
Azithromycin and Ceftriaxone Differentially Activate NLRP3 in LPS Primed Cancer Cells

 ,
, 
 ,
,  and
and
Abstract
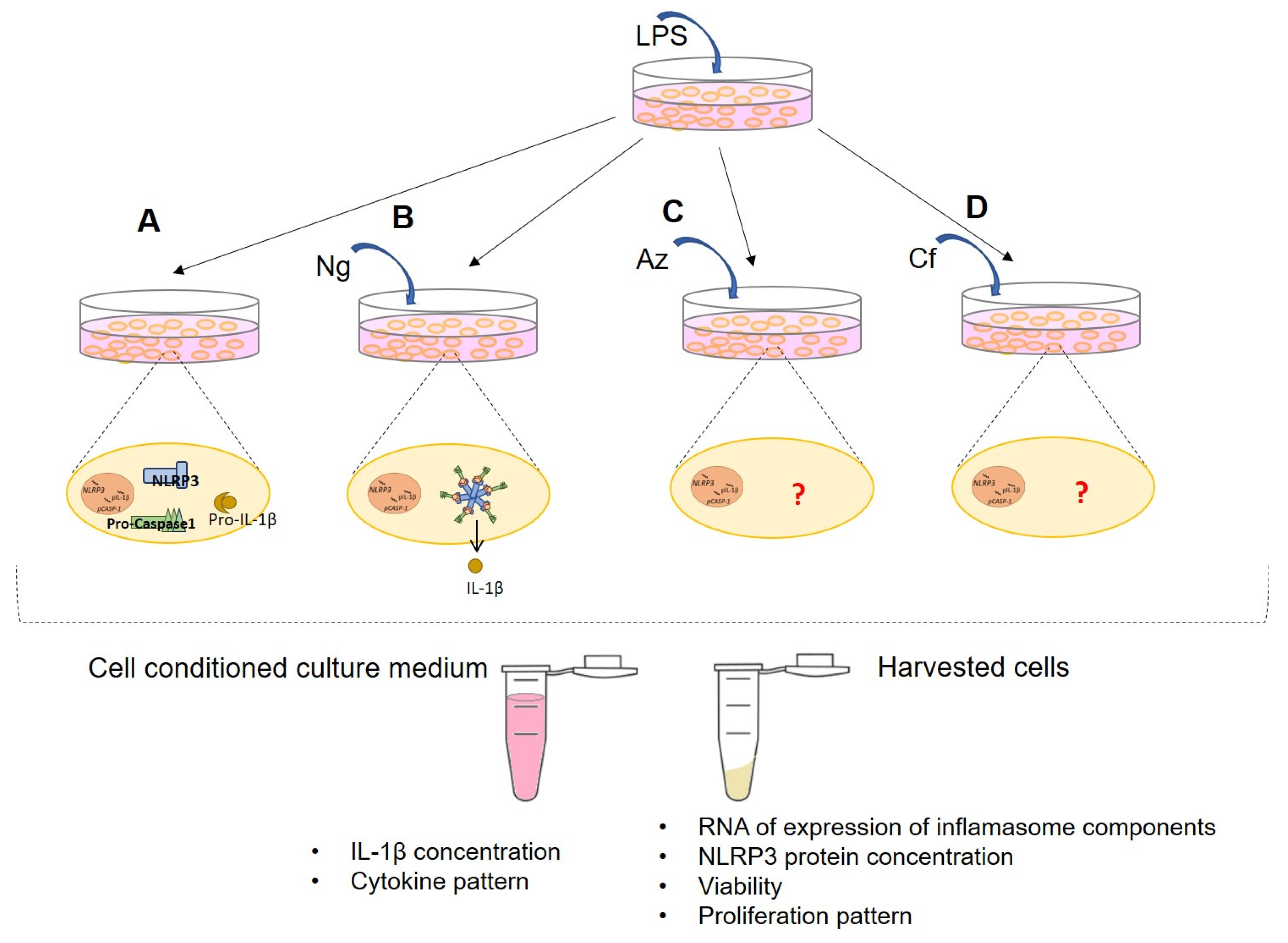
:1. Introduction
2. Results
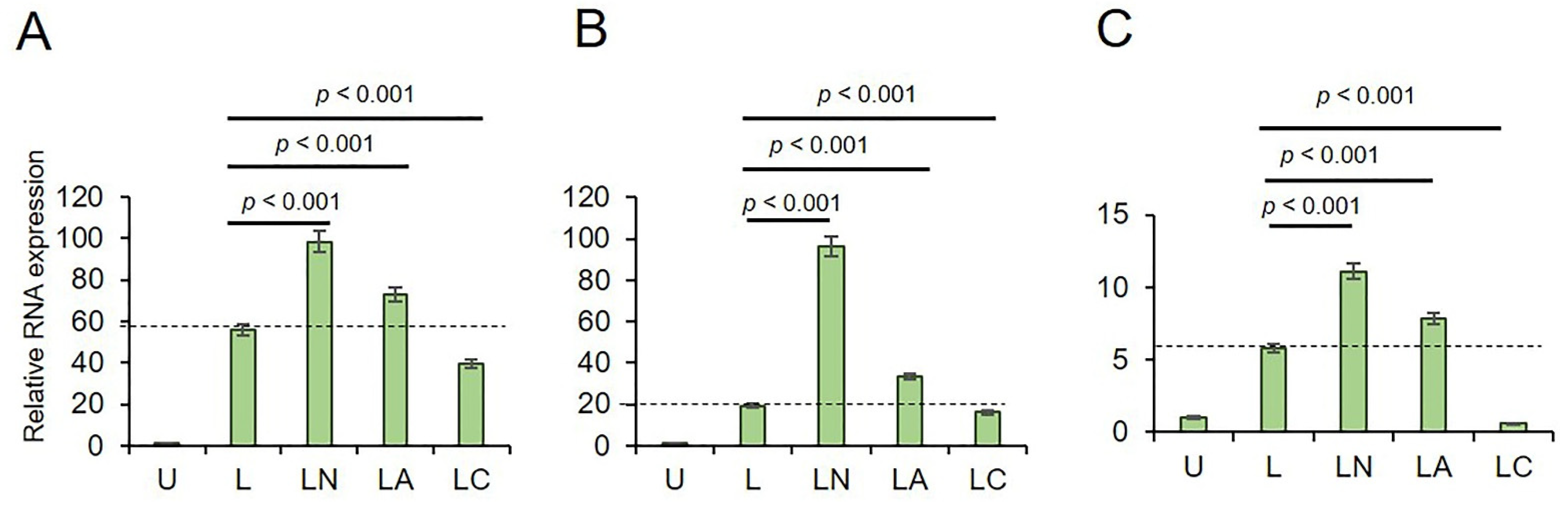
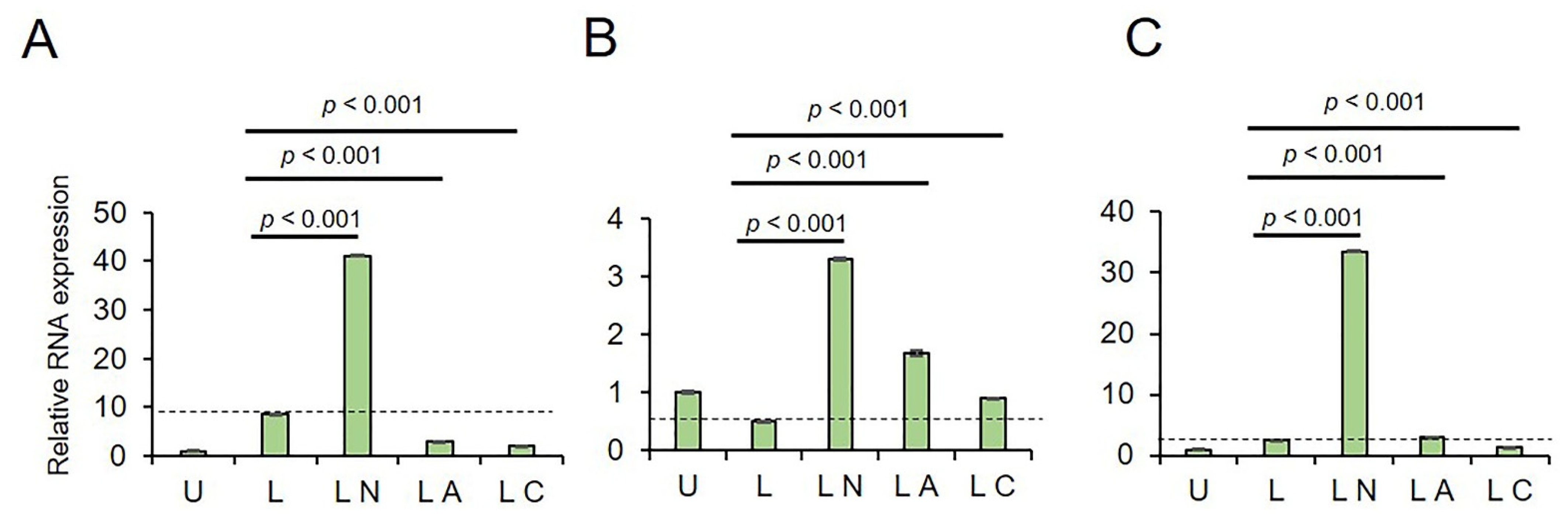
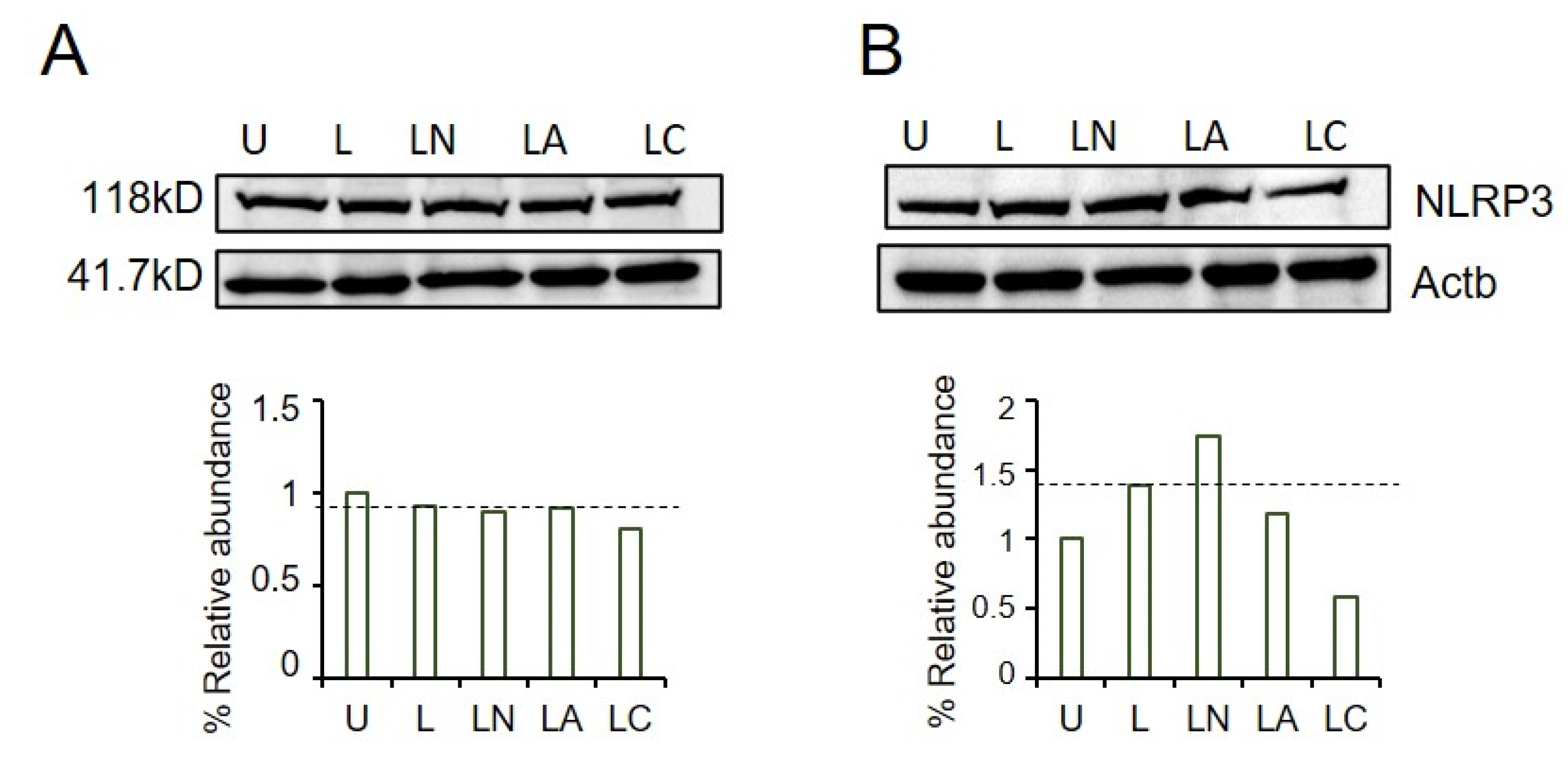
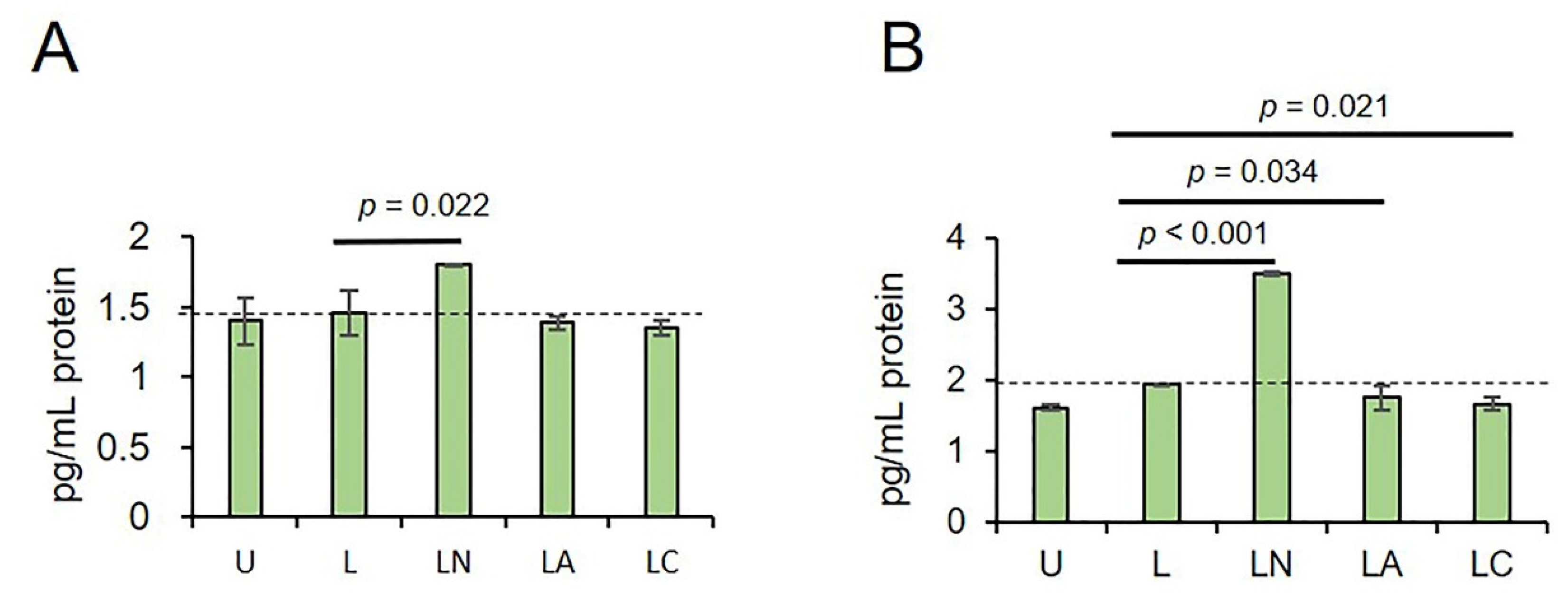
2.1. Cf and Az Suppress NLRP3 Inflammasome
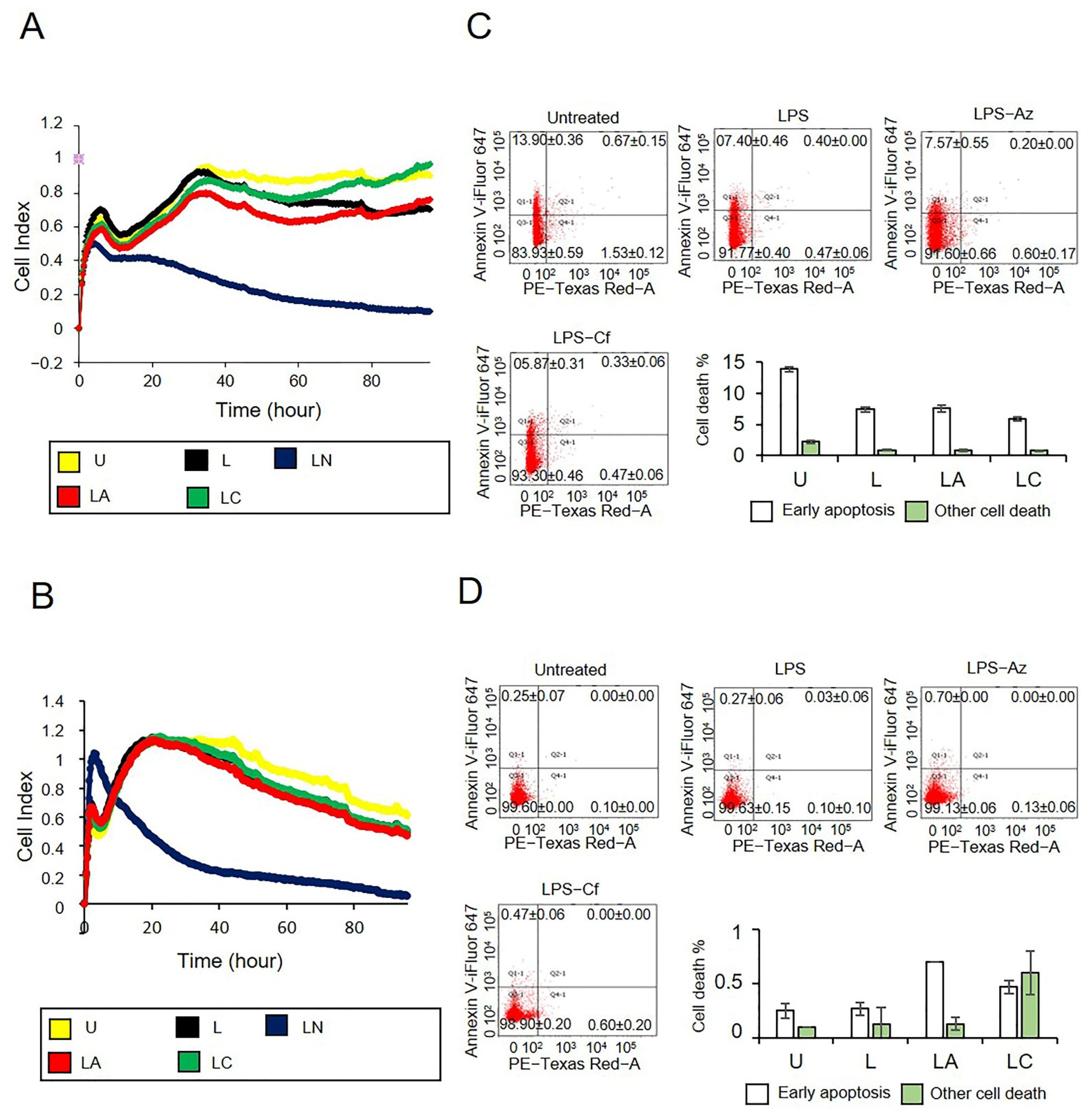
2.2. Az and Cf Did Not Affect Cell Viability
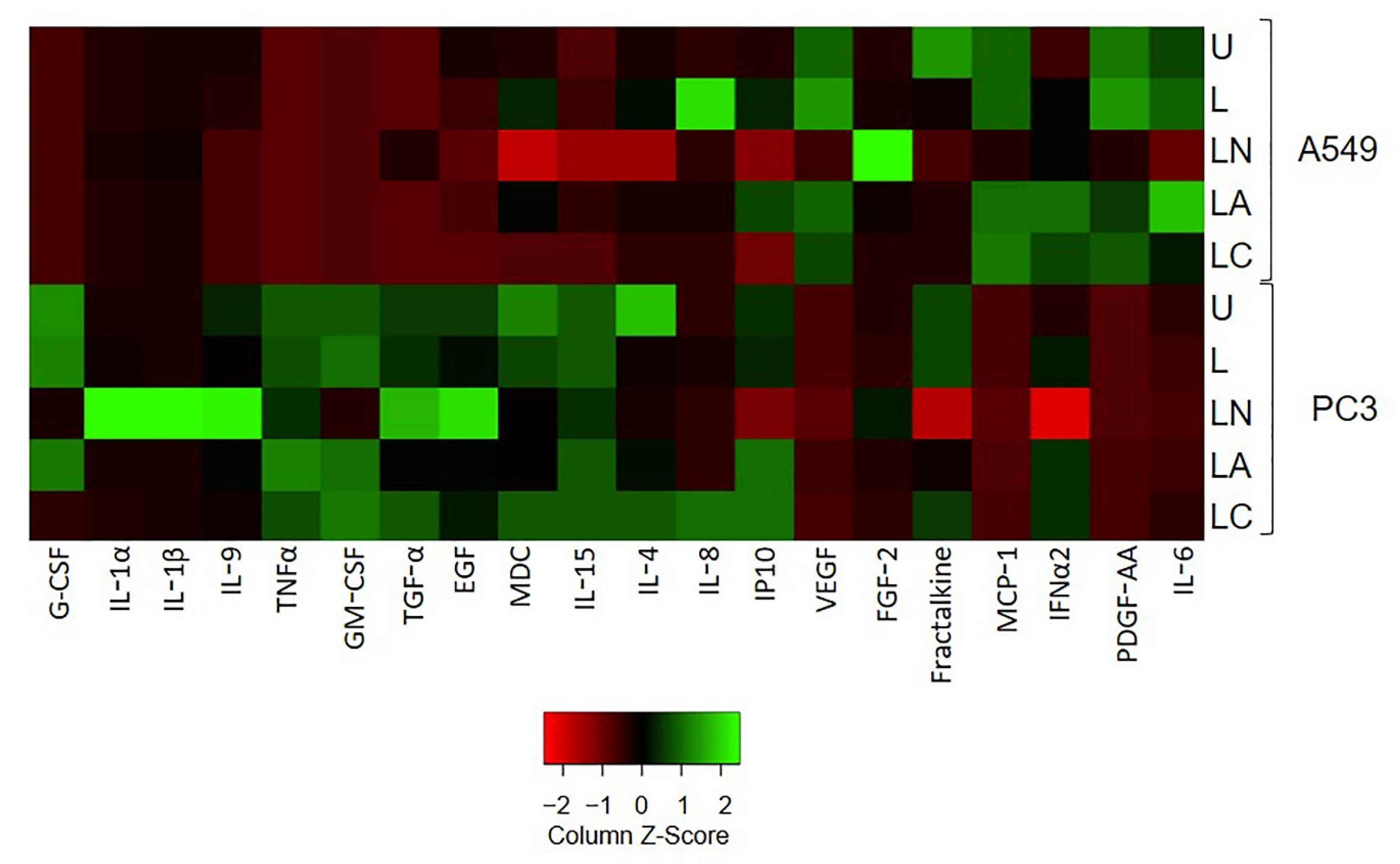
2.3. Az and Cf Differently Affect Pro-Inflammatory Cytokine Secretion
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Reagents
4.2. Real Time-qPCR
4.3. Immunoblotting
4.4. Enzyme-Linked Immunosorbent Assay (ELISA)
4.5. Real-Time Cell Proliferation Analysis
4.6. Cell Viability Assay
4.7. Multiplex Cytokine Analysis
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Hamarsheh, S.; Zeiser, R. NLRP3 Inflammasome Activation in Cancer: A Double-Edged Sword. Front. Immunol. 2020, 11, 1444. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [Green Version]
- Tartey, S.; Kanneganti, T.D. Differential role of the NLRP3 inflammasome in infection and tumorigenesis. Immunology 2019, 156, 329–338. [Google Scholar] [CrossRef] [Green Version]
- Lillo, S.; Saleh, M. Inflammasomes in Cancer Progression and Anti-Tumor Immunity. Front. Cell Dev. Biol. 2022, 10, 839041. [Google Scholar] [CrossRef]
- Lin, C.; Zhang, J. Inflammasomes in Inflammation-Induced Cancer. Front. Immunol. 2017, 8, 271. [Google Scholar] [CrossRef] [Green Version]
- Guo, B.; Fu, S.; Zhang, J.; Liu, B.; Li, Z. Targeting inflammasome/IL-1 pathways for cancer immunotherapy. Sci. Rep. 2016, 6, 36107. [Google Scholar] [CrossRef]
- Ershaid, N.; Sharon, Y.; Doron, H.; Raz, Y.; Shani, O.; Cohen, N.; Monteran, L.; Leider-Trejo, L.; Ben-Shmuel, A.; Yassin, M.; et al. NLRP3 inflammasome in fibroblasts links tissue damage with inflammation in breast cancer progression and metastasis. Nat. Commun. 2019, 10, 4375. [Google Scholar] [CrossRef] [Green Version]
- Kaplanov, I.; Carmi, Y.; Kornetsky, R.; Shemesh, A.; Shurin, G.V.; Shurin, M.R.; Dinarello, C.A.; Voronov, E.; Apte, R.N. Blocking IL-1β reverses the immunosuppression in mouse breast cancer and synergizes with anti-PD-1 for tumor abrogation. Proc. Natl. Acad. Sci. USA 2019, 116, 1361–1369. [Google Scholar] [CrossRef] [Green Version]
- Weichand, B.; Popp, R.; Dziumbla, S.; Mora, J.; Strack, E.; Elwakeel, E.; Frank, A.C.; Scholich, K.; Pierre, S.; Syed, S.N.; et al. S1PR1 on tumor-associated macrophages promotes lymphangiogenesis and metastasis via NLRP3/IL-1β. J. Exp. Med. 2017, 214, 2695–2713. [Google Scholar] [CrossRef]
- Huber, S.; Gagliani, N.; Zenewicz, L.; Huber, F.J.; Bosurgi, L.; Hu, B.; Hedl, M.; Zhang, W.; O’Connor, W.; Murphy, A.; et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature 2012, 491, 259–263. [Google Scholar] [CrossRef] [Green Version]
- Drexler, S.K.; Bonsignore, L.; Masin, M.; Tardivel, A.; Jackstadt, R.; Hermeking, H.; Schneider, P.; Gross, O.; Tschopp, J.; Yazdi, A.S. Tissue-specific opposing functions of the inflammasome adaptor ASC in the regulation of epithelial skin carcinogenesis. Proc. Natl. Acad. Sci. USA 2012, 109, 18384–18389. [Google Scholar] [CrossRef] [Green Version]
- Chow, M.T.; Sceneay, J.; Paget, C.; Wong, C.S.; Duret, H.; Tschopp, J.; Möller, A.; Smyth, M.J. NLRP3 suppresses NK cell-mediated responses to carcinogen-induced tumors and metastases. Cancer Res. 2012, 72, 5721–5732. [Google Scholar] [CrossRef] [Green Version]
- Bae, J.Y.; Lee, S.-W.; Shin, Y.-H.; Lee, J.-H.; Jahng, J.W.; Park, K. P2X7 receptor and NLRP3 inflammasome activation in head and neck cancer. Oncotarget 2017, 8, 48972–48982. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Huang, C.F.; Li, Y.C.; Deng, W.W.; Mao, L.; Wu, L.; Zhang, W.F.; Zhang, L.; Sun, Z.J. Blockage of the NLRP3 inflammasome by MCC950 improves anti-tumor immune responses in head and neck squamous cell carcinoma. Cell. Mol. Life Sci. 2018, 75, 2045–2058. [Google Scholar] [CrossRef]
- Daley, D.; Mani, V.R.; Mohan, N.; Akkad, N.; Pandian, G.S.B.; Savadkar, S.; Lee, K.B.; Torres-Hernandez, A.; Aykut, B.; Diskin, B.; et al. NLRP3 signaling drives macrophage-induced adaptive immune suppression in pancreatic carcinoma. J. Exp. Med. 2017, 214, 1711–1724. [Google Scholar] [CrossRef] [Green Version]
- Tezcan, G.; Garanina, E.E.; Alsaadi, M.; Gilazieva, Z.E.; Martinova, E.V.; Markelova, M.I.; Arkhipova, S.S.; Hamza, S.; McIntyre, A.; Rizvanov, A.A.; et al. Therapeutic Potential of Pharmacological Targeting NLRP3 Inflammasome Complex in Cancer. Front. Immunol. 2021, 11, 607881. [Google Scholar] [CrossRef]
- Alsaadi, M.; Tezcan, G.; Garanina, E.E.; Hamza, S.; McIntyre, A.; Rizvanov, A.A.; Khaiboullina, S.F. Doxycycline Attenuates Cancer Cell Growth by Suppressing NLRP3-Mediated Inflammation. Pharmaceuticals 2021, 14, 852. [Google Scholar] [CrossRef]
- Gao, Y.; Shang, Q.; Li, W.; Guo, W.; Stojadinovic, A.; Mannion, C.; Man, Y.-G.; Chen, T. Antibiotics for cancer treatment: A double-edged sword. J. Cancer 2020, 11, 5135–5149. [Google Scholar] [CrossRef]
- Höffken, G.; Niederman, M.S. Nosocomial pneumonia: The importance of a de-escalating strategy for antibiotic treatment of pneumonia in the ICU. Chest 2002, 122, 2183–2196. [Google Scholar] [CrossRef] [PubMed]
- Stevermer, J.J.; Easley, S.K. Treatment of prostatitis. Am. Fam. Physician 2000, 61, 3015–3022, Erratum in Am. Fam. Physician 2001, 63, 2129. [Google Scholar]
- Pratheeshkumar, P.; Kuttan, G. Oleanolic acid induces apoptosis by modulating p53, Bax, Bcl-2 and caspase-3 gene expression and regulates the activation of transcription factors and cytokine profile in B16F. J. Environ. Pathol. Toxicol. Oncol. 2011, 30, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Li, P.; Xue, X.; He, S.; Kuang, Y.; Zhao, H.; Chen, S.; Zhi, Q.; Guo, X. Salinomycin induces apoptosis in cisplatin-resistant colorectal cancer cells by accumulation of reactive oxygen species. Toxicol. Lett. 2013, 222, 139–145. [Google Scholar] [CrossRef] [Green Version]
- Xia, D.; Yang, X.; Liu, W.; Shen, F.; Pan, J.; Lin, Y.; Du, N.; Sun, Y.; Xi, X. Over-expression of CHAF1A in Epithelial Ovarian Cancer can promote cell proliferation and inhibit cell apoptosis. Biochem. Biophys. Res. Commun. 2017, 486, 191–197. [Google Scholar] [CrossRef]
- Sánchez-Tilló, E.; Fanlo, L.; Siles, L.; Montes-Moreno, S.; Moros, A.; Chiva-Blanch, G.; Estruch, R.; Martinez, A.; Colomer, D.; Győrffy, B.; et al. The EMT activator ZEB1 promotes tumor growth and determines differential response to chemotherapy in mantle cell lymphoma. Cell Death Differ. 2014, 21, 247–257. [Google Scholar] [CrossRef]
- Sullivan, A.; Edlund, C.; Nord, C.E. Effect of antimicrobial agents on the ecological balance of human microflora. Lancet Infect. Dis. 2001, 1, 101–114. [Google Scholar] [CrossRef]
- Ahmed, J.; Kumar, A.; Parikh, K.; Anwar, A.; Knoll, B.M.; Puccio, C.; Chun, H.; Fanucchi, M.; Lim, S.H. Use of broad-spectrum antibiotics impacts outcome in patients treated with immune checkpoint inhibitors. Oncoimmunology 2018, 7, e1507670. [Google Scholar] [CrossRef] [Green Version]
- Sommer, F.; Anderson, J.M.; Bharti, R.; Raes, J.; Rosenstiel, P. The resilience of the intestinal microbiota influences health and disease. Nat. Rev. Microbiol. 2017, 15, 630–638. [Google Scholar] [CrossRef]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Mishra, S.R.; Mahapatra, K.K.; Behera, B.P.; Patra, S.; Bhol, C.S.; Panigrahi, D.P.; Praharaj, P.P.; Singh, A.; Patil, S.; Dhiman, R.; et al. Mitochondrial dysfunction as a driver of NLRP3 inflammasome activation and its modulation through mitophagy for potential therapeutics. Int. J. Biochem. Cell Biol. 2021, 136, 106013. [Google Scholar] [CrossRef]
- Kanoh, S.; Rubin, B.K. Mechanisms of action and clinical application of macrolides as immunomodulatory medications. Clin. Microbiol. Rev. 2010, 23, 590–615. [Google Scholar] [CrossRef] [Green Version]
- Kohanski, M.A.; Tharakan, A.; London, N.R.; Lane, A.P.; Ramanathan, M., Jr. Bactericidal antibiotics promote reactive oxygen species formation and inflammation in human sinonasal epithelial cells. Int. Forum Allergy Rhinol. 2016, 6, 191–200. [Google Scholar] [CrossRef]
- Léger, L.; Budin-Verneuil, A.; Cacaci, M.; Benachour, A.; Hartke, A.; Verneuil, N. β-Lactam Exposure Triggers Reactive Oxygen Species Formation in Enterococcus faecalis via the Respiratory Chain Component DMK. Cell Rep. 2019, 29, 2184–2191.e3. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Baucom, C.; Elliott, R.L. Mitochondrial Toxicity of Azithromycin Results in Aerobic Glycolysis and DNA Damage of Human Mammary Epithelia and Fibroblasts. Antibiotics 2019, 8, 110. [Google Scholar] [CrossRef] [Green Version]
- Müller, S.; Wolf, A.J.; Iliev, I.D.; Berg, B.L.; Underhill, D.M.; Liu, G.Y. Poorly Cross-Linked Peptidoglycan in MRSA Due to mecA Induction Activates the Inflammasome and Exacerbates Immunopathology. Cell Host Microbe 2015, 18, 604–612. [Google Scholar] [CrossRef] [Green Version]
- Thacker, J.D.; Balin, B.J.; Appelt, D.M.; Sassi-Gaha, S.; Purohit, M.; Rest, R.F.; Artlett, C.M. NLRP3 inflammasome is a target for development of broad-spectrum anti-infective drugs. Antimicrob. Agents Chemother. 2012, 56, 1921–1930. [Google Scholar] [CrossRef] [Green Version]
- Paul, M.; Dickstein, Y.; Schlesinger, A.; Grozinsky-Glasberg, S.; Soares-Weiser, K.; Leibovici, L. Beta-lactam versus beta-lactam-aminoglycoside combination therapy in cancer patients with neutropenia. Cochrane Database Syst. Rev. 2013, 2013, CD003038. [Google Scholar] [CrossRef]
- Paprocka, P.; Durnaś, B.; Mańkowska, A.; Król, G.; Wollny, T.; Bucki, R. Pseudomonas aeruginosa Infections in Cancer Patients. Pathogens 2022, 11, 679. [Google Scholar] [CrossRef]
- Feng, X.; Luo, Q.; Zhang, H.; Wang, H.; Chen, W.; Meng, G.; Chen, F. The role of NLRP3 inflammasome in 5-fluorouracil resistance of oral squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2017, 36, 81. [Google Scholar] [CrossRef] [Green Version]
- Stein, G.E.; Havlichek, D.H. The new macrolide antibiotics. Azithromycin and clarithromycin. Postgrad. Med. 1992, 92, 269–282. [Google Scholar] [CrossRef]
- Wei, J.; Pan, X.; Pei, Z.; Wang, W.; Qiu, W.; Shi, Z.; Xiao, G. The beta-lactam antibiotic, ceftriaxone, provides neuroprotective potential via anti-excitotoxicity and anti-inflammation response in a rat model of traumatic brain injury. J. Trauma Acute Care Surg. 2012, 73, 654–660. [Google Scholar] [CrossRef]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; Macdonald, K.L.; Speert, D.P.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.; et al. Cutting edge: NF-κB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef]
- Broz, P.; Monack, D.M. Molecular mechanisms of inflammasome activation during microbial infections. Immunol. Rev. 2011, 243, 174–190. [Google Scholar] [CrossRef]
- He, Y.; Hara, H.; Núñez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Planillo, R.; Kuffa, P.; Martínez-Colón, G.; Smith, B.L.; Rajendiran, T.M.; Núñez, G. K⁺ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef] [Green Version]
- Yuan, A.; Chen, J.J.; Yao, P.L.; Yang, P.C. The role of interleukin-8 in cancer cells and microenvironment interaction. Front. Biosci. 2005, 10, 853–865. [Google Scholar] [CrossRef] [Green Version]
- Akl, M.R.; Nagpal, P.; Ayoub, N.M.; Tai, B.; Prabhu, S.A.; Capac, C.M.; Gliksman, M.; Goy, A.; Suh, K.S. Molecular and clinical significance of fibroblast growth factor 2 (FGF2/bFGF) in malignancies of solid and hematological cancers for personalized therapies. Oncotarget 2016, 7, 44735–44762. [Google Scholar] [CrossRef] [Green Version]
- Carmeliet, P. VEGF as a key mediator of angiogenesis in cancer. Oncology 2005, 69, 4–10. [Google Scholar] [CrossRef]
- Lamb, R.; Ozsvari, B.; Lisanti, C.L.; Tanowitz, H.B.; Howell, A.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: Treating cancer like an infectious disease. Oncotarget 2015, 6, 4569–4584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coll, R.C.; Holley, C.L.; Schroder, K. Mitochondrial DNA synthesis fuels NLRP3 activation. Cell Res. 2018, 28, 1046–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshall, W.F.; Blair, J.E. The cephalosporins. Mayo Clin. Proc. 1999, 74, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Cottagnoud, P.; Neftel, K.A. Beta-lactams act on DNA synthesis in K-562 cells. Cell Biol. Toxicol. 1986, 2, 523–529. [Google Scholar] [CrossRef]
- Fram, R.J. A comparison of the effects of cytosine arabinoside and beta-lactams on DNA synthesis and cellular proliferation. Cell Biol. Toxicol. 1986, 2, 531–539. [Google Scholar] [CrossRef]
- Hübscher, U.; Huynh, U.D.; Hässig, M.; Neftel, K.A. Effects of beta-lactams on DNA replication. Cell Biol. Toxicol. 1986, 2, 541–548. [Google Scholar] [CrossRef]
- Qiao, X.; Wang, X.; Shang, Y.; Li, Y.; Chen, S.Z. Azithromycin enhances anticancer activity of TRAIL by inhibiting autophagy and up-regulating the protein levels of DR4/5 in colon cancer cells in vitro and in vivo. Cancer Commun. 2018, 38, 43. [Google Scholar] [CrossRef] [Green Version]
- Yang, N.; Liang, Y.; Yang, P.; Ji, F. Propofol suppresses LPS-induced nuclear accumulation of HIF-1α and tumor aggressiveness in non-small cell lung cancer. Oncol. Rep. 2017, 37, 2611–2619. [Google Scholar] [CrossRef] [Green Version]
- Bosnar, M.; Čužić, S.; Bošnjak, B.; Nujić, K.; Ergović, G.; Marjanović, N.; Pašalić, I.; Hrvačić, B.; Polančec, D.; Glojnarić, I.; et al. Azithromycin inhibits macrophage interleukin-1β production through inhibition of activator protein-1 in lipopolysaccharide-induced murine pulmonary neutrophilia. Int. Immunopharmacol. 2011, 11, 424–434. [Google Scholar] [CrossRef]
- Bosnar, M.; Bosnjak, B.; Cuzic, S.; Hrvacic, B.; Marjanovic, N.; Glojnaric, I.; Culic, O.; Parnham, M.J.; Haber, V.E. Azithromycin and clarithromycin inhibit lipopolysaccharide-induced murine pulmonary neutrophilia mainly through effects on macrophage-derived granulocyte-macrophage colony-stimulating factor and interleukin-1beta. J. Pharmacol. Exp. Ther. 2009, 331, 104–113. [Google Scholar] [CrossRef] [Green Version]
- Geudens, N.; Timmermans, L.; Vanhooren, H.; Vanaudenaerde, B.M.; Vos, R.; Van De Wauwer, C.; Verleden, G.M.; Verbeken, E.; Lerut, T.; Van Raemdonck, D.E. Azithromycin reduces airway inflammation in a murine model of lung ischaemia reperfusion injury. Transpl. Int. 2008, 21, 688–695. [Google Scholar] [CrossRef]
- Li, Z.; Chen, L.; Qin, Z. Paradoxical roles of IL-4 in tumor immunity. Cell. Mol. Immunol. 2009, 6, 415–422. [Google Scholar] [CrossRef]
- Karagiannidis, I.; Salataj, E.; Egal, E.S.A.; Beswick, E.J. G-CSF in tumors: Aggressiveness, tumor microenvironment and immune cell regulation. Cytokine 2021, 142, 155479. [Google Scholar] [CrossRef]
- Yang, X.; Liu, F.; Xu, Z.; Chen, C.; Wu, X.; Li, G.; Li, J. Expression of granulocyte colony stimulating factor receptor in human colorectal cancer. Postgrad. Med. J. 2005, 81, 333–337. [Google Scholar] [CrossRef]
- Fukui, Y.; Kawashima, M.; Kawaguchi, K.; Takeuchi, M.; Hirata, M.; Kataoka, T.R.; Sakurai, T.; Kataoka, M.; Kanao, S.; Nakamoto, Y.; et al. Granulocyte-colony-stimulating factor-producing metaplastic carcinoma of the breast with significant elevation of serum interleukin-17 and vascular endothelial growth factor levels. Int. Cancer Conf. J. 2018, 7, 107–113. [Google Scholar] [CrossRef]
- Kitade, H.; Yanagida, H.; Yamada, M.; Satoi, S.; Yoshioka, K.; Shikata, N.; Kon, M. Granulocyte-colony stimulating factor producing anaplastic carcinoma of the pancreas treated by distal pancreatectomy and chemotherapy: Report of a case. Surg. Case Rep. 2015, 1, 46. [Google Scholar] [CrossRef] [Green Version]
- Wightman, S.C.; Uppal, A.; Pitroda, S.P.; Ganai, S.; Burnette, B.; Stack, M.; Oshima, G.; Khan, S.; Huang, X.; Posner, M.C.; et al. Oncogenic CXCL10 signalling drives metastasis development and poor clinical outcome. Br. J. Cancer 2015, 113, 327–335. [Google Scholar] [CrossRef]
- Chiu, J.W.; Binte Hanafi, Z.; Chew, L.C.Y.; Mei, Y.; Liu, H. IL-1α Processing, Signaling and Its Role in Cancer Progression. Cells 2021, 10, 92. [Google Scholar] [CrossRef]
- Mantovani, A.; Barajon, I.; Garlanda, C. IL-1 and IL-1 regulatory pathways in cancer progression and therapy. Immunol. Rev. 2018, 281, 57–61. [Google Scholar] [CrossRef]
- Yeh, J.; Yeh, Y.C. Transforming growth factor-α and human cancer. Biomed. Pharmacother. 1989, 43, 651–659. [Google Scholar] [CrossRef]
- Auvinen, P.K.; Lipponen, P.K.; Kataja, V.V.; Johansson, R.T.; Syrjänen, K.J. Prognostic significance of TGF-α expression in breast cancer. Acta Oncol. 1996, 35, 995–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrantini, M.; Capone, I.; Belardelli, F. Interferon-α and cancer: Mechanisms of action and new perspectives of clinical use. Biochimie 2007, 89, 884–893. [Google Scholar] [CrossRef] [PubMed]
- Paul, F.; Pellegrini, S.; Uzé, G. IFNA2: The prototypic human alpha interferon. Gene 2015, 567, 132–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergeron, A.; Chevret, S.; Granata, A.; Chevallier, P.; Vincent, L.; Huynh, A.; Tabrizi, R.; Labussiere-Wallet, H.; Bernard, M.; Chantepie, S.; et al. Effect of azithromycin on airflow decline-free survival after allogeneic hematopoietic stem cell transplant: The ALLOZITHRO randomized clinical trial. JAMA 2017, 318, 557–566. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. FDA Warns about Increased Risk of Cancer Relapse with Long-Term Use of Azithromycin (Zithromax, Zmax) Antibiotic after Donor Stem Cell Transplant: FDA Public Health Advisory. In Center for Drug Evaluation and Research; U.S. Food and Drug Administration: Washington, DC, USA, 2008. [Google Scholar]
- Yifan, Z.; Benxiang, N.; Zheng, X.; Luwei, X.; Liuhua, Z.; Yuzheng, G.; Ruipeng, J. Ceftriaxone Calcium Crystals Induce Acute Kidney Injury by NLRP3-Mediated Inflammation and Oxidative Stress Injury. Oxid. Med. Cell. Longev. 2020, 2020, 6428498. [Google Scholar] [CrossRef]
- Lee, G.-S.; Subramanian, N.; Kim, A.I.; Aksentijevich, I.; Goldbach-Mansky, R.; Sacks, D.; Germain, R.N.; Kastner, D.L.; Chae, J.J. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 2012, 492, 123–127. [Google Scholar] [CrossRef] [Green Version]
- Rio, D.C.; Ares, M., Jr.; Hannon, G.J.; Nilsen, T.W. Purification of RNA using TRIzol (TRI reagent). Cold Spring Harb. Protoc. 2010, 2010, pdb-prot5439. [Google Scholar] [CrossRef]
- Tezcan, G.; Garanina, E.E.; Zhuravleva, M.N.; Hamza, S.; Rizvanov, A.A.; Khaiboullina, S.F. Rab GTPase Mediating Regulation of NALP3 in Colorectal Cancer. Molecules 2020, 25, 4834. [Google Scholar] [CrossRef]
- Lagarkova, M.A.; Shutova, M.V.; Bogomazova, A.; Vassina, E.M.; Glazov, E.A.; Zhang, P.; Rizvanov, A.; Chestkov, I.V.; Kiselev, S. Induction of pluripotency in human endothelial cells resets epigenetic profile on genome scale. Cell Cycle 2010, 9, 937–946. [Google Scholar] [CrossRef] [Green Version]
- Babicki, S.; Arndt, D.; Marcu, A.; Liang, Y.; Grant, J.R.; Maciejewski, A.; Wishart, D.S. Heatmapper: Web-enabled heat mapping for all. Nucleic Acids Res. 2016, 44, W147–W153. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers | ||
|---|---|---|
| NLRP3 | F: | 5′-ATGAGTGCTGCTTCGACATC-3′ |
| R: | 5′-TTGTCACTCAGGTCCAGCTC-3′ | |
| Pro-CASP1 | F: | 5′-TGCCTTTCTTCTGGTCAGTG-3′ |
| R: | 5′-TGCTGAGGTGAAGGAGAGAA-3′ | |
| Pro-IL-1β | F: | 5′-TCAGCACCTCTCAAGCAGAA-3′ |
| R: | 5′-GGACTCTCTGGGTACAGCTC-3′ | |
| ACTB | F: | 5′-GACAGGATGCAGAAGGAGATTACT-3′ |
| R: | 5′-TGATCCACATCTGCTGGAAGGT-3′ | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tezcan, G.; Alsaadi, M.; Hamza, S.; Garanina, E.E.; Martynova, E.V.; Ziganshina, G.R.; Farukshina, E.R.; Rizvanov, A.A.; Khaiboullina, S.F. Azithromycin and Ceftriaxone Differentially Activate NLRP3 in LPS Primed Cancer Cells. Int. J. Mol. Sci. 2022, 23, 9484. https://doi.org/10.3390/ijms23169484
Tezcan G, Alsaadi M, Hamza S, Garanina EE, Martynova EV, Ziganshina GR, Farukshina ER, Rizvanov AA, Khaiboullina SF. Azithromycin and Ceftriaxone Differentially Activate NLRP3 in LPS Primed Cancer Cells. International Journal of Molecular Sciences. 2022; 23(16):9484. https://doi.org/10.3390/ijms23169484
Chicago/Turabian StyleTezcan, Gulcin, Mohammad Alsaadi, Shaimaa Hamza, Ekaterina E. Garanina, Ekaterina V. Martynova, Gulshat R. Ziganshina, Elina R. Farukshina, Albert A. Rizvanov, and Svetlana F. Khaiboullina. 2022. "Azithromycin and Ceftriaxone Differentially Activate NLRP3 in LPS Primed Cancer Cells" International Journal of Molecular Sciences 23, no. 16: 9484. https://doi.org/10.3390/ijms23169484
APA StyleTezcan, G., Alsaadi, M., Hamza, S., Garanina, E. E., Martynova, E. V., Ziganshina, G. R., Farukshina, E. R., Rizvanov, A. A., & Khaiboullina, S. F. (2022). Azithromycin and Ceftriaxone Differentially Activate NLRP3 in LPS Primed Cancer Cells. International Journal of Molecular Sciences, 23(16), 9484. https://doi.org/10.3390/ijms23169484