
Identification of Potential Cytochrome P450 3A5 Inhibitors: An Extensive Virtual Screening through Molecular Docking, Negative Image-Based Screening, Machine Learning and Molecular Dynamics Simulation Studies

 , , and
, , and
Abstract
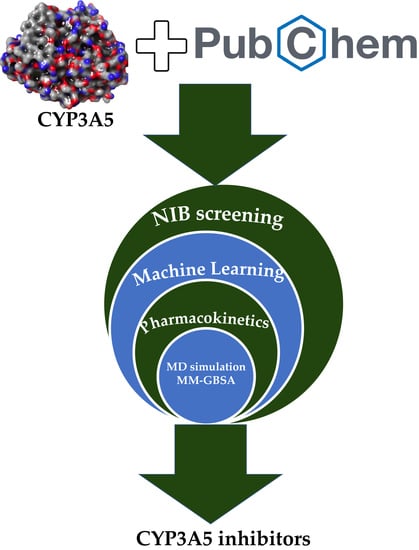
:
1. Introduction
2. Results and Discussion
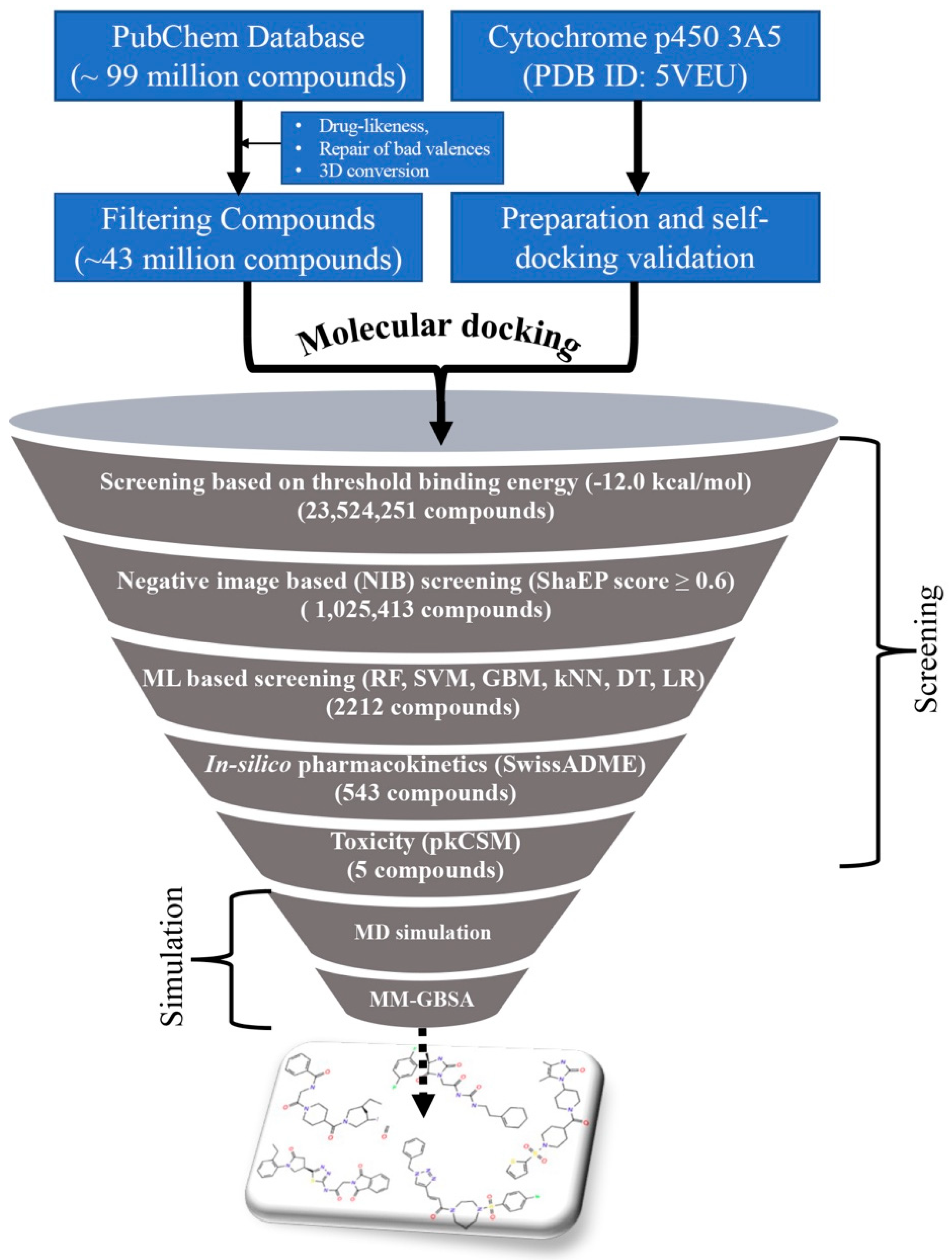
2.1. Virtual Screening
2.1.1. Molecular Docking
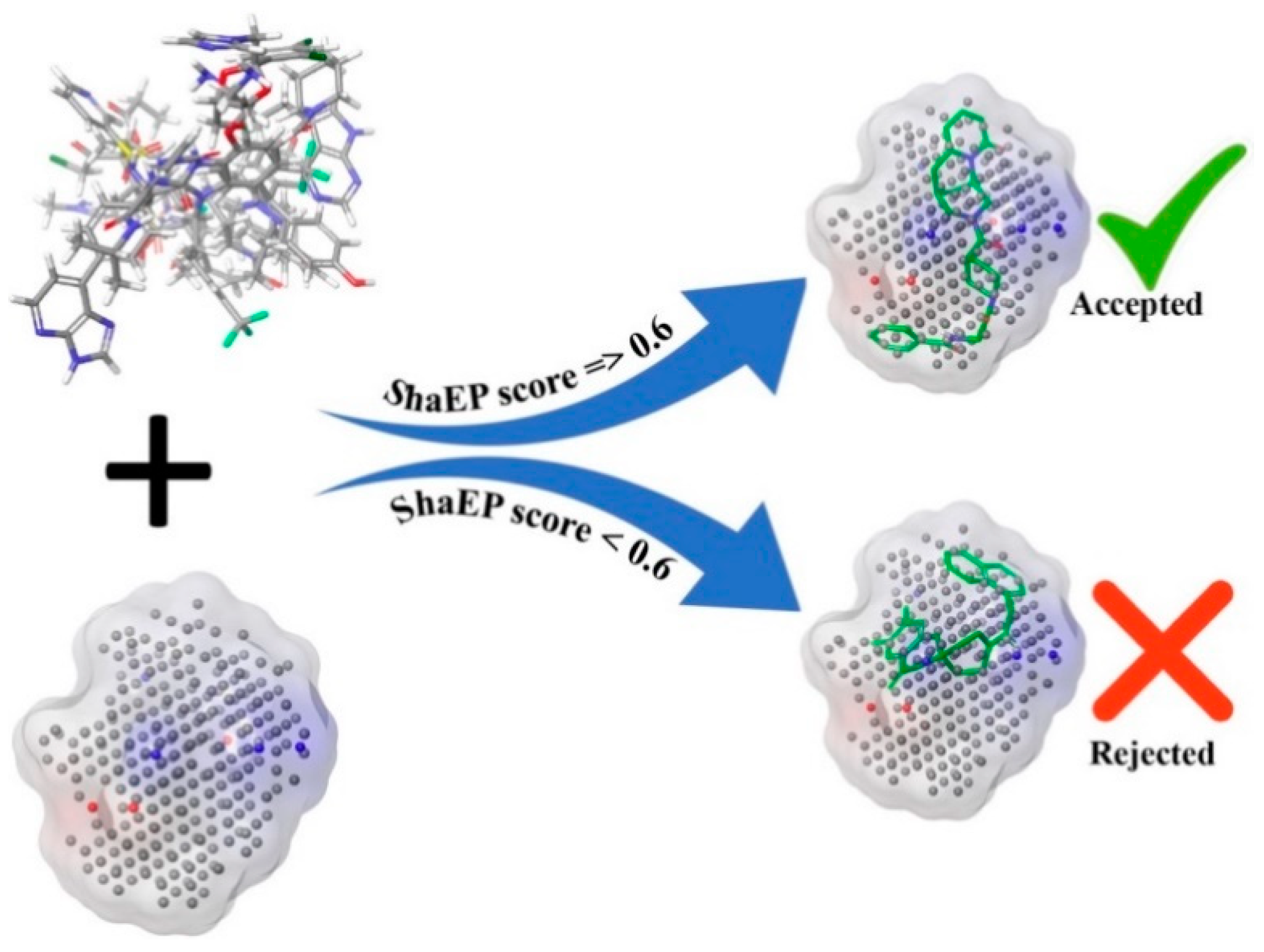
2.1.2. Negative Image-Based (NIB) Screening
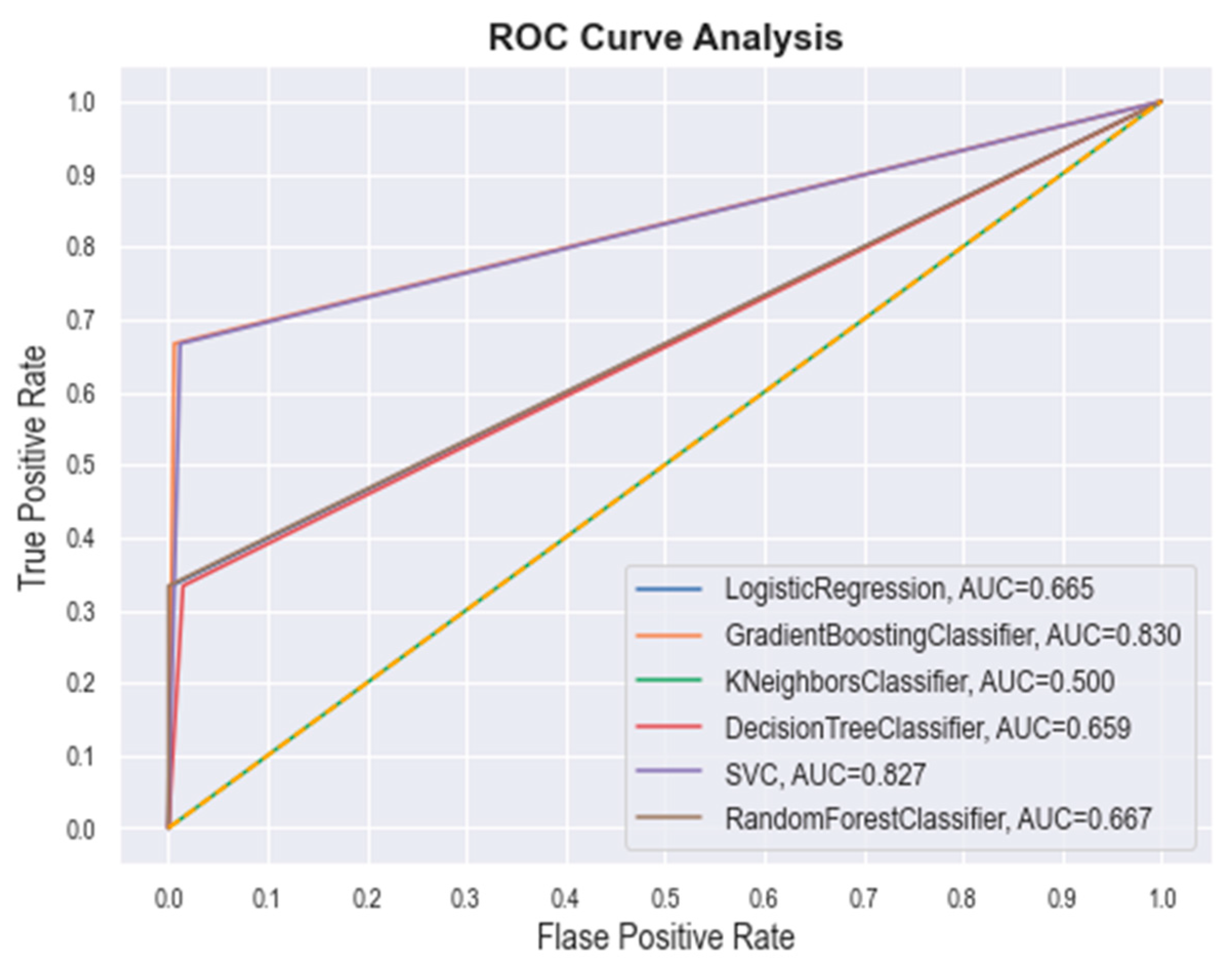
2.1.3. Machine Learning-(ML) Based Screening
2.1.4. In Silico Pharmacokinetics and Toxicity-Based Screening
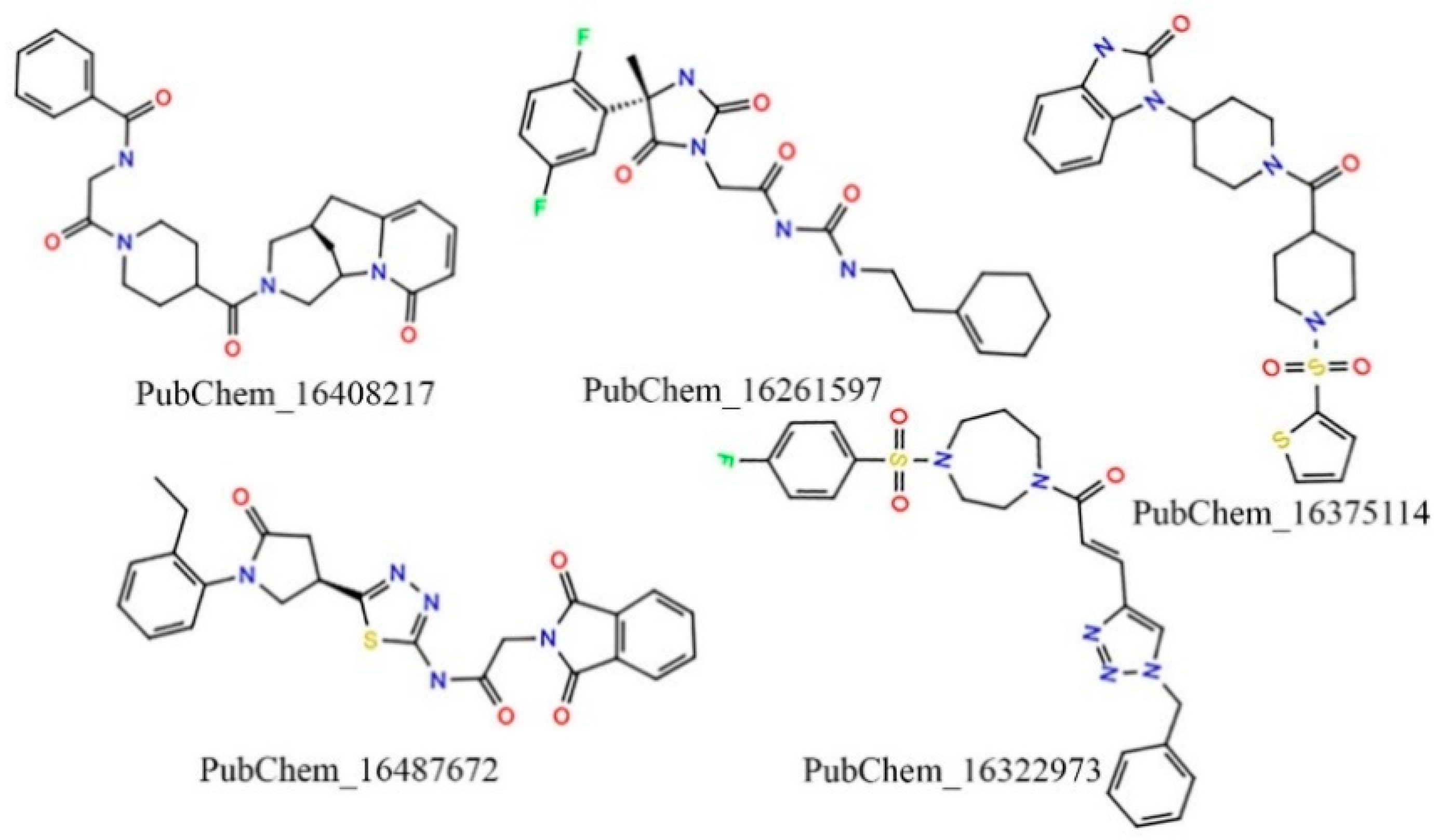
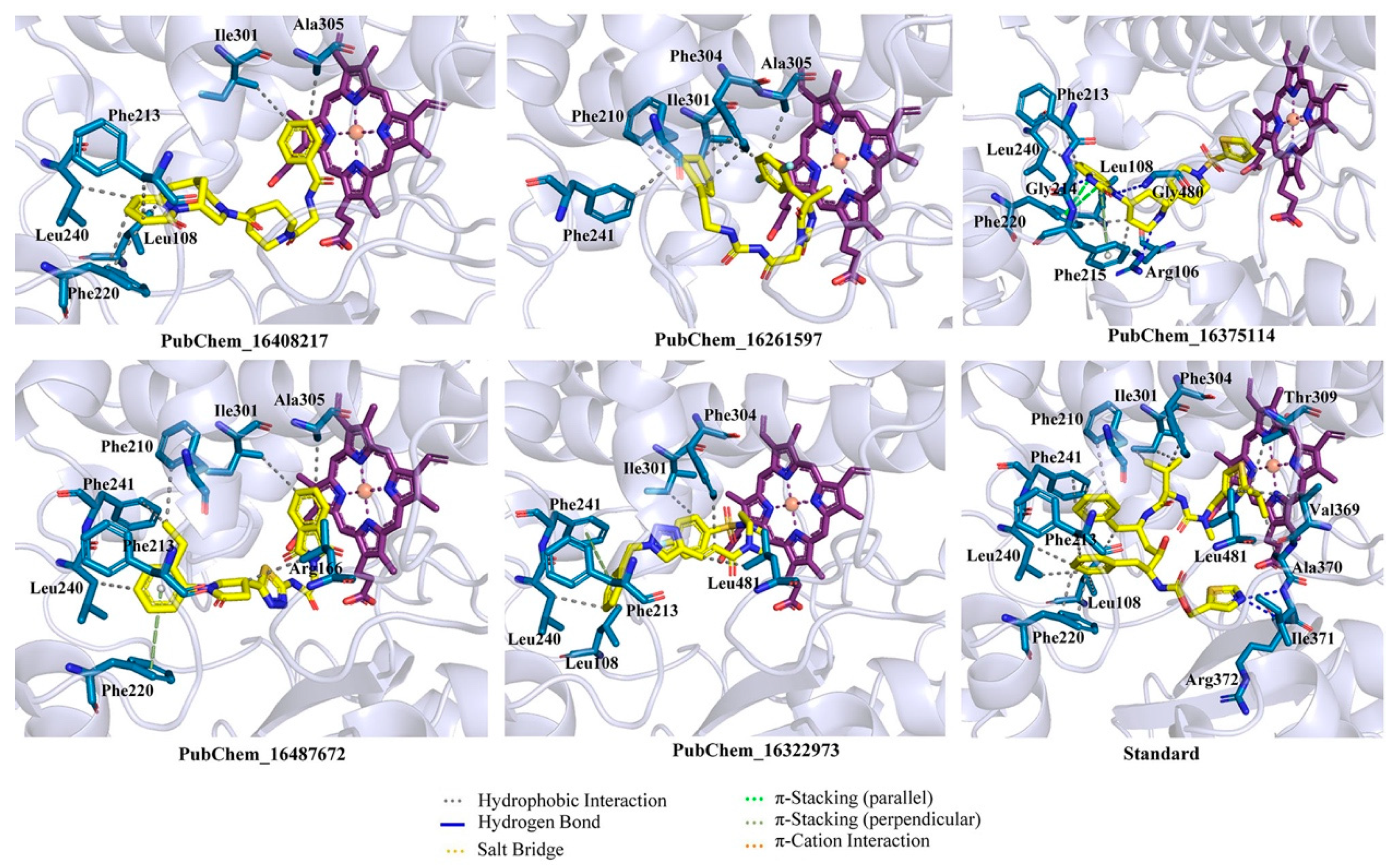
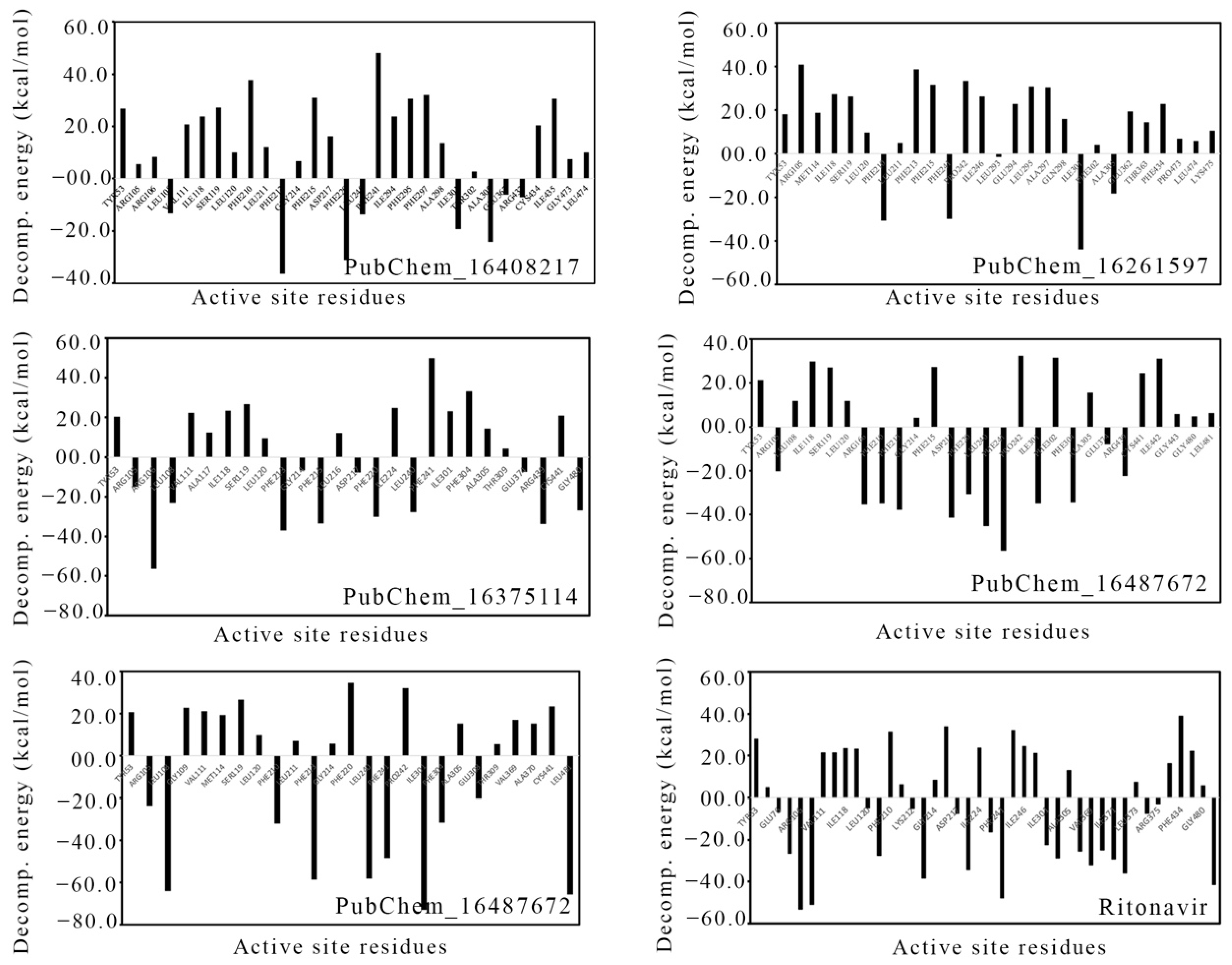
2.2. Binding Interaction Analysis
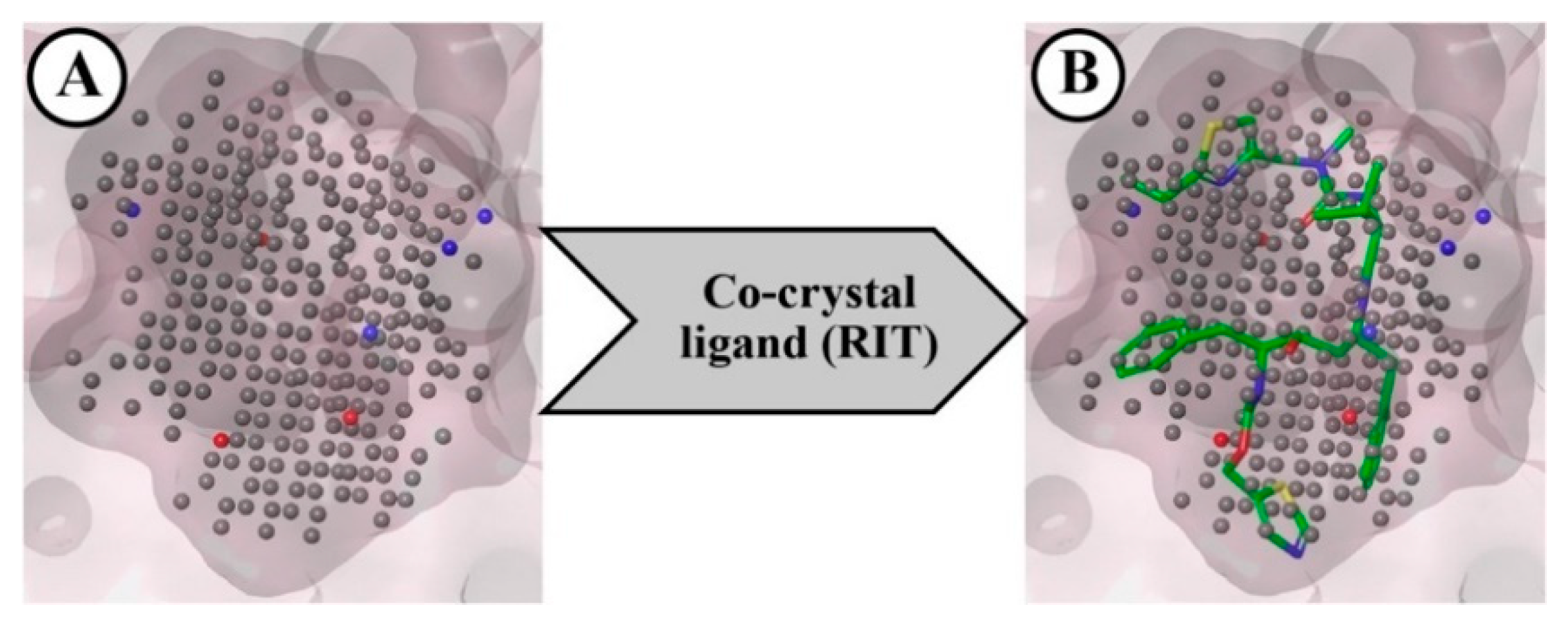
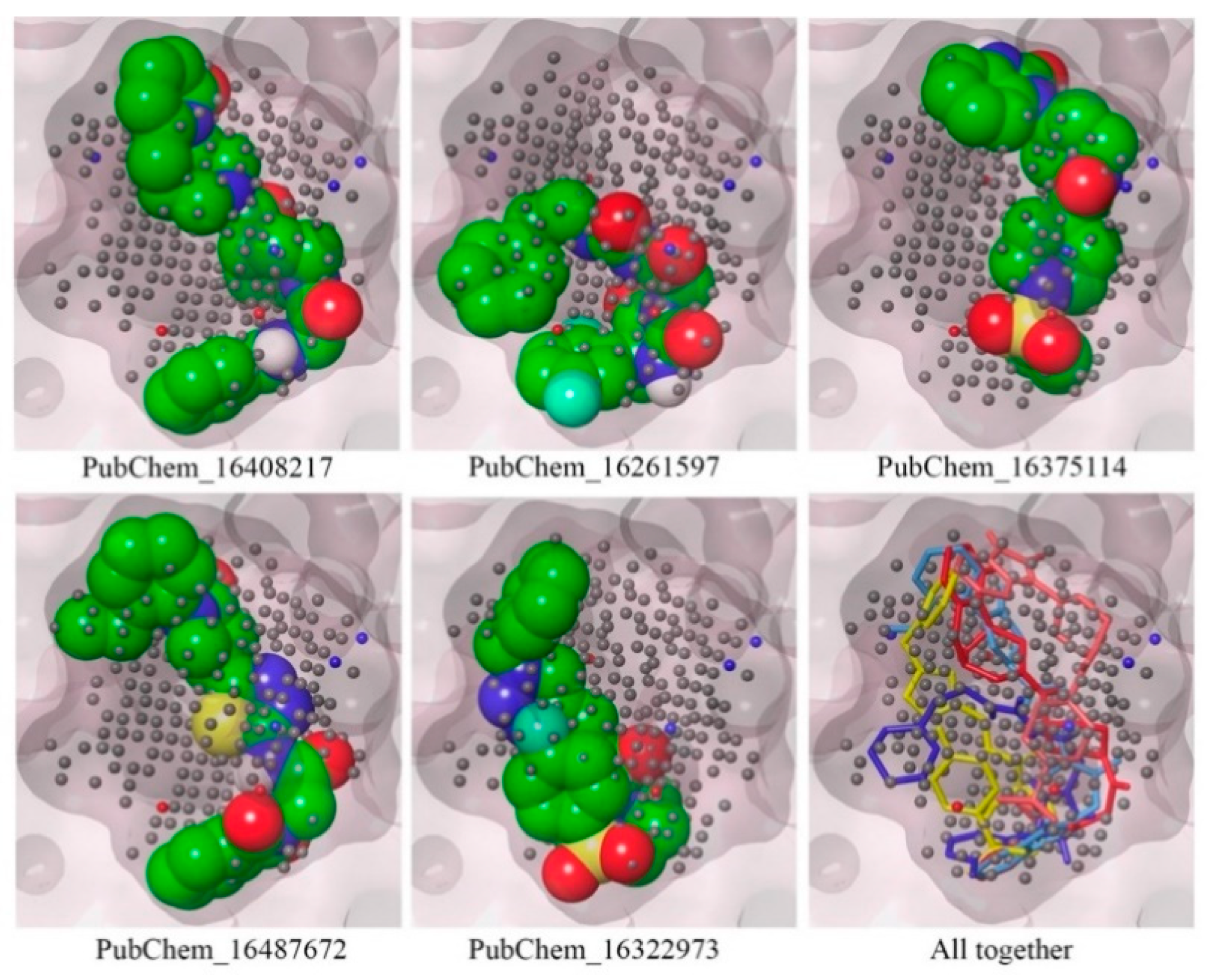
2.3. Mapping of Proposed Molecules on NIB Model
2.4. In Silico Pharmacokinetic and Drug-Likeness Assessment
2.5. Toxicity Assessment
2.6. Molecular Dynamics Simulation
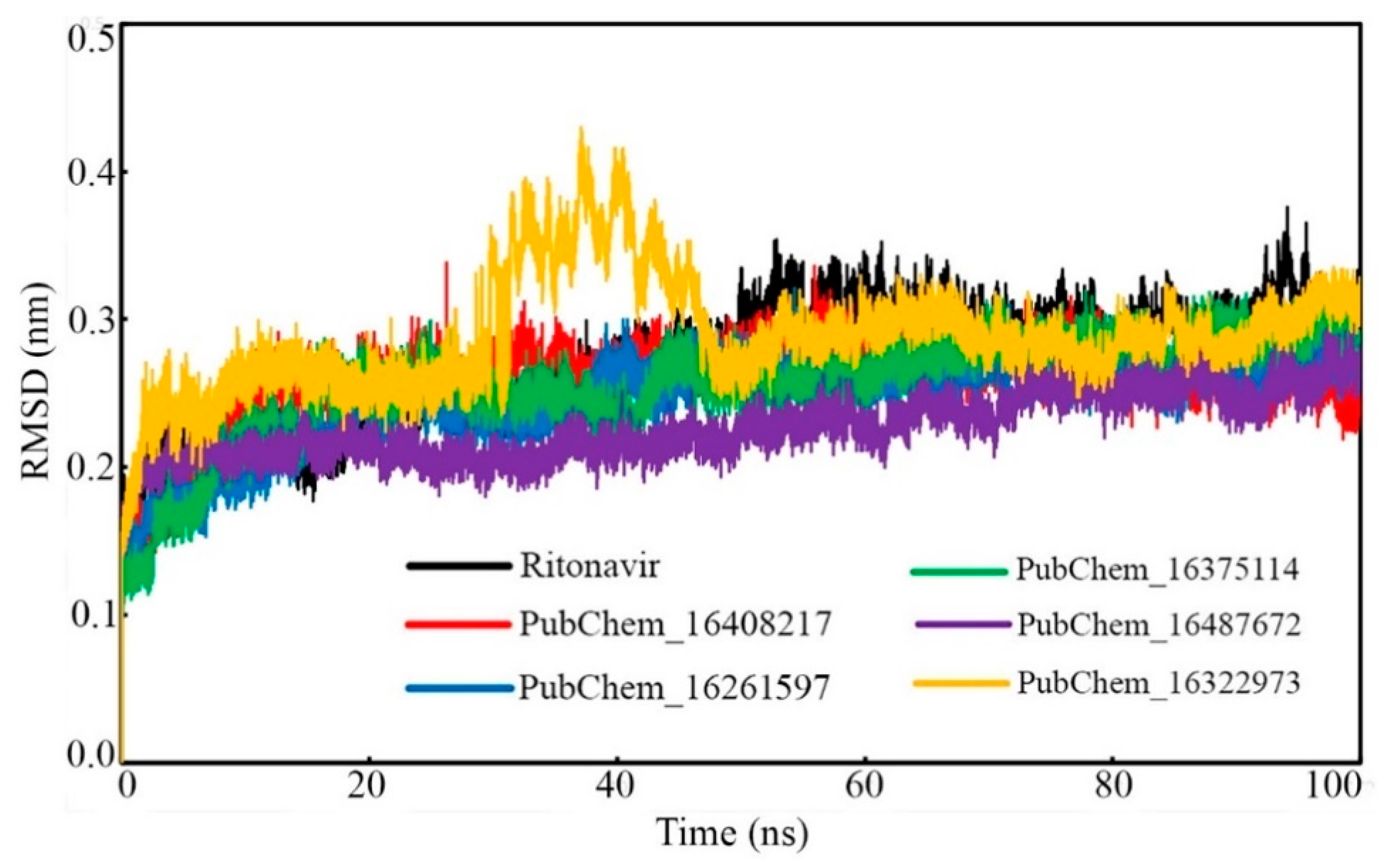
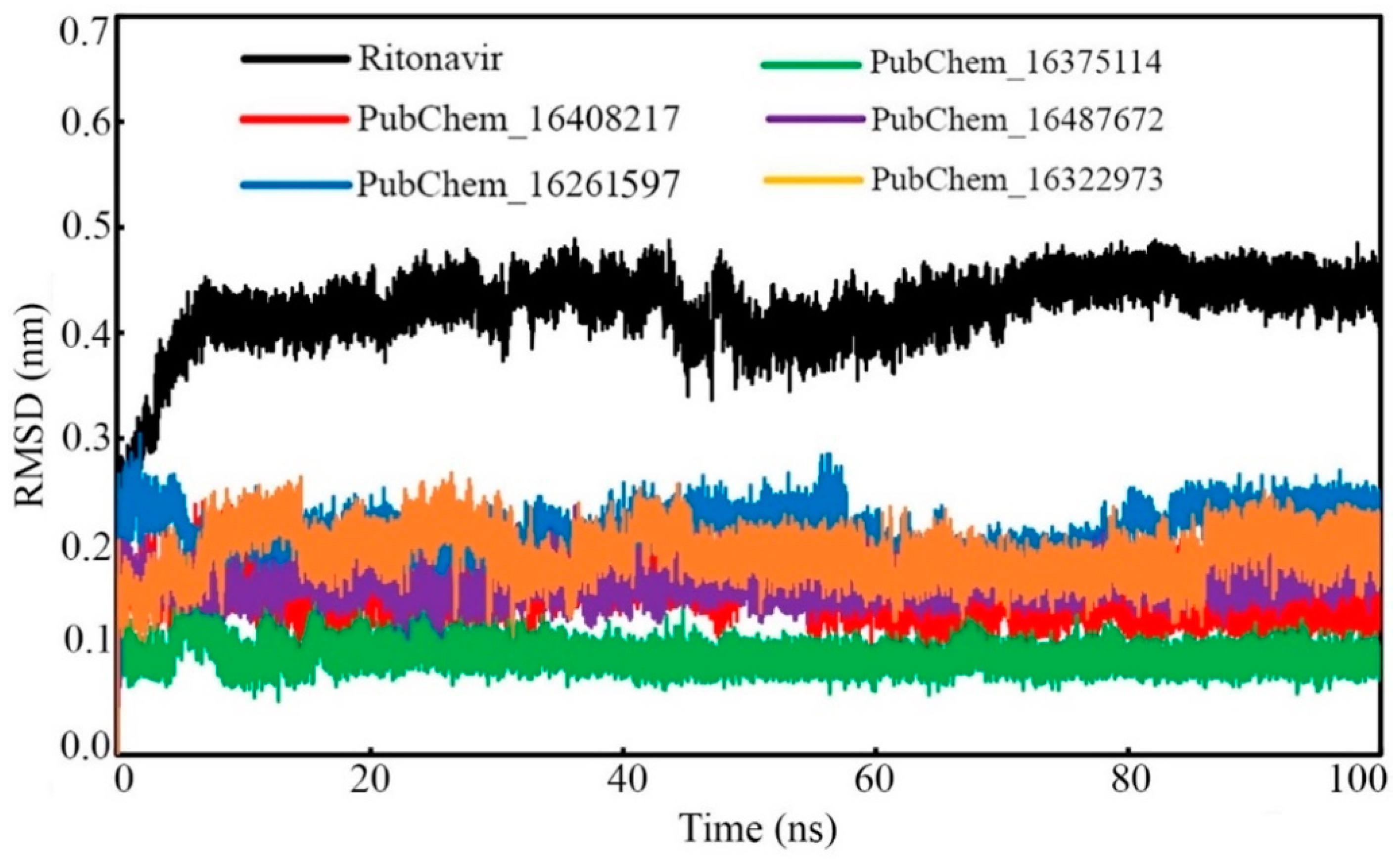
2.6.1. Root-Mean-Square Deviation (RMSD)
Protein Backbone RMSD
Ligand RMSD
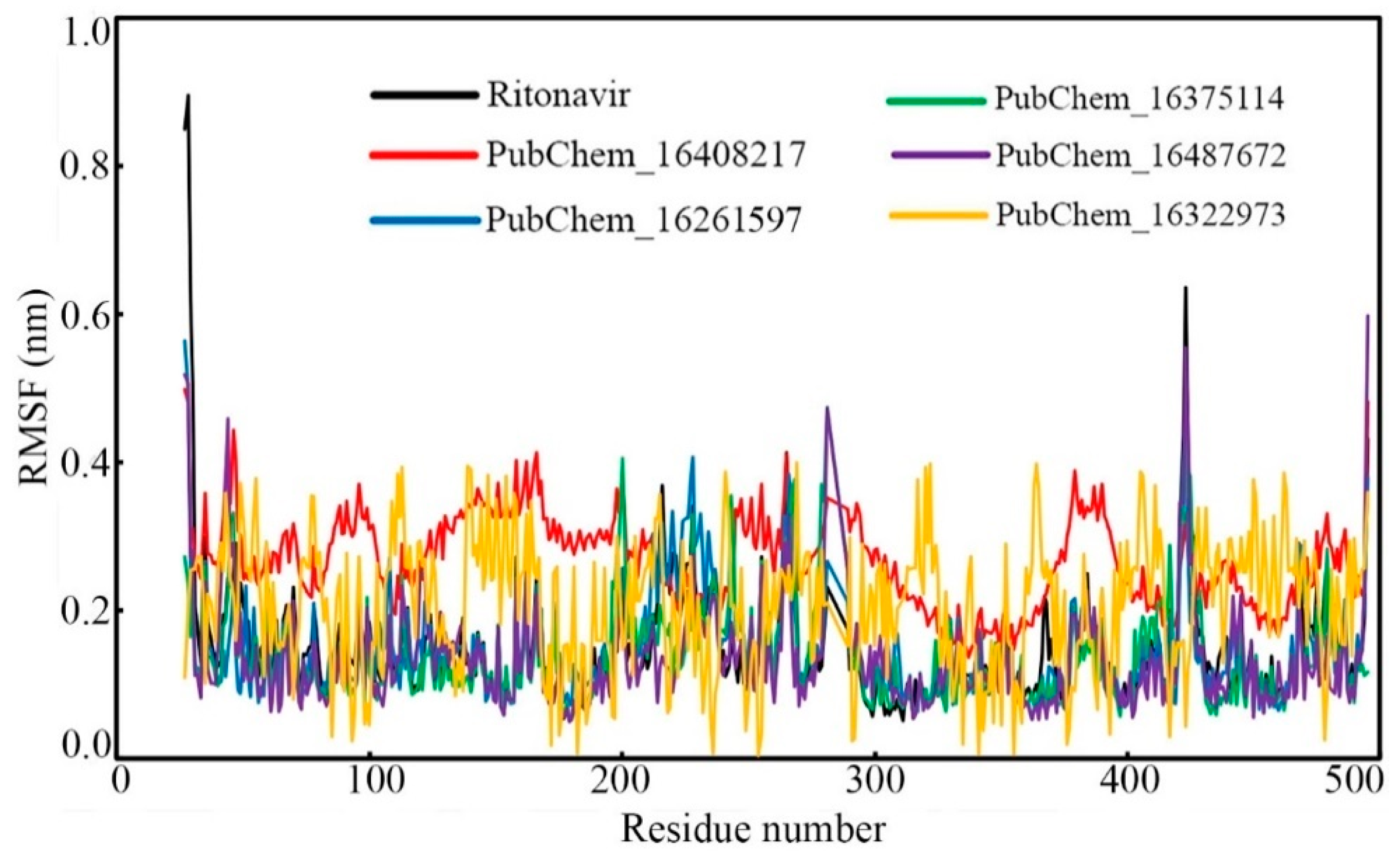
2.6.2. Root-Mean-Square Fluctuation (RMSF)
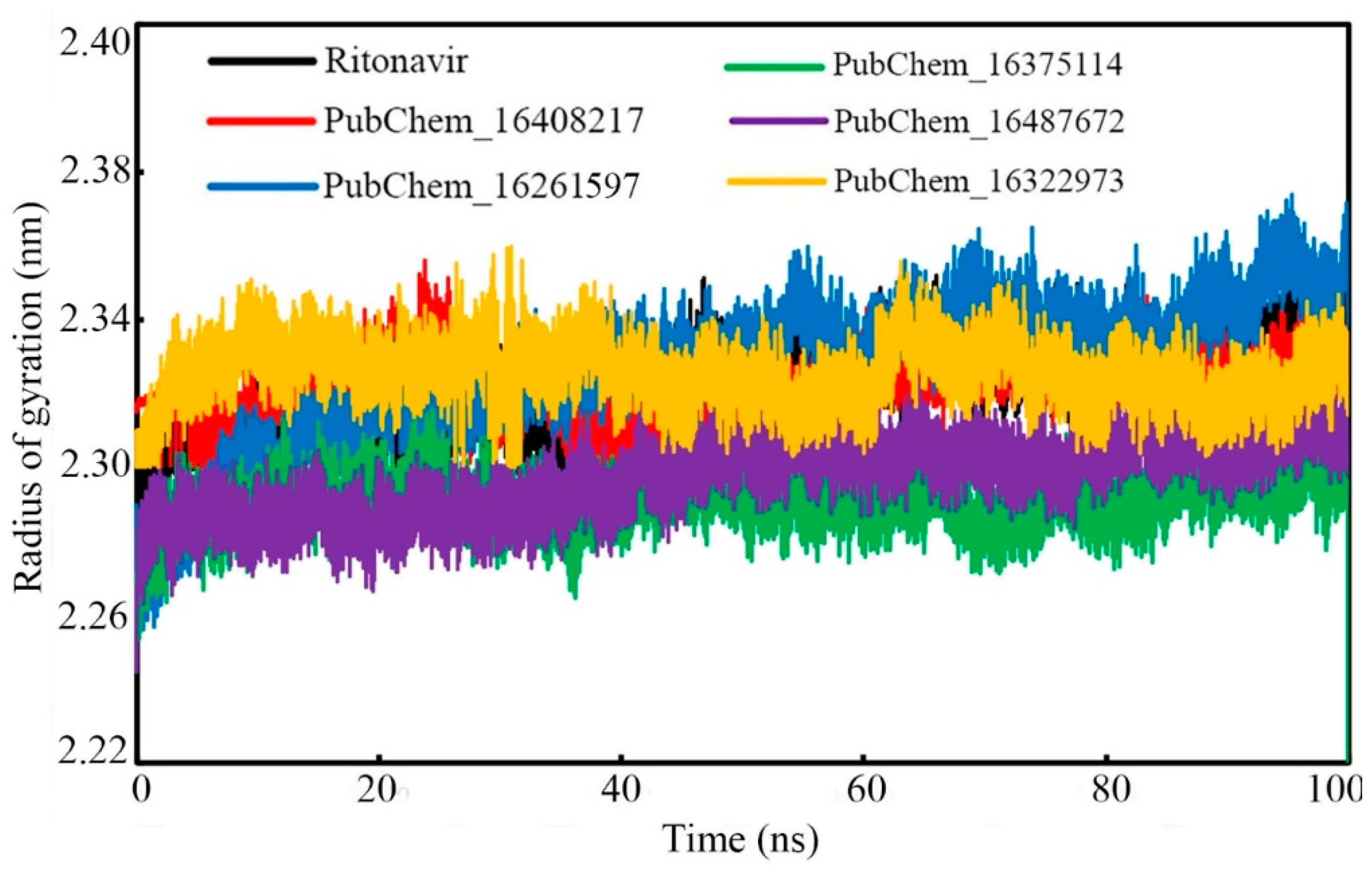
2.6.3. Radius of Gyration (RoG)
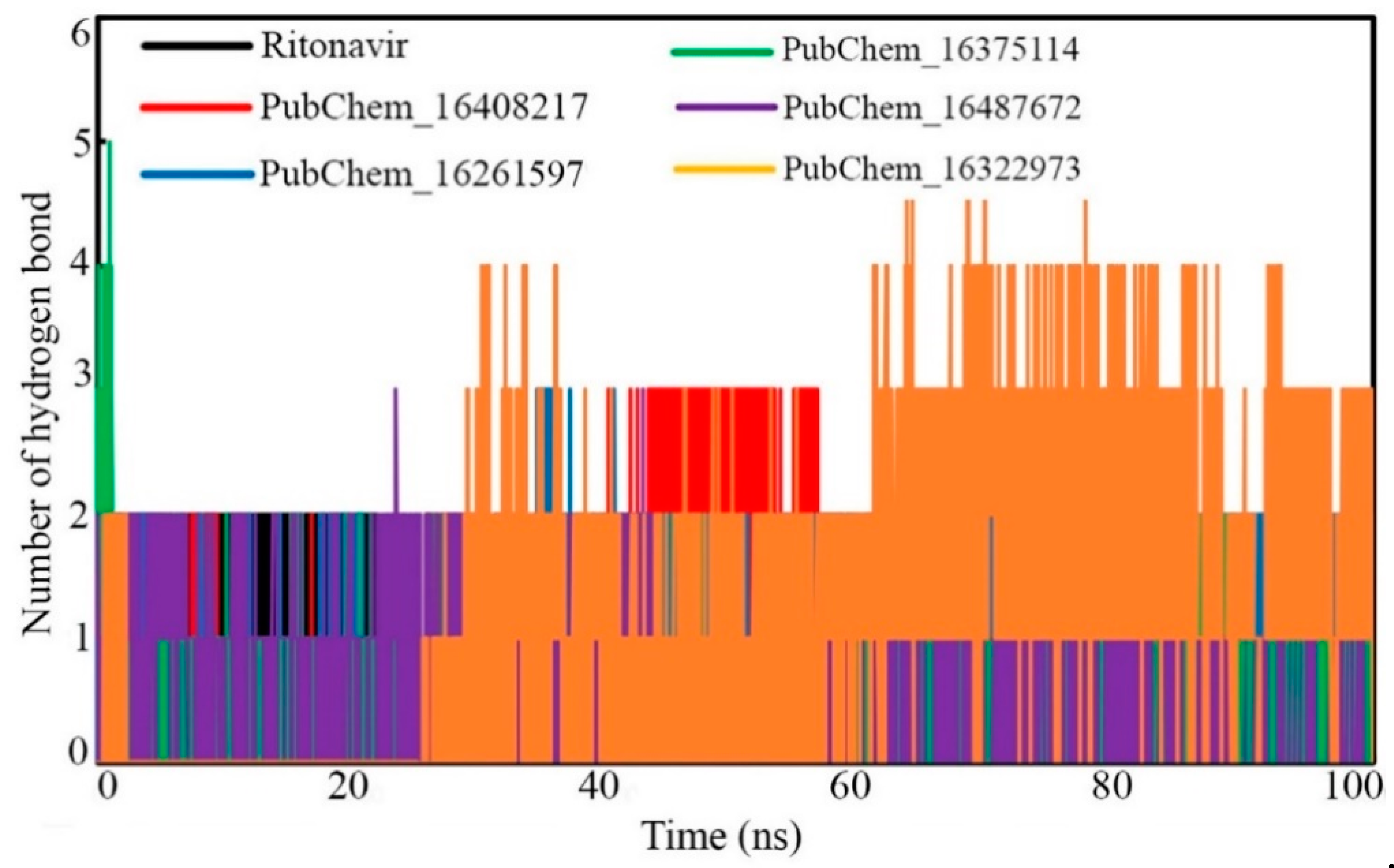
2.6.4. Hydrogen Bond Analysis
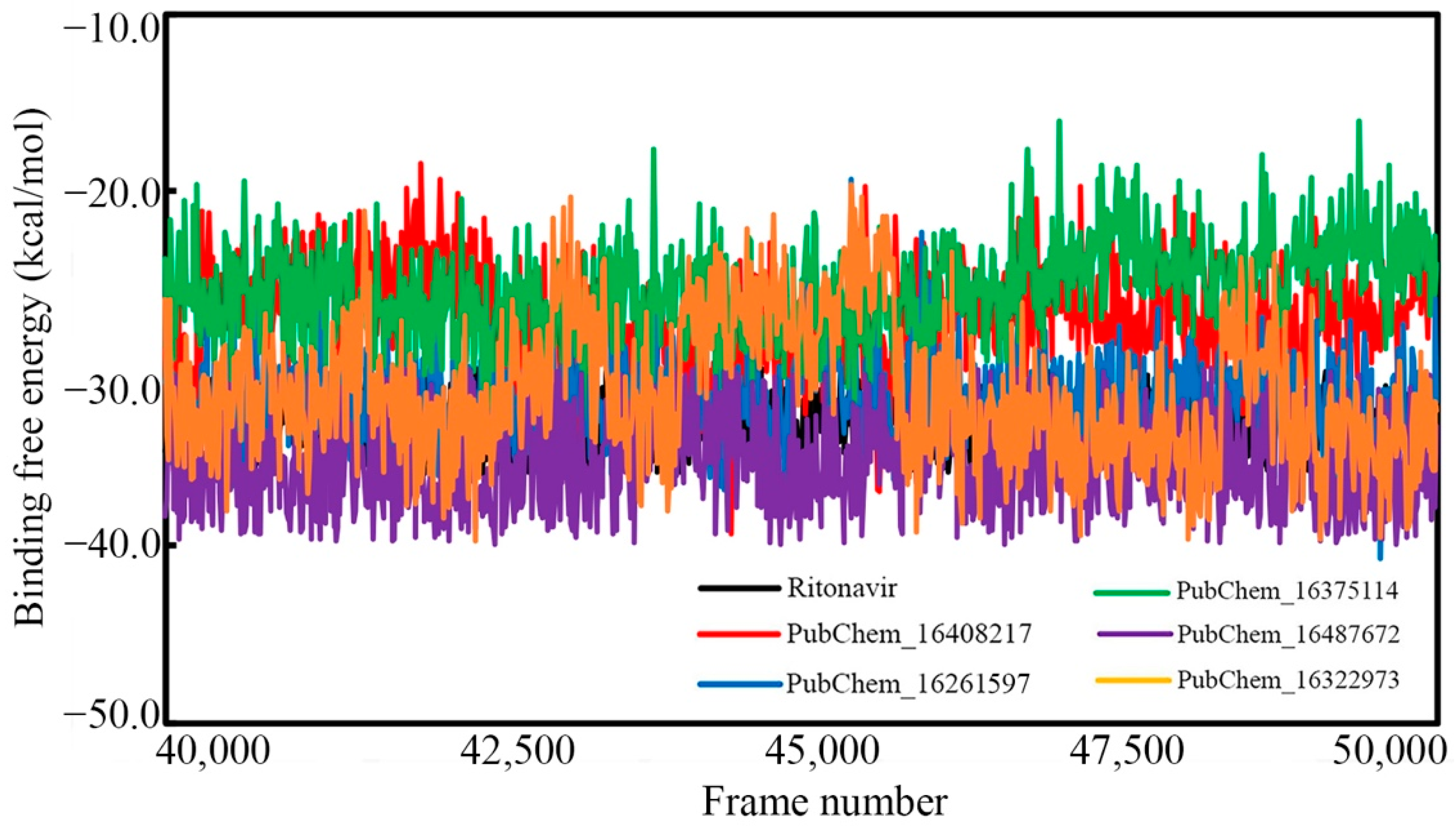
2.7. Binding Free Energy Using MM-GBSA Approach
3. Materials and Methods
3.1. Ligand Preparation
3.2. Protein Preparation
3.3. Molecular Docking
3.4. Negative Image-Based (NIB) Screening
3.5. Machine Learning-(ML) Based Screening
3.6. In Silico Pharmacokinetics and Toxicity Assessment
3.7. Molecular Dynamics (MD) Simulation
3.8. Molecular Mechanics Generalized Born Surface Area- (MM-GBSA) Based Binding Energy Calculation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References and Notes
- Bonam, S.R.; Sekar, M.; Guntuku, G.S.; Nerella, S.G.; Pawar, A.K.M.; Challa, S.R.; Eswara, G.K.; Mettu, S. Role of pharmaceutical sciences in future drug discovery. Futur. Drug Discov. 2021, 3, 1–15. [Google Scholar] [CrossRef]
- Tanrikulu, Y.; Krüger, B.; Proschak, E. The holistic integration of virtual screening in drug discovery. Drug Discov. Today 2013, 18, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Graff, D.E.; Shakhnovich, E.I.; Coley, C.W. Accelerating high-throughput virtual screening through molecular pool-based active learning. Chem. Sci. 2021, 12, 7866–7881. [Google Scholar] [CrossRef] [PubMed]
- Walters, W.P.; Wang, R. New trends in virtual screening. J. Chem. Inf. Model. 2020, 60, 4109–4111. [Google Scholar] [CrossRef]
- Mohs, R.C.; Greig, N.H. Drug discovery and development: Role of basic biological research. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2017, 3, 651–657. [Google Scholar] [CrossRef]
- de Souza Neto, L.R.; Moreira-Filho, J.T.; Neves, B.J.; Maidana, R.L.B.R.; Guimarães, A.C.R.; Furnham, N.; Andrade, C.H.; Silva, F.P. In silico Strategies to Support Fragment-to-Lead Optimization in Drug Discovery. Front. Chem. 2020, 8, 93. [Google Scholar] [CrossRef]
- Bhunia, S.S.; Saxena, M.; Saxena, A.K. Ligand- and Structure-Based Virtual Screening in Drug Discovery. In Topics in Medicinal Chemistry; Springer: Cham, Switzerland, 2021; Volume 37, pp. 281–339. [Google Scholar]
- Varela-Rial, A.; Majewski, M.; De Fabritiis, G. Structure based virtual screening: Fast and slow. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2022, 12, 1–17. [Google Scholar] [CrossRef]
- Bender, B.J.; Gahbauer, S.; Luttens, A.; Lyu, J.; Webb, C.M.; Stein, R.M.; Fink, E.A.; Balius, T.E.; Carlsson, J.; Irwin, J.J.; et al. A practical guide to large-scale docking. Nat. Protoc. 2021, 16, 4799–4832. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Venkatachalam, C.M.; Jiang, X.; Oldfield, T.; Waldman, M. LigandFit: A novel method for the shape-directed rapid docking of ligands to protein active sites. J. Mol. Graph. Model. 2003, 21, 289–307. [Google Scholar] [CrossRef]
- Rarey, M.; Kramer, B.; Lengauer, T.; Klebe, G. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 1996, 261, 470–489. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.C.; Nissink, J.W.M.; Taylor, R. Protein-ligand docking and virtual screening with GOLD. In Virtual Screening in Drug Discovery; CRC Press: Boca Raton, FL, USA, 2005; pp. 379–415. [Google Scholar]
- Molecular Operating Environment (MOE), 2013.08; Chemical Computing Group Inc.: Montreal, QC, Canada, 2013.
- Ahn, Y.; Jun, Y. Measurement of pain-like response to various NICU stimulants for high-risk infants. Early Hum. Dev. 2007, 83, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.L.; Shen, C.; Xiong, G.L.; Ding, J.J.; Lu, A.P.; Hou, T.J.; Cao, D.S. Improving docking-based virtual screening ability by integrating multiple energy auxiliary terms from molecular docking scoring. J. Chem. Inf. Model. 2020, 60, 4216–4230. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Ahinko, M.; Kurkinen, S.T.; Niinivehmas, S.P.; Pentikäinen, O.T.; Postila, P.A. A practical perspective: The effect of ligand conformers on the negative image-based screening. Int. J. Mol. Sci. 2019, 20, 2779. [Google Scholar] [CrossRef]
- McDonnell, A.M.; Dang, C.H. Basic Review of the Cytochrome P450 System. J. Adv. Pract. Oncol. 2013, 4, 263–268. [Google Scholar]
- Hakkola, J.; Hukkanen, J.; Turpeinen, M.; Pelkonen, O. Inhibition and induction of CYP enzymes in humans: An update. Arch. Toxicol. 2020, 94, 3671–3722. [Google Scholar] [CrossRef]
- Williams, J.A.; Hyland, R.; Jones, B.C.; Smith, D.A.; Hurst, S.; Goosen, T.C.; Peterkin, V.; Koup, J.R.; Ball, S.E. Drug-drug interactions for UDP-glucuronosyltransferase substrates: A pharmacokinetic explanation for typically observed low exposure (AUC 1/AUC) ratios. Drug Metab. Dispos. 2004, 32, 1201–1208. [Google Scholar] [CrossRef]
- Wienkers, L.C.; Heath, T.G. Predicting in vivo drug interactions from in vitro drug discovery data. Nat. Rev. Drug Discov. 2005, 4, 825–833. [Google Scholar] [CrossRef]
- Danielson, P.Á. The Cytochrome P450 Superfamily: Biochemistry, Evolution and Drug Metabolism in Humans. Curr. Drug Metab. 2005, 3, 561–597. [Google Scholar] [CrossRef] [PubMed]
- Dennison, J.B.; Kulanthaivel, P.; Barbuch, R.J.; Renbarger, J.L.; Ehlhardt, W.J.; Hall, S.D. Selective metabolism of vincristine in vitro by CYP3A5. Drug Metab. Dispos. 2006, 34, 1317–1327. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Hendrix, C.W.; Bumpus, N.N. Cytochrome P450 3A5 plays a prominent role in the oxidative metabolism of the anti-human immunodeficiency virus drug maraviroc. Drug Metab. Dispos. 2012, 40, 2221–2230. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.R.; Raza, A.; Firasat, S.; Abid, A. CYP3A5 gene polymorphisms and their impact on dosage and trough concentration of tacrolimus among kidney transplant patients: A systematic review and meta-analysis. Pharm. J. 2020, 20, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Hebert, M.F.; Isoherranen, N.; Davis, C.L.; Marsh, C.; Shen, D.D.; Thummel, K.E. Effect of CYP3A5 polymorphism on tacrolimus metabolic clearance in vitro. Drug Metab. Dispos. 2006, 34, 836–847. [Google Scholar] [CrossRef]
- Patel, J.K.; Kobashigawa, J.A. Tacrolimus in heart transplant recipients: An overview. BioDrugs 2007, 21, 139–143. [Google Scholar] [CrossRef]
- Zhang, Y.P.; Zuo, X.C.; Huang, Z.J.; Cai, J.J.; Wen, J.; Duan, D.D.; Yuan, H. CYP3A5 polymorphism, amlodipine and hypertension. J. Hum. Hypertens. 2014, 28, 145–149. [Google Scholar] [CrossRef]
- Wu, J.J.; Cao, Y.F.; Feng, L.; He, Y.Q.; Hong, J.Y.; Dou, T.Y.; Wang, P.; Hao, D.C.; Ge, G.B.; Yang, L. A Naturally Occurring Isoform-Specific Probe for Highly Selective and Sensitive Detection of Human Cytochrome P450 3A5. J. Med. Chem. 2017, 60, 3804–3813. [Google Scholar] [CrossRef]
- Niwa, T.; Narita, K.; Okamoto, A.; Murayama, N.; Yamazaki, H. Comparison of steroid hormone hydroxylations by and docking to human cytochromes P450 3A4 and 3A5. J. Pharm. Pharm. Sci. 2019, 22, 332–339. [Google Scholar] [CrossRef]
- Niwa, T.; Murayama, N.; Imagawa, Y.; Yamazaki, H. Comparison of catalytic and inhibitory properties in human drug-metabolizing cytochrome P450 3A4 and 3A5 for various compounds including endogenous steroid hormones and azole antifungals by molecular docking simulation. Drug Metab. Pharmacokinet. 2017, 32, S38. [Google Scholar] [CrossRef]
- Dai, Z.R.; Ning, J.; Sun, G.B.; Wang, P.; Zhang, F.; Ma, H.Y.; Zou, L.W.; Hou, J.; Wu, J.J.; Ge, G.B.; et al. Cytochrome P450 3A enzymes are key contributors for hepatic metabolism of bufotalin, a natural constitute in Chinese medicine Chansu. Front. Pharmacol. 2019, 10, 52. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Buchman, C.D.; Seetharaman, J.; Miller, D.J.; Huber, A.D.; Wu, J.; Chai, S.C.; Garcia-Maldonado, E.; Wright, W.C.; Chenge, J.; et al. Unraveling the Structural Basis of Selective Inhibition of Human Cytochrome P450 3A5. J. Am. Chem. Soc. 2021, 143, 18467–18480. [Google Scholar] [CrossRef]
- Vainio, M.J.; Puranen, J.S.; Johnson, M.S. ShaEP: Molecular overlay based on shape and electrostatic potential. J. Chem. Inf. Model. 2009, 49, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Gilson, M.K.; Liu, T.; Baitaluk, M.; Nicola, G.; Hwang, L.; Chong, J. BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016, 44, D1045–D1053. [Google Scholar] [CrossRef]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef] [PubMed]
- Yap, C.W. PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef]
- Quinlan, J.R. Simplifying decision trees. Int. J. Man. Mach. Stud. 1987, 27, 221–234. [Google Scholar] [CrossRef]
- Sperandei, S. Understanding logistic regression analysis. Biochem. Medica 2014, 24, 12–18. [Google Scholar] [CrossRef]
- Altman, N.S. An introduction to kernel and nearest-neighbor nonparametric regression. Am. Stat. 1992, 46, 175–185. [Google Scholar]
- Ho, T.K. Random decision forests. In Proceedings of the International Conference on Document Analysis and Recognition, ICDAR; IEEE: Manhattan, NY, USA, 1995; Volume 1, pp. 278–282. [Google Scholar]
- Cortes, C.; Vapnik, V. Support-Vector Networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Friedman, J.H. Greedy function approximation: A gradient boosting machine. Ann. Stat. 2001, 29, 1189–1232. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Saba, N.; Bhuyan, R.; Nandy, S.; Seal, A. Differential Interactions of Cytochrome P450 3A5 and 3A4 with Chemotherapeutic Agent-Vincristine: A Comparative Molecular Dynamics Study. Anticancer. Agents Med. Chem. 2015, 15, 475–483. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Polêto, M.D.; Rusu, V.H.; Grisci, B.I.; Dorn, M.; Lins, R.D.; Verli, H. Aromatic rings commonly used in medicinal chemistry: Force fields comparison and interactions with water toward the design of New Chemical Entities. Front. Pharmacol. 2018, 9, 395. [Google Scholar] [CrossRef]
- Vázquez, J.; López, M.; Gibert, E.; Herrero, E.; Javier Luque, F. Merging ligand-based and structure-based methods in drug discovery: An overview of combined virtual screening approaches. Molecules 2020, 25, 4723. [Google Scholar] [CrossRef]
- Shoichet, B.K. Virtual screening of chemical libraries. Nature 2004, 432, 862–865. [Google Scholar] [CrossRef]
- Schneider, G. Virtual screening: An endless staircase? Nat. Rev. Drug Discov. 2010, 9, 273–276. [Google Scholar] [CrossRef]
- Westermaier, Y.; Barril, X.; Scapozza, L. Virtual screening: An in silico tool for interlacing the chemical universe with the proteome. Methods 2015, 71, 44–57. [Google Scholar] [CrossRef]
- Liu, X.; Shi, D.; Zhou, S.; Liu, H.; Liu, H.; Yao, X. Molecular dynamics simulations and novel drug discovery. Expert Opin. Drug Discov. 2018, 13, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Torres, P.H.M.; Sodero, A.C.R.; Jofily, P.; Silva-Jr, F.P. Key topics in molecular docking for drug design. Int. J. Mol. Sci. 2019, 20, 4574. [Google Scholar] [CrossRef] [PubMed]
- Kontoyianni, M. Docking and virtual screening in drug discovery. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2017; Volume 1647, pp. 255–266. [Google Scholar]
- Jokinen, E.M.; Postila, P.A.; Ahinko, M.; Niinivehmas, S.; Pentikäinen, O.T. Fragment- and negative image-based screening of phosphodiesterase 10A inhibitors. Chem. Biol. Drug Des. 2019, 94, 1799–1812. [Google Scholar] [CrossRef] [PubMed]
- Adeshina, Y.O.; Deeds, E.J.; Karanicolas, J. Machine learning classification can reduce false positives in structure-based virtual screening. Proc. Natl. Acad. Sci. USA 2020, 117, 18477–18488. [Google Scholar] [CrossRef]
- Gupta, A.; Zhou, H.X. Machine Learning-Enabled Pipeline for Large-Scale Virtual Drug Screening. J. Chem. Inf. Model. 2021, 61, 4236–4244. [Google Scholar] [CrossRef]
- Ekins, S.; Mestres, J.; Testa, B. In silico pharmacology for drug discovery: Methods for virtual ligand screening and profiling. Br. J. Pharmacol. 2007, 152, 9–20. [Google Scholar] [CrossRef]
- Rim, K.T. In silico prediction of toxicity and its applications for chemicals at work. Toxicol. Environ. Health Sci. 2020, 12, 191–202. [Google Scholar] [CrossRef]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Manallack, D.T.; Prankerd, R.J.; Yuriev, E.; Oprea, T.I.; Chalmers, D.K. The significance of acid/base properties in drug discovery. Chem. Soc. Rev. 2013, 42, 485–496. [Google Scholar] [CrossRef]
- Gasteiger, J.; Marsili, M. A new model for calculating atomic charges in molecules. Tetrahedron Lett. 1978, 19, 3181–3184. [Google Scholar] [CrossRef]
- Berman, H.; Henrick, K.; Nakamura, H. Announcing the worldwide Protein Data Bank. Nat. Struct. Biol. 2003, 10, 980. [Google Scholar] [CrossRef]
- Hsu, M.H.; Savas, U.; Johnson, E.F. The X-ray crystal structure of the human mono-oxygenase cytochrome P450 3A5-ritonavir complex reveals active site differences between P450s 3A4 and 3A5. Mol. Pharmacol. 2018, 93, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Cross, J.B.; Thompson, D.C.; Rai, B.K.; Baber, J.C.; Fan, K.Y.; Hu, Y.; Humblet, C. Comparison of several molecular docking programs: Pose prediction and virtual screening accuracy. J. Chem. Inf. Model. 2009, 49, 1455–1474. [Google Scholar] [CrossRef] [PubMed]
- Erickson, J.A.; Jalaie, M.; Robertson, D.H.; Lewis, R.A.; Vieth, M. Lessons in Molecular Recognition: The Effects of Ligand and Protein Flexibility on Molecular Docking Accuracy. J. Med. Chem. 2004, 47, 45–55. [Google Scholar] [CrossRef]
- Mishra, A. AWS Lambda. In Machine Learning in the AWS Cloud; John Wiley & Sons: Hoboken, NJ, USA, 2019; pp. 237–255. [Google Scholar]
- Al-Sayyed, R.M.H.; Hijawi, W.A.; Bashiti, A.M.; Al Jarah, I.; Obeid, N.; Adwan, O.Y. An Investigation of Microsoft Azure and Amazon web services from users’ perspectives. Int. J. Emerg. Technol. Learn. 2019, 14, 218–241. [Google Scholar] [CrossRef]
- Islam, M.A.; Subramanyam Rallabandi, V.P.; Mohammed, S.; Srinivasan, S.; Natarajan, S.; Dudekula, D.B.; Park, J. Screening of β1-and β2-adrenergic receptor modulators through advanced pharmacoinformatics and machine learning approaches. Int. J. Mol. Sci. 2021, 22, 11191. [Google Scholar] [CrossRef]
- Niinivehmas, S.P.; Salokas, K.; Lätti, S.; Raunio, H.; Pentikäinen, O.T. Ultrafast protein structure-based virtual screening with Panther. J. Comput. Aided. Mol. Des. 2015, 29, 989–1006. [Google Scholar] [CrossRef] [PubMed]
- McNemar, Q. Note on the sampling error of the difference between correlated proportions or percentages. Psychometrika 1947, 12, 153–157. [Google Scholar] [CrossRef]
- Weininger, D. SMILES, a Chemical Language and Information System: 1: Introduction to Methodology and Encoding Rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Lindahl, A.; Hess VD, S.; van der Spoel, D. GROMACS 2021.3 Source Code 2021. Zenodo April 2020, 302020. [Google Scholar]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. Charmm-Gui: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; De Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2016, 14, 71–73. [Google Scholar] [CrossRef]
- He, X.; Man, V.H.; Yang, W.; Lee, T.S.; Wang, J. A fast and high-quality charge model for the next generation general AMBER force field. J. Chem. Phys. 2020, 153, 114502. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. Gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Sitkoff, D.; Sharp, K.A.; Honig, B. Accurate calculation of hydration free energies using macroscopic solvent models. J. Phys. Chem. 1994, 98, 1978–1988. [Google Scholar] [CrossRef]
- Tan, C.; Tan, Y.H.; Luo, R. Implicit nonpolar solvent models. J. Phys. Chem. B 2007, 111, 12263–12274. [Google Scholar] [CrossRef]
- Gohlke, H.; Kiel, C.; Case, D.A. Insights into protein-protein binding by binding free energy calculation and free energy decomposition for the Ras-Raf and Ras-RalGDS complexes. J. Mol. Biol. 2003, 330, 891–913. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Landrum, G. RDKit: Open-Source Cheminformatics Software. Available online: http://Www.Rdkit.Org/ (accessed on 20 December 2021).
- Steffen, C.; Thomas, K.; Huniar, U.; Hellweg, A.; Rubner, O.; Schroer, A. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar]
- ChemAxon Marvin Sketch.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classifier | Precision | Recall | F-Score | Accuracy | Specificity |
|---|---|---|---|---|---|
| SVM | 0.83 ± 0.01 | 0.80 ± 0.02 | 0.78 ± 0.02 | 0.82 ± 0.01 | 0.81 ± 0.01 |
| RF | 0.74 ± 0.01 | 0.61 ± 0.01 | 0.57 ± 0.01 | 0.67 ± 0.01 | 0.74 ± 0.01 |
| KNN | 0.54 ± 0.01 | 0.51 ± 0.02 | 0.48 ± 0.02 | 0.52 ± 0.01 | 0.54 ± 0.01 |
| GBM | 0.84 ± 0.02 | 0.81 ± 0.02 | 0.76 ± 0.01 | 0.82 ± 0.01 | 0.83 ± 0.02 |
| DT | 0.73 ± 0.01 | 0.61 ± 0.01 | 0.54 ± 0.01 | 0.67 ± 0.01 | 0.71 ± 0.01 |
| LR | 0.69 ± 0.01 | 0.62 ± 0.02 | 0.52 ± 0.01 | 0.66 ± 0.01 | 0.68 ± 0.01 |
| Mols. | Sol | GI | BBB | SA | HA | AHA | RB | HBA | HBD | MR | TPSA |
|---|---|---|---|---|---|---|---|---|---|---|---|
| PubChem_16408217 | Soluble | High | No | 4.46 | 34 | 12 | 7 | 4 | 1 | 133.24 | 91.72 |
| PubChem_16261597 | Soluble | High | No | 5.40 | 31 | 6 | 8 | 6 | 3 | 112.01 | 107.61 |
| PubChem_16375114 | Soluble | High | No | 4.11 | 32 | 14 | 5 | 4 | 1 | 130.87 | 132.10 |
| PubChem_16487672 | Soluble | High | No | 3.83 | 34 | 17 | 7 | 6 | 1 | 133.76 | 140.81 |
| PubChem_16322973 | Soluble | High | No | 4.12 | 33 | 17 | 7 | 6 | 0 | 128.04 | 96.78 |
| Mols | AMES Toxicity | MTD (Human) | ORAT (LD50) | Minnow Toxicity | SS |
|---|---|---|---|---|---|
| PubChem_16408217 | No | −0.695 | 2.758 | 3.037 | No |
| PubChem_16261597 | No | 0.139 | 2.501 | 3.995 | No |
| PubChem_16375114 | No | −0.065 | 2.739 | 1.961 | No |
| PubChem_16487672 | No | −0.138 | 2.13 | 0.889 | No |
| PubChem_16322973 | No | 0.375 | 2.622 | −0.227 | No |
| Ritonavir | M1 | M2 | M3 | M4 | M5 | ||
|---|---|---|---|---|---|---|---|
| Backbone RMSD (nm) | Min. | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| Max. | 0.376 | 0.338 | 0.321 | 0.322 | 0.297 | 0.430 | |
| Average | 0.269 | 0.258 | 0.247 | 0.256 | 0.227 | 0.288 | |
| RMSF (nm) | Min. | 0.051 | 0.136 | 0.054 | 0.052 | 0.050 | 0.003 |
| Max. | 0.895 | 0.498 | 0.563 | 0.473 | 0.598 | 0.398 | |
| Average | 0.150 | 0.266 | 0.146 | 0.137 | 0.133 | 0.208 | |
| Ligand RMSD (nm) | Min. | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| Max. | 0.489 | 0.248 | 0.304 | 0.190 | 0.234 | 0.267 | |
| Average | 0.423 | 0.151 | 0.211 | 0.094 | 0.162 | 0.191 | |
| RoG (nm) | Min. | 2.256 | 2.300 | 2.254 | 2.205 | 2.245 | 2.300 |
| Max. | 2.357 | 2.363 | 2.374 | 2.316 | 2.328 | 2.360 | |
| Average | 2.322 | 2.323 | 2.327 | 2.291 | 2.297 | 2.325 |
| Molecule | Total ΔGbind (kcal/mol) | Standard Deviation |
|---|---|---|
| Ritonavir | −33.015 | ±1.578 |
| PubChem_16408217 | −27.165 | ±2.840 |
| PubChem_16261597 | −31.826 | ±2.437 |
| PubChem_16375114 | −25.557 | ±2.617 |
| PubChem_16487672 | −35.386 | ±2.633 |
| PubChem_16322973 | −31.378 | ±3.918 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Islam, M.A.; Dudekula, D.B.; Rallabandi, V.P.S.; Srinivasan, S.; Natarajan, S.; Chung, H.; Park, J. Identification of Potential Cytochrome P450 3A5 Inhibitors: An Extensive Virtual Screening through Molecular Docking, Negative Image-Based Screening, Machine Learning and Molecular Dynamics Simulation Studies. Int. J. Mol. Sci. 2022, 23, 9374. https://doi.org/10.3390/ijms23169374
Islam MA, Dudekula DB, Rallabandi VPS, Srinivasan S, Natarajan S, Chung H, Park J. Identification of Potential Cytochrome P450 3A5 Inhibitors: An Extensive Virtual Screening through Molecular Docking, Negative Image-Based Screening, Machine Learning and Molecular Dynamics Simulation Studies. International Journal of Molecular Sciences. 2022; 23(16):9374. https://doi.org/10.3390/ijms23169374
Chicago/Turabian StyleIslam, Md Ataul, Dawood Babu Dudekula, V. P. Subramanyam Rallabandi, Sridhar Srinivasan, Sathishkumar Natarajan, Hoyong Chung, and Junhyung Park. 2022. "Identification of Potential Cytochrome P450 3A5 Inhibitors: An Extensive Virtual Screening through Molecular Docking, Negative Image-Based Screening, Machine Learning and Molecular Dynamics Simulation Studies" International Journal of Molecular Sciences 23, no. 16: 9374. https://doi.org/10.3390/ijms23169374
APA StyleIslam, M. A., Dudekula, D. B., Rallabandi, V. P. S., Srinivasan, S., Natarajan, S., Chung, H., & Park, J. (2022). Identification of Potential Cytochrome P450 3A5 Inhibitors: An Extensive Virtual Screening through Molecular Docking, Negative Image-Based Screening, Machine Learning and Molecular Dynamics Simulation Studies. International Journal of Molecular Sciences, 23(16), 9374. https://doi.org/10.3390/ijms23169374