresh unfixed sample, 10,000 cells from the DSP-fixed sample, and 5000 cells from the DSP-fixed/DTT de-crosslinked sample (
Figure 5A). We observed some cell loss, but further controls indicated that RNA quality remained unaffected.
Second, the number of UMIs per cell was compared. UMIs represent “tags” that are added to DNA fragments upon library preparation and used to identify the input DNA fragment. UMIs are important because they provide error corrections and, therefore, increase the accuracy of sequencing. The lowest threshold was set to 500 UMIs per cell, based on the default recommendations by Seurat, and is indicated by the vertical line at 500 UMIs in the graph (
Figure 5B). This threshold indicates the minimum requirement for each sample to be considered for further analysis. The peaks of all three samples exceeded the threshold, and those of the unfixed and DSP-fixed samples largely overlapped, whereas that of the DSP-fixed/DTT de-crosslinked sample was slightly lower. We overlaid the number of genes detected per cell in a histogram (
Figure 5C) and visualized them in a boxplot (
Figure 5D). The minimal count level was set at 300, and again, we observed an overlap between the unfixed and DSP-fixed samples, and a slightly lower peak in the DSP-fixed/DTT de-crosslinked sample. However, all three samples showed one high peak above 300, which is expected with high-quality data.
To further assess the quality of the samples, we evaluated the proportion of UMIs among the detected genes. The minimum requirements were set at 300 UMIs and 500 genes (
Figure 5E). The cells that did not meet the threshold are displayed in the bottom left quadrant for each sample, indicating poor quality. High-quality cells should contain a high number of UMIs and genes. Almost all the cells met the threshold, and the few cells that did not were excluded from further analysis (
Figure 5E). Next, we examined the ratio of mitochondrial gene counts present in any of the cells to assess possible contamination with dying or dead cells that may have passed through cell sorting or died afterwards. The threshold for mitochondrial ratio was set to a maximum possible score of 0.2 (
Figure 5F). However, we did not observe any mitochondrial genes in the samples, indicating that cell death or contamination with dead cells was unlikely. The final part of sample QC involves assessing the complexity at the gene level by relating the detected genes to the UMIs. This indicates whether genes are present in many UMIs, which is a requirement for highly complex cells. The minimal threshold value was set at 0.8 to ensure that there would be no contamination with low-complexity cell types (
Figure 5G). All three samples passed QC cutoffs, with genes per UMI scores of 0.85 and an overall peak above 0.9, indicating that all samples contained similarly complex populations of RNA, without major outliers. Finally, we filtered the cell counts at the cellular and genetic levels (
Figure 5H). We filtered out cells that had <500 UMIs per cell, <300 genes per cell, cells that presented low genes per UMI, and all cells that had a mitochondrial ratio > 0.2. We also removed genes that were expressed in fewer than 10 cells based on our data as they lowered the average complexity of the other cells in which they were not expressed.
Overall, QC demonstrated that the data were of good quality as they met the minimum requirements for passing all QC tests performed. Since all three samples exceeded all minimum QC standards, all samples were considered to have sufficient sequencing quality. Thus, neither DSP fixation nor DSP-DTT de-crosslinking seemed to drastically alter RNA quality, further suggesting that these techniques may be valuable as methods for fixing fresh cells from tissue samples for subsequent cell sorting and scRNA-seq. However, a loss of cells was observed, particularly in the DSP-fixed/DTT de-crosslinked sample, which we address in further detail in the Discussion section. As DTT de-crosslinking did not improve RNA quality and decreased cell number and RNA, it is not reasonable to include this extra step in the protocol, which requires additional time and effort.
2.5. Analysis of scRNA-Seq Data
QC of the scRNA-seq data showed that all samples were suitable for further downstream analysis. Next, we investigated whether the fixation protocol caused qualitative, i.e., differences in biological analysis between samples. A preliminary analysis was performed using the Seurat package in R [
13]. We aimed to discriminate any variance that might have arisen in the dispersion or clustering of cells, which were represented by uniform manifold approximation and projections (UMAP). We compared both the DSP-crosslinked cells and the DSP-DTT treated cells to unfixed cells and the DSP-fixed sample to the DSP-fixed/DTT de-crosslinked sample (
Figure 6A–C).
First, in the analysis of cell dispersion, we found a considerable similarity in the superposition obtained under all conditions investigated in both independent experiments (
Figure 6 and
Supplemental Figure S3). The greatest degree of overlap was exhibited by the DSP-fixed cells and the non-fixed cells (
Figure 6A and
Supplemental Figure S3A). Nonetheless, the overlay in the other two groups was also high (
Figure 6B,C and
Supplemental Figure S3B,C). The observed divergence in overlap may be due to differences in cell numbers in the DSP-DTT-treated group, as the DSP-fixed and non-fixed groups showed the highest cell numbers. Clustering analysis of the cells also showed similarity in all three groups, with very similar proportions of cell types and few differences (
Figure 6A–C and
Supplemental Figure S3A–C). Cell dispersion and clustering analysis of the merged datasets also showed a significant similarity in the superposition and the clustering (
Supplemental Figure S4A,B). In addition, proportion analysis, which indicates the percentage of cells in each sample (merged data) assigned to each cluster, showed no significant differences between the unfixed and DSP-fixed samples (
Supplemental Figure S4C,D). The only significant changes were observed between the DSP fixed and the DSP-fixed/DTT de-crosslinked samples for the Mast cell and DC cluster (
Supplemental Figure S4C,D). Together, these results indicate that DSP fixation does not significantly alter cell viability, clustering, or dispersion of cells. DSP crosslinking was visibly more similar to unfixed cells than to DSP-fixed/DTT de-crosslinked cells in terms of cell clustering. We also performed pseudo-bulk and cluster-specific differential gene expression analysis to assess if either DSP fixation or DSP-DTT treatment affected sequencing results of the samples. Comparison between the three groups revealed a minimal number of differentially expressed genes (DEGs) in pseudo-bulk (
Supplemental Figure S5A) and for all cell clusters (
Supplemental Figure S5B and Table S1).
In summary, we concluded that DSP fixation does not affect cell sorting, RNA quality, and sequencing results and DTT de-crosslinking does not provide additional benefits.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}