
The Arrival of Gene Therapy for Patients with Hemophilia A




Abstract
:1. Introduction
2. Currently Available Therapies and Their Limitations
2.1. Exogenous Coagulation Factors
Factor VIII
2.2. Emicizumab
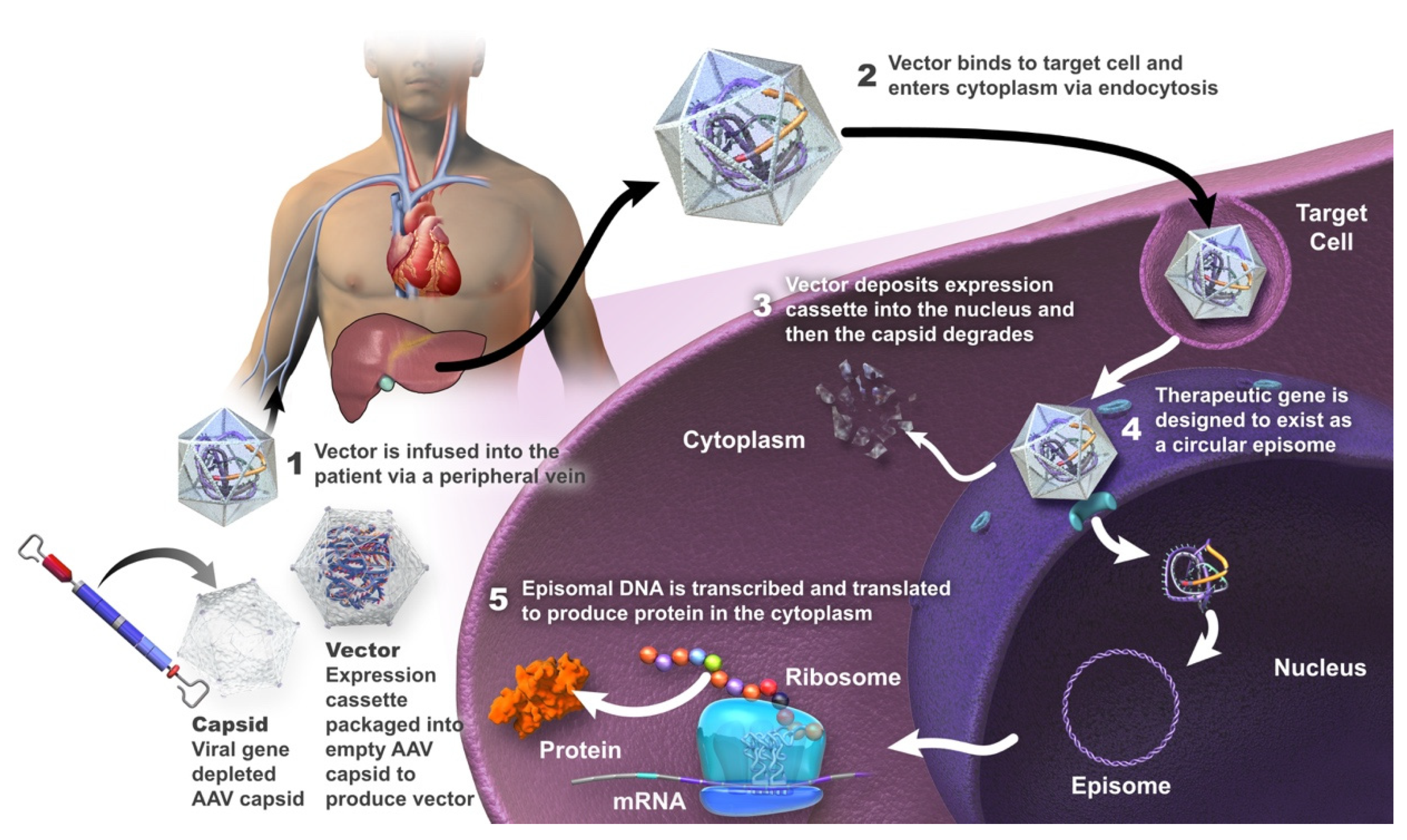
3. Gene Therapy in Hemophilia A
Adeno-Associated Viral Vectors
4. Hemophilia A Clinical Trials Involving Gene Therapy
4.1. Valoctocogene Roxaparvovec
4.2. Dirloctocogene Samoparvovec
4.3. SPK-8016
4.4. Giroctocogene Fitelparvovec
4.5. TAK-754
4.6. BAY 2599023
4.7. AAV8-HLP-hFVIII-V3
5. Opportunities Associated with Gene Therapy in Hemophilia A
6. Challenges of Gene Therapy in Hemophilia A
6.1. FVIII Structure
6.2. FVIII Expression
6.3. Infusion-Related Adverse Reactions
6.4. Anti-AAV Neutralizing Antibodies
6.5. Elevation of Liver Enzymes
6.6. Oncogenesis
6.7. Durability and Variability of Transgene Expression
6.8. Patient Expectations
7. Assessment of Efficacy of FVIII Expression
8. Implementing Gene Therapy in a Real-World Setting
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arruda, V.R.; Weber, J.; Samelson-Jones, B.J. Gene Therapy for Inherited Bleeding Disorders. Semin. Thromb. Hemost. 2021, 47, 161–173. [Google Scholar] [CrossRef]
- Berntorp, E.; Fischer, K.; Hart, D.P.; Mancuso, M.E.; Stephensen, D.; Shapiro, A.D.; Blanchette, V. Haemophilia. Nat. Rev. Dis. Prim. 2021, 7, 45. [Google Scholar] [CrossRef]
- Castaman, G.; Matino, D. Hemophilia A and B: Molecular and clinical similarities and differences. Haematologica 2019, 104, 1702–1709. [Google Scholar] [CrossRef]
- Perrin, G.Q.; Herzog, R.W.; Markusic, D.M. Update on clinical gene therapy for hemophilia. Blood 2019, 133, 407–414. [Google Scholar] [CrossRef]
- Peyvandi, F.; Garagiola, I.; Young, G. The past and future of haemophilia: Diagnosis, treatments, and its complications. Lancet 2016, 388, 187–197. [Google Scholar] [CrossRef]
- Pipe, S.W.; Gonen-Yaacovi, G.; Segurado, O.G. Hemophilia A gene therapy: Current and next-generation approaches. Expert Opin. Biol. Ther. 2022, 1–17. [Google Scholar] [CrossRef]
- Everett, L.A.; Cleuren, A.; Khoriaty, R.N.; Ginsburg, D. Murine coagulation factor VIII is synthesized in endothelial cells. Blood 2014, 123, 3697–3705. [Google Scholar] [CrossRef]
- Fahs, S.A.; Hille, M.T.; Shi, Q.; Weiler, H.; Montgomery, R.R. A conditional knockout mouse model reveals endothelial cells as the principal and possibly exclusive source of plasma factor VIII. Blood 2014, 123, 3706–3713. [Google Scholar] [CrossRef]
- Shahani, T.; Covens, K.; Lavend’Homme, R.; Jazouli, N.; Sokal, E.; Peerlinck, K.; Jacquemin, M. Human liver sinusoidal endothelial cells but not hepatocytes contain factor VIII. J. Thromb. Haemost. 2014, 12, 36–42. [Google Scholar] [CrossRef]
- Blanchette, V.S.; Key, N.S.; Ljung, L.R.; Manco-Johnson, M.J.; van den Berg, H.M.; Srivastava, A. Definitions in hemophilia: Communication from the SSC of the ISTH. J. Thromb. Haemost. 2014, 12, 1935–1939. [Google Scholar] [CrossRef]
- Collins, P.W.; Blanchette, V.S.; Fischer, K.; Björkman, S.; Oh, M.; Fritsch, S.; Schroth, P.; Spotts, G.; Astermark, J.; Ewenstein, B.; et al. Break-through bleeding in relation to predicted factor VIII levels in patients receiving prophylactic treatment for severe hemophilia A. J. Thromb. Haemost. 2009, 7, 413–420. [Google Scholar] [CrossRef]
- Uijl, I.E.M.D.; Fischer, K.; Van Der Bom, J.G.; Grobbee, D.E.; Rosendaal, F.R.; Plug, I. Analysis of low frequency bleeding data: The association of joint bleeds according to baseline FVIII activity levels. Haemophilia 2011, 17, 41–44. [Google Scholar] [CrossRef]
- Castaman, G.; Linari, S. Prophylactic versus on-demand treatments for hemophilia: Advantages and drawbacks. Expert Rev. Hematol. 2018, 11, 567–576. [Google Scholar] [CrossRef]
- Mahlangu, J.; Oldenburg, J.; Paz-Priel, I.; Negrier, C.; Niggli, M.; Mancuso, M.E.; Schmitt, C.; Jiménez-Yuste, V.; Kempton, C.; Dhalluin, C.; et al. Emicizumab Prophylaxis in Patients Who Have Hemophilia A without Inhibitors. N. Engl. J. Med. 2018, 379, 811–822. [Google Scholar] [CrossRef]
- Mannucci, P.M.; Cortesi, P.A.; Di Minno, M.N.D.; Sanò, M.; Mantovani, L.G.; Di Minno, G. Comparative analysis of the pivotal studies of extended half-life recombinant FVIII products for treatment of haemophilia A. Haemophilia 2021, 27, e422–e433. [Google Scholar] [CrossRef]
- Ar, M.C.; Balkan, C.; Kavaklı, K. Extended Half-Life Coagulation Factors: A New Era in the Management of Haemophilia Patients. Turk. J. Hematol. 2019, 36, 141–154. [Google Scholar] [CrossRef]
- Darby, S.C.; Keeling, D.M.; Spooner, R.J.D.; Kan, S.W.; Giangrande, P.L.F.; Collins, P.W.; Hill, F.G.H.; Hay, C.R.M.; Organisation, U.H.C.D. The incidence of factor VIII and factor IX inhibitors in the hemophilia population of the UK and their effect on subsequent mortality, 1977–1999. J. Thromb. Haemost. 2004, 2, 1047–1054. [Google Scholar] [CrossRef]
- Witmer, C.; Young, G. Factor VIII inhibitors in hemophilia A: Rationale and latest evidence. Ther. Adv. Hematol. 2013, 4, 59–72. [Google Scholar] [CrossRef]
- Rosendaal, F.R.; Palla, R.; Garagiola, I.; Mannucci, P.M.; Peyvandi, F. Genetic risk stratification to reduce inhibitor development in the early treatment of hemophilia A: A SIPPET analysis. Blood 2017, 130, 1757–1759. [Google Scholar] [CrossRef]
- Oldenburg, J. Optimal treatment strategies for hemophilia: Achievements and limitations of current prophylactic regimens. Blood 2015, 125, 2038–2044. [Google Scholar] [CrossRef]
- Olivieri, M.; Kurnik, K.; Pfluger, T.; Bidlingmaier, C. Identification and long-term observation of early joint damage by magnetic resonance imaging in clinically asymptomatic joints in patients with haemophilia A or B despite prophylaxis. Haemophilia 2012, 18, 369–374. [Google Scholar] [CrossRef]
- Blair, H.A. Emicizumab: A Review in Haemophilia, A. Drugs 2019, 79, 1697–1707. [Google Scholar] [CrossRef]
- Franchini, M.; Marano, G.; Pati, I.; Candura, F.; Profili, S.; Veropalumbo, E.; Masiello, F.; Catalano, L.; Piccinini, V.; Vaglio, S.; et al. Emicizumab for the treatment of haemophilia A: A narrative review. Blood Transfus. 2019, 17, 223–228. [Google Scholar] [CrossRef]
- Kitazawa, T.; Igawa, T.; Sampei, Z.; Muto, A.; Kojima, T.; Soeda, T.; Yoshihashi, K.; Okuyama-Nishida, Y.; Saito, H.; Tsunoda, H.; et al. A bispecific antibody to factors IXa and X restores factor VIII hemostatic activity in a hemophilia A model. Nat. Med. 2012, 18, 1570–1574. [Google Scholar] [CrossRef]
- Sampei, Z.; Igawa, T.; Soeda, T.; Okuyama-Nishida, Y.; Moriyama, C.; Wakabayashi, T.; Tanaka, E.; Muto, A.; Kojima, T.; Kitazawa, T.; et al. Identification and Multidimensional Optimization of an Asymmetric Bispecific IgG Antibody Mimicking the Function of Factor VIII Cofactor Activity. PLoS ONE 2013, 8, e57479. [Google Scholar] [CrossRef]
- Oldenburg, J.; Mahlangu, J.N.; Kim, B.; Schmitt, C.; Callaghan, M.U.; Young, G.; Santagostino, E.; Kruse-Jarres, R.; Negrier, C.; Kessler, C.; et al. Emicizumab Prophylaxis in Hemophilia A with Inhibitors. N. Engl. J. Med. 2017, 377, 809–818. [Google Scholar] [CrossRef]
- Pipe, S.W.; Shima, M.; Lehle, M.; Shapiro, A.; Chebon, S.; Fukutake, K.; Key, N.S.; Portron, A.; Schmitt, C.; Podolak-Dawidziak, M.; et al. Efficacy, safety, and pharmacokinetics of emicizumab prophylaxis given every 4 weeks in people with haemophilia A (HAVEN 4): A multicentre, open-label, non-randomised phase 3 study. Lancet Haematol. 2019, 6, e295–e305. [Google Scholar] [CrossRef]
- Anguela, X.M.; High, K.A. Entering the Modern Era of Gene Therapy. Annu. Rev. Med. 2019, 70, 273–288. [Google Scholar] [CrossRef]
- Kumar, S.R.; Markusic, D.M.; Biswas, M.; High, K.A.; Herzog, R.W. Clinical development of gene therapy: Results and lessons from recent successes. Mol. Ther. Methods Clin. Dev. 2016, 3, 16034. [Google Scholar] [CrossRef]
- Maestro, S.; Weber, N.D.; Zabaleta, N.; Aldabe, R.; Gonzalez-Aseguinolaza, G. Novel vectors and approaches for gene therapy in liver diseases. JHEP Rep. 2021, 3, 100300. [Google Scholar] [CrossRef]
- High, K.A.; Roncarolo, M.G. Gene Therapy. N. Engl. J. Med. 2019, 381, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Lisowski, L.; Staber, J.M.; Wright, J.F.; Valentino, L.A. The intersection of vector biology, gene therapy, and hemophilia. Res. Pract. Thromb. Haemost. 2021, 5, e12586. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, E.; Morfini, M.; Valentino, L. Recent Advances in the Treatment of Hemophilia: A Review. Biol. Targets Ther. 2021, 15, 221–235. [Google Scholar] [CrossRef] [PubMed]
- Miesbach, W.; Pasi, K.J.; Pipe, S.W.; Hermans, C.; O’Mahony, B.; Guelcher, C.; Steiner, B.; Skinner, M.W. Evolution of haemophilia integrated care in the era of gene therapy: Treatment centre’s readiness in United States and EU. Haemophilia 2021, 27, 511–514. [Google Scholar] [CrossRef]
- Rodríguez-Merchán, E.; De Pablo-Moreno, J.; Liras, A. Gene Therapy in Hemophilia: Recent Advances. Int. J. Mol. Sci. 2021, 22, 7647. [Google Scholar] [CrossRef]
- Rodriguez-Santana, I.; DasMahapatra, P.; Burke, T.; Hakimi, Z.; Bartelt-Hofer, J.; Nazir, J.; O’Hara, J. Differential humanistic and economic burden of mild, moderate and severe haemophilia in european adults: A regression analysis of the CHESS II study. Orphanet J. Rare Dis. 2022, 17, 148. [Google Scholar] [CrossRef]
- Zhou, Z.-Y.; Koerper, M.A.; Johnson, K.A.; Riske, B.; Baker, J.R.; Ullman, M.; Curtis, R.G.; Poon, J.-L.; Lou, M.; Nichol, M. Burden of illness: Direct and indirect costs among persons with hemophilia A in the United States. J. Med. Econ. 2015, 18, 457–465. [Google Scholar] [CrossRef]
- Uijl, I.E.M.D.; Bunschoten, E.P.M.; Roosendaal, G.; Schutgens, R.E.G.; Biesma, D.H.; Grobbee, D.E.; Fischer, K. Clinical severity of haemophilia A: Does the classification of the 1950s still stand? Haemophilia 2011, 17, 849–853. [Google Scholar] [CrossRef]
- Doshi, B.S.; Arruda, V.R. Gene therapy for hemophilia: What does the future hold? Ther. Adv. Hematol. 2018, 9, 273–293. [Google Scholar] [CrossRef]
- Mancuso, M.E.; Mahlangu, J.N.; Pipe, S.W. The changing treatment landscape in haemophilia: From standard half-life clotting factor concentrates to gene editing. Lancet 2021, 397, 630–640. [Google Scholar] [CrossRef]
- Mannucci, P.M.; Tuddenham, E.G. The Hemophilias—From Royal Genes to Gene Therapy. N. Engl. J. Med. 2001, 344, 1773–1779. [Google Scholar] [CrossRef] [PubMed]
- Peyvandi, F.; Garagiola, I. Clinical advances in gene therapy updates on clinical trials of gene therapy in haemophilia. Haemophilia 2019, 25, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Nathwani, A.C.; Tuddenham, E.G.D. Haemophilia, the journey in search of a cure. 1960–2020. Br. J. Haematol. 2020, 191, 573–578. [Google Scholar]
- Olgasi, C.; Borsotti, C.; Merlin, S.; Bergmann, T.; Bittorf, P.; Adewoye, A.B.; Wragg, N.; Patterson, K.; Calabria, A.; Benedicenti, F.; et al. Efficient and safe correction of hemophilia A by lentiviral vector-transduced BOECs in an implantable device. Mol. Ther. Methods Clin. Dev. 2021, 23, 551–566. [Google Scholar] [CrossRef] [PubMed]
- Hacein-Bey-Abina, S.; Garrigue, A.; Wang, G.P.; Soulier, J.; Lim, A.; Morillon, E.; Clappier, E.; Caccavelli, L.; Delabesse, E.; Beldjord, K.; et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Investig. 2008, 118, 3132–3142. [Google Scholar] [CrossRef] [PubMed]
- Hernández, R.J.; Calabria, A.; Sanvito, F.; De Mattia, F.; Farinelli, G.; Scala, S.; Visigalli, I.; Carriglio, N.; De Simone, M.; Vezzoli, M.; et al. Hematopoietic Tumors in a Mouse Model of X-linked Chronic Granulomatous Disease after Lentiviral Vector-Mediated Gene Therapy. Mol. Ther. 2021, 29, 86–102. [Google Scholar] [CrossRef]
- Schlimgen, R.; Howard, J.; Wooley, D.; Thompson, M.; Baden, L.R.; Yang, O.O.; Christiani, D.C.; Mostoslavsky, G.; Diamond, D.V.; Duane, E.G.; et al. Risks Associated with Lentiviral Vector Exposures and Prevention Strategies. J. Occup. Environ. Med. 2016, 58, 1159–1166. [Google Scholar] [CrossRef]
- Colella, P.; Ronzitti, G.; Mingozzi, F. Emerging Issues in AAV-Mediated In Vivo Gene Therapy. Mol. Ther. Methods Clin. Dev. 2018, 8, 87–104. [Google Scholar] [CrossRef]
- Mendell, J.R.; Al-Zaidy, S.A.; Rodino-Klapac, L.R.; Goodspeed, K.; Gray, S.J.; Kay, C.N.; Boye, S.L.; Boye, S.E.; George, L.A.; Salabarria, S.; et al. Current Clinical Applications of In Vivo Gene Therapy with AAVs. Mol. Ther. 2021, 29, 464–488. [Google Scholar] [CrossRef]
- Dunbar, C.E.; High, K.A.; Joung, J.K.; Kohn, D.B.; Ozawa, K.; Sadelain, M. Gene therapy comes of age. Science 2018, 359, eaan4672. [Google Scholar] [CrossRef]
- Goverdhana, S.; Puntel, M.; Xiong, W.; Zirger, J.; Barcia, C.; Curtin, J.; Soffer, E.; Mondkar, S.; King, G.; Hu, J.; et al. Regulatable gene expression systems for gene therapy applications: Progress and future challenges. Mol. Ther. 2005, 12, 189–211. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Samulski, R.J. Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet. 2020, 21, 255–272. [Google Scholar] [CrossRef]
- Li, H.; Malani, N.; Hamilton, S.R.; Schlachterman, A.; Bussadori, G.; Edmonson, S.E.; Shah, R.; Arruda, V.R.; Mingozzi, F.; Wright, J.F.; et al. Assessing the potential for AAV vector genotoxicity in a murine model. Blood 2011, 117, 3311–3319. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, G.N.; Everett, J.K.; Kafle, S.; Roche, A.M.; Raymond, H.E.; Leiby, J.; Wood, C.; Assenmacher, C.-A.; Merricks, E.P.; Long, C.T.; et al. A long-term study of AAV gene therapy in dogs with hemophilia A identifies clonal expansions of transduced liver cells. Nat. Biotechnol. 2021, 39, 47–55. [Google Scholar] [CrossRef]
- Rosas, L.E.; Grieves, J.L.; Zaraspe, K.; La Perle, K.; Fu, H.; McCarty, D.M. Patterns of scAAV Vector Insertion Associated with Oncogenic Events in a Mouse Model for Genotoxicity. Mol. Ther. 2012, 20, 2098–2110. [Google Scholar] [CrossRef] [PubMed]
- Chapin, J.; Allen, G.; Alvarez-Roman, M.; Ayash-Rashkovsky, M.; Jaime, F.; Maggiore, C.; Mingot-Castellano, M.; Rajavel, K.; Rauch, A.; Susen, S. Results from a phase 1/2 safety and dose escalation study of TAK-754, an AAV8 vector with a codon-optimized B-domain-deleted factor VIII transgene in severe hemophilia A. Haemophilia 2021, 27, 122. [Google Scholar]
- European Haemophilia Consortium; National Hemophilia Foundation; World Federation of Hemophilia. FDA Places the Pfizer/Sangamo Therapeutics Phase 3 AFFINE Haemophilia a Gene Therapy Study on Clinical Hold. Available online: https://www.hemophilia.org/sites/default/files/document/files/Pfizer-GT-pause-clean.pdf (accessed on 22 November 2021).
- George, L.A.; Monahan, P.E.; Eyster, M.E.; Sullivan, S.K.; Ragni, M.V.; Croteau, S.E.; Rasko, J.E.; Recht, M.; Samelson-Jones, B.J.; MacDougall, A.; et al. Multiyear Factor VIII Expression after AAV Gene Transfer for Hemophilia A. N. Engl. J. Med. 2021, 385, 1961–1973. [Google Scholar] [CrossRef]
- High, K.A.; George, L.A.; Eyster, M.E.; Sullivan, S.K.; Ragni, M.V.; Croteau, S.E.; Samelson-Jones, B.J.; Evans, M.; Joseney-Antoine, M.; Macdougall, A. A phase 1/2 trial of investigational SPK-8011 in hemophilia a demonstrates durable expression and prevention of bleeds. Blood 2018, 132, 487. [Google Scholar] [CrossRef]
- Leavitt, A.D.; Konkle, B.A.; Stine, K.; Visweshwar, N.; Harrington, T.J.; Giermasz, A.; Arkin, S.; Fang, A.; Plonski, F.; Smith, L. Updated follow-up of the Alta Study, a phase 1/2 study of giroctocogene fitelparvovec (SB-525) gene therapy in adults with severe hemophilia A. Blood 2020, 136, 12. [Google Scholar] [CrossRef]
- Nathwani, A.C.; Tuddenham, E.; Chowdary, P.; McIntosh, J.; Lee, D.; Rosales, C.; Phillips, M.; Pie, J.; Junfang, Z.; Meagher, M.M. GO-8: Preliminary results of a Phase I/II dose escalation trial of gene therapy for haemophilia A using a novel human factor VIII variant. Blood 2018, 132, 489. [Google Scholar] [CrossRef]
- Ozelo, M.; Mahlangu, J.; Pasi, K.; Giermasz, A.; Leavitt, A.D.; Laffan, M.; Symington, E.; Quon, D.V.; Wang, J.D.; Peerlinck, K.; et al. Efficacy and safety of valoctocogene roxaparvovec adeno-associated virus gene transfer for severe hemophilia A: Results from the phase 3 GENEr8-1 trial. Res. Pract. Thromb. Haemost. 2021, 5 (Suppl. 2), 89. [Google Scholar]
- Ozelo, M.C.; Mahlangu, J.; Pasi, K.J.; Giermasz, A.; Leavitt, A.D.; Laffan, M.; Symington, E.; Quon, D.V.; Wang, J.-D.; Peerlinck, K.; et al. Valoctocogene Roxaparvovec Gene Therapy for Hemophilia A. N. Engl. J. Med. 2022, 386, 1013–1025. [Google Scholar] [CrossRef]
- Pasi, K.J.; Laffan, M.; Rangarajan, S.; Robinson, T.M.; Mitchell, N.; Lester, W.; Symington, E.; Madan, B.; Yang, X.; Kim, B.; et al. Persistence of haemostatic response following gene therapy with valoctocogene roxaparvovec in severe haemophilia A. Haemophilia 2021, 27, 947–956. [Google Scholar] [CrossRef]
- Pasi, K.J.; Rangarajan, S.; Mitchell, N.; Lester, W.; Symington, E.; Madan, B.; Laffan, M.; Russell, C.B.; Li, M.; Pierce, G.F.; et al. Multiyear Follow-up of AAV5-hFVIII-SQ Gene Therapy for Hemophilia A. N. Engl. J. Med. 2020, 382, 29–40. [Google Scholar] [CrossRef]
- Pipe, S.; Becka, M.; Detering, E.; Vanevski, K.; Lissitchkov, T. First-in-human Gene Therapy Study of AAVhu37 Capsid Vector Technology in Severe Hemophilia A. Blood 2019, 134, 4630. [Google Scholar] [CrossRef]
- Pipe, S.; Hay, C.; Sheehan, J.; Lissitchkov, T.; Leebeek, F.; Coppens, M.; Detering, E.; Ribeiro, S.; Vanevski, K. First-in-human gene therapy study of AAVhu37 capsid vector technology in severe hemophilia A: Safety and FVIII activity results. Res. Pract. Thromb. Haemost. 2020, 4, 27–28. [Google Scholar]
- Pipe, S.W.; Sheehan, J.P.; Coppens, M.; Eichler, H.; Linardi, C.; Wiegmann, S.; Hay, C.R.; Lissitchkov, T. First-in-Human Dose-Finding Study of AAVhu37 Vector-Based Gene Therapy: BAY 2599023 Has Stable and Sustained Expression of FVIII over 2 Years. Blood 2021, 138, 3971. [Google Scholar] [CrossRef]
- Rangarajan, S.; Walsh, L.; Lester, W.; Perry, D.; Madan, B.; Laffan, M.; Yu, H.; Vettermann, C.; Pierce, G.F.; Wong, W.Y.; et al. AAV5–Factor VIII Gene Transfer in Severe Hemophilia A. N. Engl. J. Med. 2017, 377, 2519–2530. [Google Scholar] [CrossRef]
- Sullivan, S.; Barrett, J.; Drelich, D.; Tarantino, M.; MacDougall, A.; Joseney-Antoine, M.; Wachtel, K.; Jaworski, K.; Curran, M.; Kuranda, K. SPK-8016: Preliminary results from a phase 1/2 clinical trial of gene therapy for hemophilia A. Haemophilia 2021, 136, 129–130. [Google Scholar]
- Visweshwar, N.; Harrington, T.J.; Leavitt, A.D.; Konkle, B.A.; Giermasz, A.; Stine, K.; Rupon, J.; Di Russo, G.; Tseng, L.-J.; Resa, M.D.L.A.; et al. Updated Results of the Alta Study, a Phase 1/2 Study of Giroctocogene Fitelparvovec (PF-07055480/SB-525) Gene Therapy in Adults with Severe Hemophilia a. Blood 2021, 138, 564. [Google Scholar] [CrossRef]
- Nathwani, A.C. Gene therapy for hemophilia. Hematol. Am. Soc. Hematol. Educ. Program 2019, 2019, 1–8. [Google Scholar] [CrossRef]
- Zhang, F.; Yan, X.; Li, M.M.; Hua, B.; Xiao, X.; Monahan, P.E.; Sun, J. Exploring the Potential Feasibility of Intra-Articular Adeno-Associated Virus-Mediated Gene Therapy for Hemophilia Arthropathy. Hum. Gene Ther. 2020, 31, 448–458. [Google Scholar] [CrossRef]
- Nathwani, A.C.; Tuddenham, E.G.D.; Rangarajan, S.; Rosales, C.; McIntosh, J.; Linch, D.C.; Chowdary, P.; Riddell, A.; Pie, A.J.; Harrington, C.; et al. Adenovirus-Associated Virus Vector–Mediated Gene Transfer in Hemophilia B. N. Engl. J. Med. 2011, 365, 2357–2365. [Google Scholar] [CrossRef] [PubMed]
- Mingozzi, F.; High, K.A. Immune responses to AAV vectors: Overcoming barriers to successful gene therapy. Blood 2013, 122, 23–36. [Google Scholar] [CrossRef]
- Lenting, P.J.; van Mourik, J.A.; Mertens, K. The life cycle of coagulation factor VIII in view of its structure and function. Blood 1998, 92, 3983–3996. [Google Scholar] [CrossRef] [PubMed]
- Lind, P.; Larsson, K.; Spira, J.; Sydow-Backman, M.; Almstedt, A.; Gray, E.; Sandberg, H. Novel Forms of B-Domain-Deleted Recombinant Factor VIII Molecules. Construction and Biochemical Characterization. Eur. J. Biochem. 1995, 232, 19–27. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, J.; Lenting, P.J.; Rosales, C.; Lee, D.; Rabbanian, S.; Raj, D.; Patel, N.; Tuddenham, E.G.D.; Christophe, O.D.; McVey, J.H.; et al. Therapeutic levels of FVIII following a single peripheral vein administration of rAAV vector encoding a novel human factor VIII variant. Blood 2013, 121, 3335–3344. [Google Scholar] [CrossRef]
- Pittman, D.D.; Alderman, E.M.; Tomkinson, K.N.; Wang, J.H.; Giles, A.R.; Kaufman, R.J. Biochemical, immunological, and in vivo functional characterization of B-domain-deleted factor VIII. Blood 1993, 81, 2925–2935. [Google Scholar]
- Batty, P.; Lillicrap, D. Hemophilia Gene Therapy: Approaching the First Licensed Product. HemaSphere 2021, 5, e540. [Google Scholar] [CrossRef]
- Lisowski, L.; Dane, A.P.; Chu, K.; Zhang, Y.; Cunningham, S.C.; Wilson, E.M.; Nygaard, S.; Grompe, M.; Alexander, I.E.; Kay, M.A. Selection and evaluation of clinically relevant AAV variants in a xenograft liver model. Nature 2014, 506, 382–386. [Google Scholar] [CrossRef]
- Nathwani, A.C.; Gray, J.T.; Ng, C.Y.C.; Zhou, J.; Spence, Y.; Waddington, S.; Tuddenham, E.G.D.; Kemball-Cook, G.; McIntosh, J.; Boon-Spijker, M.; et al. Self-complementary adeno-associated virus vectors containing a novel liver-specific human factor IX expression cassette enable highly efficient transduction of murine and nonhuman primate liver. Blood 2006, 107, 2653–2661. [Google Scholar] [CrossRef] [PubMed]
- Leebeek, F.W.G.; Miesbach, W. Gene therapy for hemophilia: A review on clinical benefit, limitations, and remaining issues. Blood 2021, 138, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Chuah, M.K.; Petrus, I.; De Bleser, P.; Le Guiner, C.; Gernoux, G.; Adjali, O.; Nair, N.; Willems, J.; Evens, H.; Rincon, M.Y.; et al. Liver-Specific Transcriptional Modules Identified by Genome-Wide In Silico Analysis Enable Efficient Gene Therapy in Mice and Non-Human Primates. Mol. Ther. 2014, 22, 1605–1613. [Google Scholar] [CrossRef]
- Verdera, H.C.; Kuranda, K.; Mingozzi, F. AAV Vector Immunogenicity in Humans: A Long Journey to Successful Gene Transfer. Mol. Ther. 2020, 28, 723–746. [Google Scholar] [CrossRef]
- Weber, T. Anti-AAV Antibodies in AAV Gene Therapy: Current Challenges and Possible Solutions. Front. Immunol. 2021, 12, 658399. [Google Scholar] [CrossRef]
- Stanford, S.; Pink, R.; Creagh, D.; Clark, A.; Lowe, G.; Curry, N.; Pasi, J.; Perry, D.; Fong, S.; Hayes, G.; et al. Adenovirus-associated antibodies in UK cohort of hemophilia patients: A seroprevalence study of the presence of adenovirus-associated virus vector-serotypes AAV5 and AAV8 neutralizing activity and antibodies in patients with hemophilia A. Res. Pract. Thromb. Haemost. 2019, 3, 261–267. [Google Scholar] [CrossRef] [Green Version]
- Boutin, S.; Monteilhet, V.; Veron, P.; Leborgne, C.; Benveniste, O.; Montus, M.F.; Masurier, C. Prevalence of Serum IgG and Neutralizing Factors Against Adeno-Associated Virus (AAV) Types 1, 2, 5, 6, 8, and 9 in the Healthy Population: Implications for Gene Therapy Using AAV Vectors. Hum. Gene Ther. 2010, 21, 704–712. [Google Scholar] [CrossRef]
- Halbert, C.L.; Miller, A.D.; McNamara, S.; Emerson, J.; Gibson, R.L.; Ramsey, B.; Aitken, M.L. Prevalence of Neutralizing Antibodies Against Adeno-Associated Virus (AAV) Types 2, 5, and 6 in Cystic Fibrosis and Normal Populations: Implications for Gene Therapy Using AAV Vectors. Hum. Gene Ther. 2006, 17, 440–447. [Google Scholar] [CrossRef]
- Kruzik, A.; Fetahagic, D.; Hartlieb, B.; Dorn, S.; Koppensteiner, H.; Horling, F.M.; Scheiflinger, F.; Reipert, B.M.; de la Rosa, M. Prevalence of Anti-Adeno-Associated Virus Immune Responses in International Cohorts of Healthy Donors. Mol. Ther. Methods Clin. Dev. 2019, 14, 126–133. [Google Scholar] [CrossRef]
- Jeune, V.L.; Joergensen, J.A.; Hajjar, R.J.; Weber, T. Pre-existing Anti–Adeno-Associated Virus Antibodies as a Challenge in AAV Gene Therapy. Hum. Gene Ther. Methods 2013, 24, 59–67. [Google Scholar] [CrossRef]
- Long, B.R.; Veron, P.; Kuranda, K.; Hardet, R.; Mitchell, N.; Hayes, G.M.; Wong, W.Y.; Lau, K.; Li, M.; Hock, M.B.; et al. Early Phase Clinical Immunogenicity of Valoctocogene Roxaparvovec, an AAV5-Mediated Gene Therapy for Hemophilia A. Mol. Ther. 2021, 29, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Handyside, B.; Murphy, R.; Sihn, C.-R.; Xie, L.; Vitelli, C.; Harmon, D.; Sisó, S.; Liu, S.; Bullens, S.; et al. Prednisolone Does Not Regulate Factor VIII Expression in Mice Receiving AAV5-hFVIII-SQ: Valoctocogene Roxaparvovec. Mol. Ther. Methods Clin. Dev. 2020, 17, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Sabatino, D.E.; Lange, A.M.; Altynova, E.S.; Sarkar, R.; Zhou, S.; Merricks, E.P.; Franck, H.G.; Nichols, T.C.; Arruda, V.R.; Kazazian, H.H., Jr. Efficacy and Safety of Long-term Prophylaxis in Severe Hemophilia A Dogs Following Liver Gene Therapy Using AAV Vectors. Mol. Ther. 2011, 19, 442–449. [Google Scholar] [CrossRef]
- GlobalNewswire. Uniqure Announces Findings from Reported Case of Hepatocellular Carcinoma (HCC) in Hemophilia B Gene Therapy Program. Available online: https://www.globenewswire.com/news-release/2021/03/29/2200653/0/en/uniQure-Announces-Findings-from-Reported-Case-of-Hepatocellular-Carcinoma-HCC-in-Hemophilia-B-Gene-Therapy-Program.html (accessed on 23 May 2022).
- BioMarin. A Statement for the Haemophilia Community from BioMarin Regarding a Serious Adverse event, Deemed Unrelated, by the Independent Data Safety Monitoring Committee, to the BioMarin Haemophilia a Gene Therapy Phase 1/2 Trial (Clinical Studies Are Ongoing). Available online: https://www.ehc.eu/wp-content/uploads/BioMarin-Haemophilia-EMEA-Program-Update-for-Patient-Associations-04FEB22-MMRC-BMN27-00217.pdf (accessed on 20 May 2022).
- Konkle, B.A.; Recht, M.; Hilger, A.; Marks, P. The critical need for postmarketing surveillance in gene therapy for haemophilia. Haemophilia 2021, 27, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Pierce, G.F.; Kaczmarek, R.; Noone, D.; O’Mahony, B.; Page, D.; Skinner, M.W. Gene therapy to cure haemophilia: Is robust scientific inquiry the missing factor? Haemophilia 2020, 26, 931–933. [Google Scholar] [CrossRef]
- Spark Therapeutics. Spark Therapeutics’ SPK-8011 Suggests Stable and Durable Factor Viii Expression in Largest Phase 1/2 Gene Therapy Study in Hemophilia A to Date. Available online: https://www.globenewswire.com/news-release/2021/07/21/2266633/0/en/Spark-Therapeutics-SPK-8011-Suggests-Stable-and-Durable-Factor-VIII-Expression-in-Largest-Phase-1-2-Gene-Therapy-Study-in-Hemophilia-A-to-Date.html (accessed on 5 November 2021).
- Batty, P.; Lillicrap, D. Gene therapy for hemophilia: Current status and laboratory consequences. Int. J. Lab. Hematol. 2021, 43, 117–123. [Google Scholar] [CrossRef]
- Miesbach, W.; Klamroth, R. The Patient Experience of Gene Therapy for Hemophilia: Qualitative Interviews with Trial Patients. Patient Prefer. Adherence 2020, 14, 767–770. [Google Scholar] [CrossRef]
- Woollard, L.; Gorman, R.; Rosenfelt, D.J. Improving patient informed consent for haemophilia gene therapy: The case for change. Ther. Adv. Rare Dis. 2021, 2, 26330040211047244. [Google Scholar] [CrossRef]
- Sidonio, R.F.; Pipe, S.W.; Callaghan, M.U.; Valentino, L.A.; Monahan, P.E.; Croteau, S.E. Discussing investigational AAV gene therapy with hemophilia patients: A guide. Blood Rev. 2021, 47, 100759. [Google Scholar] [CrossRef]
- Bowyer, A.E.; Lowe, A.E.; Tiefenbacher, S. Laboratory issues in gene therapy and emicizumab. Haemophilia 2021, 27, 142–147. [Google Scholar] [CrossRef]
- Rosen, S.; Tiefenbacher, S.; Robinson, M.; Huang, M.; Srimani, J.; MacKenzie, D.; Christianson, T.; Pasi, K.J.; Rangarajan, S.; Symington, E.; et al. Activity of transgene-produced B-domain–deleted factor VIII in human plasma following AAV5 gene therapy. Blood 2020, 136, 2524–2534. [Google Scholar] [CrossRef]
- George, L.A.; Sullivan, S.K.; Giermasz, A.; Rasko, J.E.; Samelson-Jones, B.J.; Ducore, J.; Cuker, A.; Sullivan, L.M.; Majumdar, S.; Teitel, J.; et al. Hemophilia B Gene Therapy with a High-Specific-Activity Factor IX Variant. N. Engl. J. Med. 2017, 377, 2215–2227. [Google Scholar] [CrossRef]
- Jacquemin, M.; Vodolazkaia, A.; Toelen, J.; Schoeters, J.; Van Horenbeeck, I.; Vanlinthout, I.; Debasse, M.; Peerlinck, K.; Vodolozkaia, A. Measurement of B-domain-deleted ReFacto AF activity with a product-specific standard is affected by choice of reagent and patient-specific factors. Haemophilia 2018, 24, 675–682. [Google Scholar] [CrossRef]
- Ham, R.M.T.; Walker, S.M.; Soares, M.O.; Frederix, G.W.; Leebeek, F.W.; Fischer, K.; Coppens, M.; Palmer, S.J. Modeling Benefits, Costs, and Affordability of a Novel Gene Therapy in Hemophilia A. HemaSphere 2022, 6, e679. [Google Scholar] [CrossRef]
- European Association for Haemophilia and Allied Disorders. EAHAD-EHC Joint Statement on: Promoting Hub-and-Spoke Model for the Treatment of Haemophilia and Rare Bleeding Disorders Using Gene Therapies. Available online: http://eahad.org/wp-content/uploads/2020/05/Hub-and-Spoke.pdf (accessed on 20 May 2022).
- Miesbach, W.; Chowdary, P.; Coppens, M.; Hart, D.P.; Jimenez-Yuste, V.; Klamroth, R.; Makris, M.; Noone, D.; Peyvandi, F. Delivery of AAV-based gene therapy through haemophilia centres—A need for re-evaluation of infrastructure and comprehensive care: A Joint publication of EAHAD and EHC. Haemophilia 2021, 27, 967–973. [Google Scholar] [CrossRef]
- Di Minno, G.; Castaman, G.; De Cristofaro, R.; Brunetti-Pierri, N.; Pastore, L.; Castaldo, G.; Trama, U.; Di Minno, M. Progress, and prospects in the therapeutic armamentarium of persons with congenital hemophilia. Defining the place for liver-directed gene therapy. Blood Rev. 2022, 101011. [Google Scholar] [CrossRef]

{kind=link}
| Gene Therapy | Adenovirus Serotype | Clinical Stage (Trial Name; ClinicalTrials.gov Identifier) | Starting Year/Status on ClinicalTrials.gov | Company |
|---|---|---|---|---|
| Valoctocogene roxaparvovec (AAV5-hFVIII-SQ; BMN 270) | AAV5 | Phase 3 (GENEr8-1; NCT03370913; NCT03392974) [63] | 2017, active, not recruiting; 2018, active, not recruiting | BioMarin |
| Phase ½ (NCT02576795) [64,65,69] | 2015, active, not recruiting | |||
| Dirloctocogene samoparvovec (SPK 8011) | AAV3 | Phase ½ (NCT03003533; NCT03432520) [58] | 2016, recruiting; 2018, enrolling by invitation | Spark Therapeutics; Roche |
| SPK-8016 | AAV | Phase ½ (NCT03734588) [70] | 2018, recruiting | Spark Therapeutics; Roche |
| Giroctocogene fitelparvovec (SB-525; PF-07055480) | rAAV6 | Phase 3 (AFFINE; NCT04370054) [57] [on hold] | 2020, active, not recruiting | Pfizer (Sangamo) |
| Phase ½ (NCT03061201) [60,71] | 2017, active, not recruiting | |||
| TAK-754 (previously SHP654 and BAX 888) | AAV8 | Phase ½ (NCT03370172) [56] | 2017, active, not recruiting | Takeda-Shire |
| BAY 2599023 | AAVhu37 | Phase ½ (NCT03588299) [67] | 2018, recruiting | Bayer-Ultragenix Pharmaceutics |
| AAV8-HLP-hFVIII-V3 | AAV8 | Phase ½ (GO-8; NCT03001830) [61] | 2016, recruiting | University College, London |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castaman, G.; Di Minno, G.; De Cristofaro, R.; Peyvandi, F. The Arrival of Gene Therapy for Patients with Hemophilia A. Int. J. Mol. Sci. 2022, 23, 10228. https://doi.org/10.3390/ijms231810228
Castaman G, Di Minno G, De Cristofaro R, Peyvandi F. The Arrival of Gene Therapy for Patients with Hemophilia A. International Journal of Molecular Sciences. 2022; 23(18):10228. https://doi.org/10.3390/ijms231810228
Chicago/Turabian StyleCastaman, Giancarlo, Giovanni Di Minno, Raimondo De Cristofaro, and Flora Peyvandi. 2022. "The Arrival of Gene Therapy for Patients with Hemophilia A" International Journal of Molecular Sciences 23, no. 18: 10228. https://doi.org/10.3390/ijms231810228
APA StyleCastaman, G., Di Minno, G., De Cristofaro, R., & Peyvandi, F. (2022). The Arrival of Gene Therapy for Patients with Hemophilia A. International Journal of Molecular Sciences, 23(18), 10228. https://doi.org/10.3390/ijms231810228