
Aspiletrein A Induces Apoptosis Cell Death via Increasing Reactive Oxygen Species Generation and AMPK Activation in Non-Small-Cell Lung Cancer Cells

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
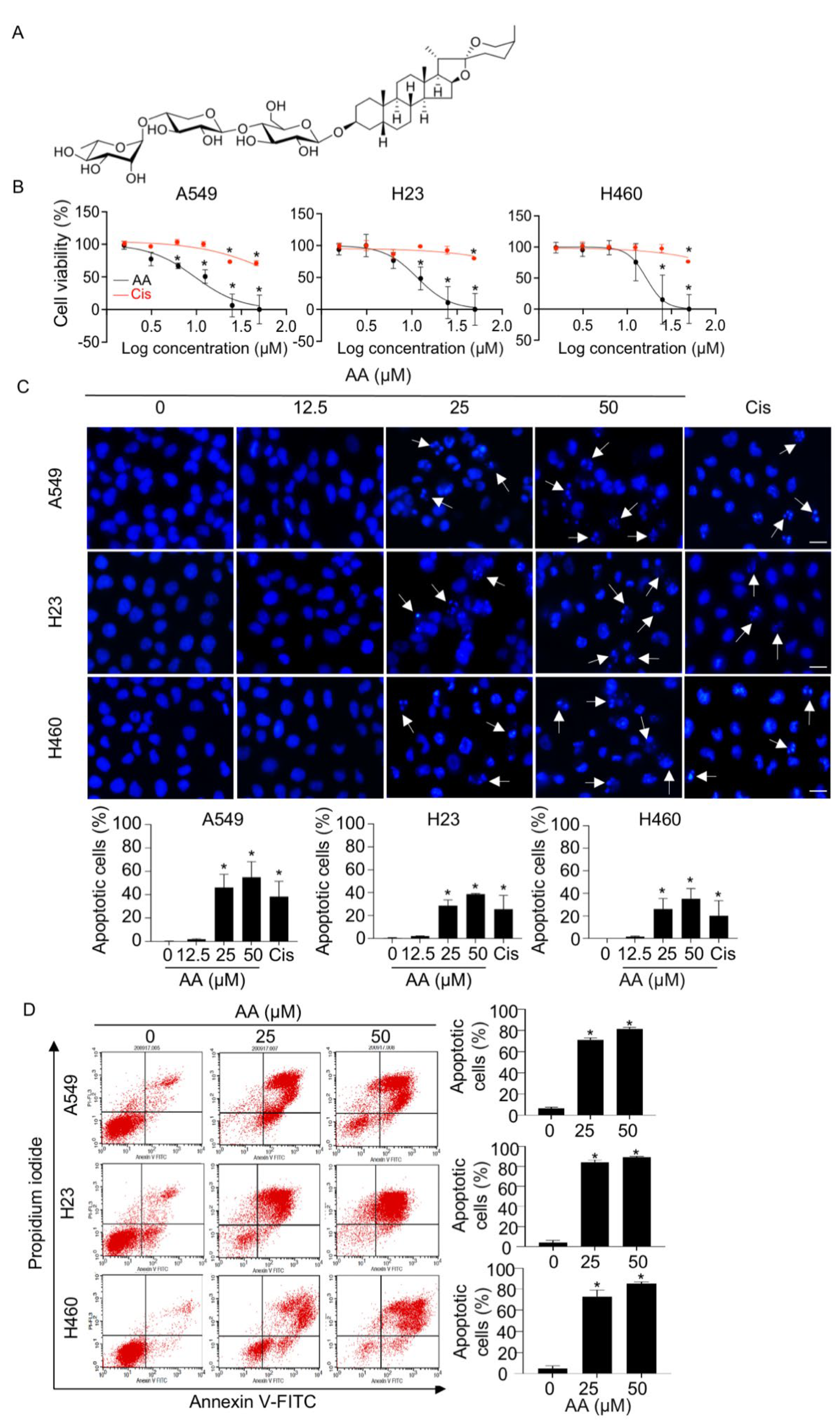
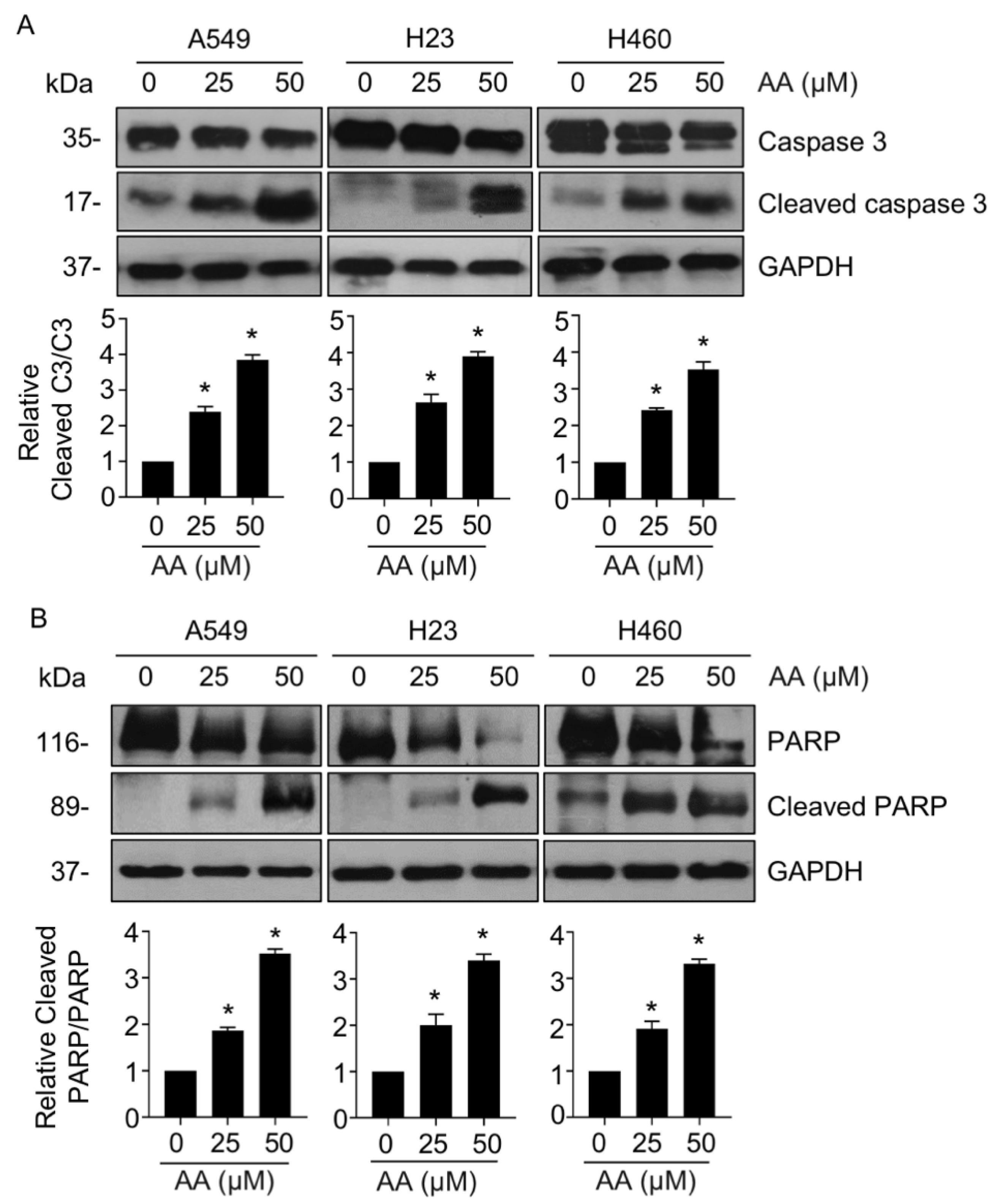
2.1. AA Mediates Cell Death via an Apoptotic Mechanism in NSCLC
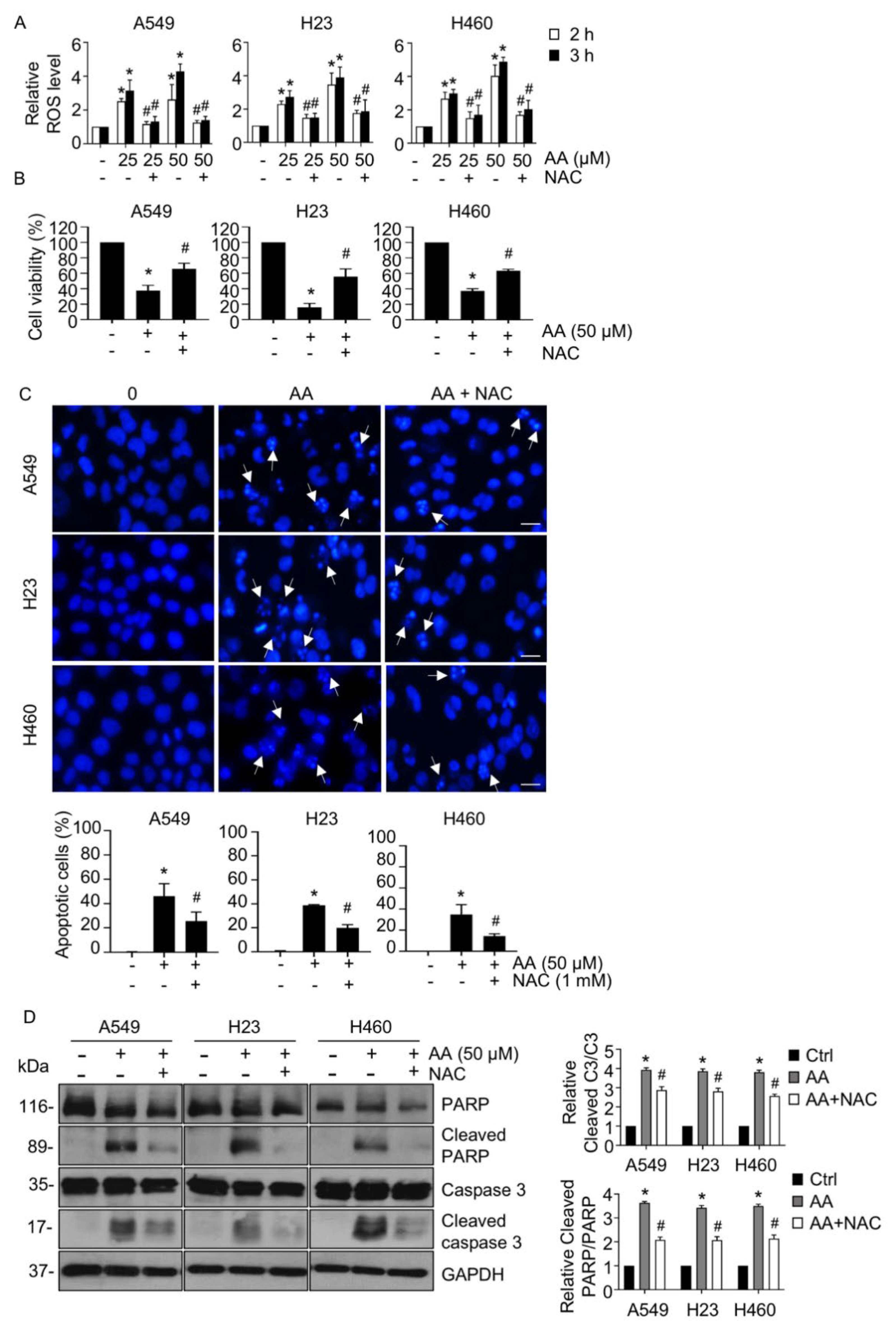
2.2. AA Induced Apoptosis by Increasing ROS Generation
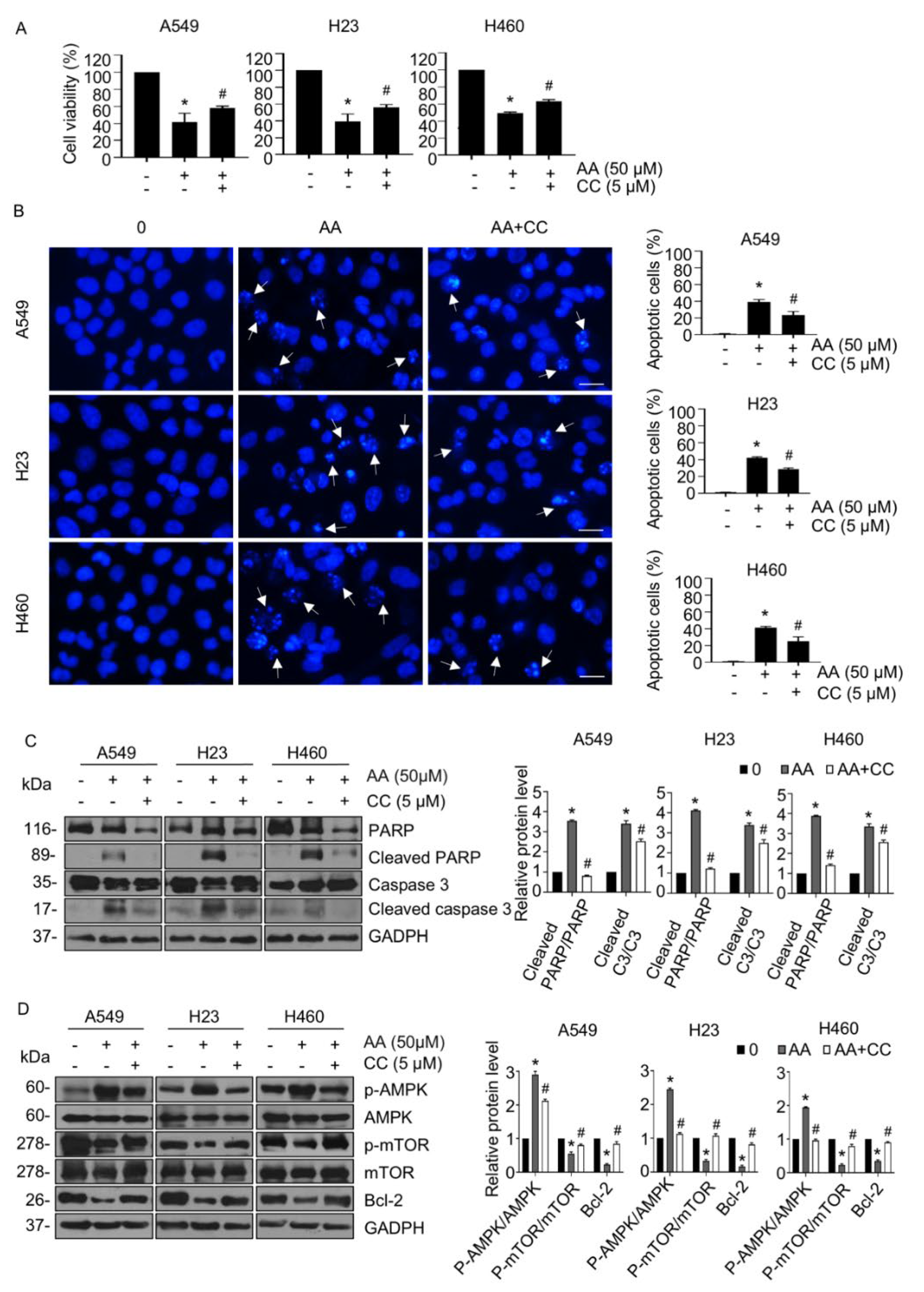
2.3. AA Induced Apoptosis through Activation of AMPK Signaling in a ROS-Dependent Manner
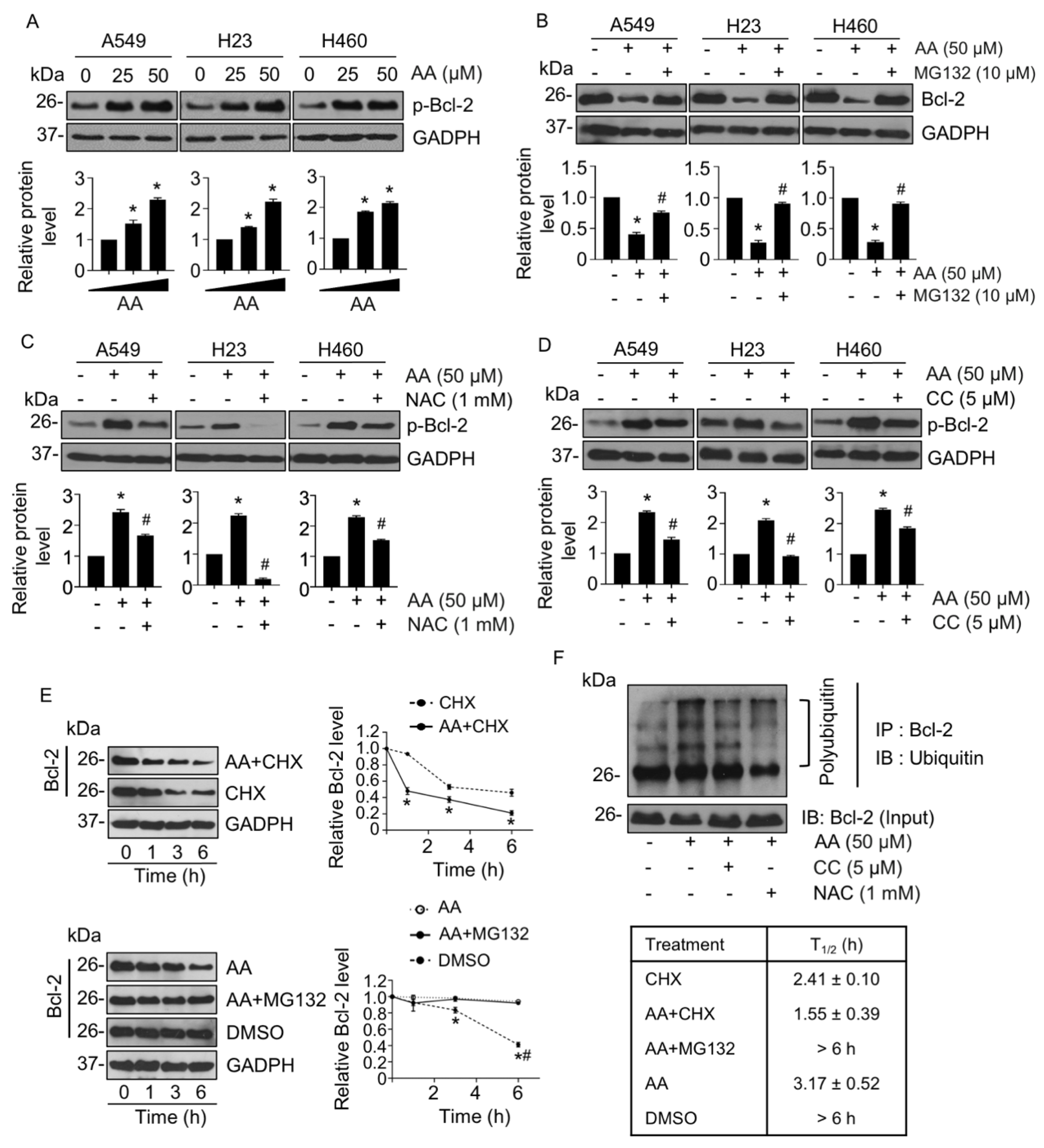
2.4. AA-Mediated Anti-Apoptotic Bcl-2 Proteasomal Degradation
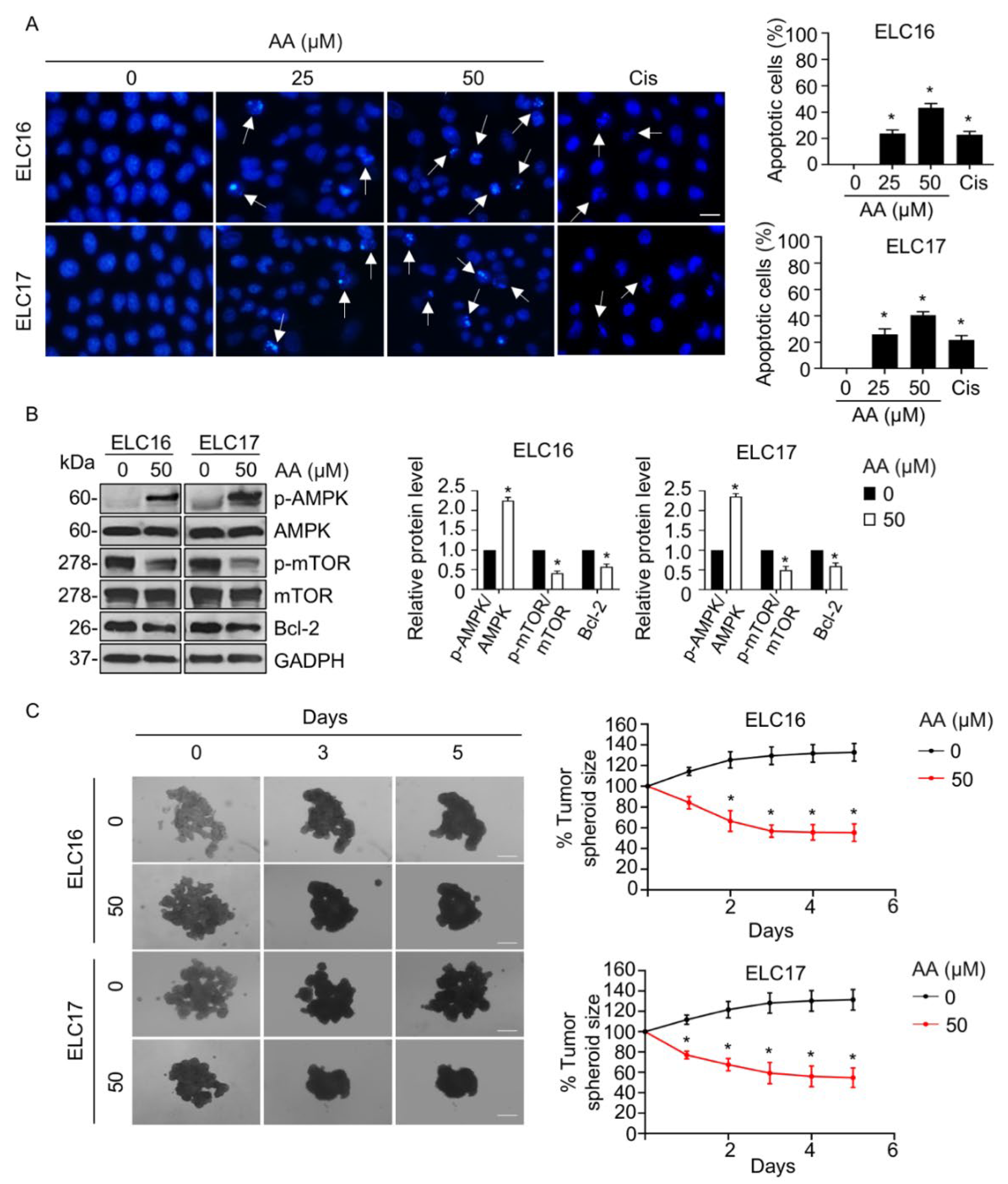
2.5. AA-Mediated Apoptosis in Patient-Derived Malignant Lung Cancer Cells
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Ethical Approval
4.3. Cell Culture
4.4. Cell Viability Assay
4.5. Apoptosis Evaluation
4.6. Measurement of Reactive Oxygen Species (ROS)
4.7. Western Blot Analysis
4.8. Immunoprecipitation
4.9. In Vitro Three-Dimensional (3D) Tumor Spheroid Formation Assay
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AMPK | AMP-activated protein kinase |
| AA | aspiletrein A |
| CC | compound C (Dorsomorphin) |
| CHX | cycloheximide |
| DCFH2-DA | 2′,7′-dichlorofluorescin diacetate |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| NAC | N-Acetylcysteine |
| NSCLC | non-small-cell lung cancer cells |
| PARP | poly-ADP-ribose polymerase |
| PI | propidium iodide |
| ROS | reactive oxygen species |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E. Cancer Statistics 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Shanker, M.; Willcutts, D.; Roth, J.A.; Ramesh, R. Drug resistance in lung cancer. Lung Cancer 2010, 1, 23–36. [Google Scholar] [PubMed]
- Knight, S.B.; Crosbie, P.A.; Balata, H.; Chudziak, J.; Hussell, T.; Dive, C. Progress and prospects of early detection in lung cancer. Open Biol. 2017, 7, 170070. [Google Scholar] [CrossRef]
- Wadowska, K.; Bil-lula, I.; Trembecki, Ł.; Śliwińska-Mossoń, M. Genetic markers in lung cancer diagnosis: A review. Int. J. Mol. Sci. 2020, 21, 4569. [Google Scholar] [CrossRef] [PubMed]
- Neal, R.D.; Hamilton, W.; Rogers, T.K. Lung cancer. BMJ 2014, 349, g6560. [Google Scholar] [CrossRef]
- Wood, S.L.; Pernemalm, M.; Crosbie, P.A.; Whetton, A.D. The role of the tumor-microenvironment in lung cancer-metastasis and its relationship to potential therapeutic targets. Cancer Treat. Rev. 2014, 40, 558–566. [Google Scholar] [CrossRef]
- Yang, S.R.; Schultheis, A.M.; Yu, H.; Mandelker, D.; Ladanyi, M.; Büttner, R. Precision medicine in non-small cell lung cancer: Current applications and future directions. Semin. Cancer Biol. 2022, 84, 184–198. [Google Scholar] [CrossRef]
- Kapeleris, J.; Kulasinghe, A.; Warkiani, M.E.; Vela, I.; Kenny, L.; O’Byrne, K.; Punyadeera, C. The prognostic role of circulating tumor cells (CTCs) in lung cancer. Front Oncol. 2018, 8, 311. [Google Scholar] [CrossRef]
- Liang, R.; Li, X.; Li, W.; Zhu, X.; Li, C. DNA methylation in lung cancer patients: Opening a “window of life” under precision medicine. Biomed. Pharmacother. 2021, 144, 112202. [Google Scholar] [CrossRef]
- Huang, F.X.; Chen, H.J.; Zheng, F.X.; Gao, Z.Y.; Sun, P.F.; Peng, Q.; Liu, Y.; Deng, X.; Huang, Y.H.; Zhao, C.; et al. LncRNA BLACAT1 is involved in chemoresistance of non-small cell lung cancer cells by regulating autophagy. Int. J. Oncol. 2019, 54, 339–347. [Google Scholar] [CrossRef] [PubMed]
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.M.; Cory, S. The BCL-2 arbiters of apoptosis and their growing role as cancer targets. Cell Death Differ. 2018, 25, 27–36. [Google Scholar] [CrossRef]
- Neophytou, C.M.; Trougakos, I.P.; Erin, N.; Papageorgis, P. Apoptosis deregulation and the development of cancer multi-drug resistance. Cancers 2021, 13, 4363. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, P.; Medema, J.P. BCL-2 family deregulation in colorectal cancer: Potential for BH3 mimetics in therapy. Apoptosis 2020, 25, 305–320. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK a nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef]
- Xie, G.; Sun, L.; Li, Y.; Chen, B.; Wang, C. Periplocin inhibits the growth of pancreatic cancer by inducing apoptosis via AMPK-mTOR signaling. Cancer Med. 2021, 10, 325–336. [Google Scholar] [CrossRef]
- Kwan, H.T.; Chan, D.W.; Cai, P.C.; Mak, C.S.; Yung, M.M.; Leung, T.H.; Wong, O.G.; Cheung, A.N.; Ngan, H.Y. AMPK activators suppress cervical cancer cell growth through inhibition of DVL3 mediated Wnt/β-catenin signaling activity. PLoS ONE 2013, 8, e53597. [Google Scholar] [CrossRef]
- Hui, G.D.; Xiu, W.Y.; Yong, C.; Yuan, C.B.; Jun, Z.; Jun, G.J.; Jun, Y.J.; Xiang, X.X.; Wei, H.S.; Feng, M.L. AMP-activated protein kinase α1 serves a carcinogenic role via regulation of vascular endothelial growth factor expression in patients with non-small cell lung cancer. Oncol. Lett. 2019, 17, 4329–4334. [Google Scholar] [CrossRef]
- Li, J.Y.; Luo, Z.Q. LCAL1 enhances lung cancer survival via inhibiting AMPK-related antitumor functions. Mol. Cell Biochem. 2019, 457, 11–20. [Google Scholar] [CrossRef]
- Xia, Y.C.; Zha, J.H.; Sang, Y.H.; Yin, H.; Xu, G.Q.; Zhen, Y.; Zhang, Y.; Yu, B.T. AMPK activation by ASP4132 inhibits non-small cell lung cancer cell growth. Cell Death Dis. 2021, 12, 365. [Google Scholar] [CrossRef] [PubMed]
- Storozhuk, Y.; Hopmans, S.N.; Sanli, T.; Barron, C.; Tsiani, E.; Cutz, J.; Pond, G.; Wright, J.; Singh, G.; Tsakiridis, T. Metformin inhibits growth and enhances radiation response of non-small cell lung cancer (NSCLC) through ATM and AMPK. Br. J. Cancer. 2021, 108, 2021–2032. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.S.; Han, Q.S.; Jia, Z.R.; Chen, C.S.; Qiao, C.; Liu, Q.Q.; Zhang, Y.M.; Wang, K.W.; Wang, J.; Xiao, K.; et al. PPARα agonist fenofibrate relieves acquired resistance to gefitinib in non-small cell lung cancer by promoting apoptosis via PPARα/AMPK/AKT/FoxO1 pathway. Acta Pharmacol. Sin. 2022, 43, 167–176. [Google Scholar] [CrossRef]
- Nguyen, H.M.; Nguyen, H.T.; Seephan, S.; Do, H.B.; Nguyen, H.T.; Ho, D.V.; Pongrakhananon, V. Antitumor activities of Aspiletrein A, a steroidal saponin from Aspidistra letreae, on non-small cell lung cancer cells. BMC Complement. Med. Ther. 2021, 21, 87. [Google Scholar] [CrossRef] [PubMed]
- Ho, D.V.; Hoang, H.N.T.; Vo, H.Q.; Nguyen, K.V.; Pham, T.V.; Le, A.T.; Van Phan, K.; Nguyen, H.M.; Morita, H.; Nguyen, H.T. Three new steroidal saponins from Aspidistra letreae plants and their cytotoxic activities. J. Nat. Med. 2020, 74, 591–598. [Google Scholar] [CrossRef]
- Wang, N.; Zhang, Q.; Luo, L.; Ning, B.; Fang, Y. β-asarone inhibited cell growth and promoted autophagy via P53/Bcl-2/Bclin-1 and P53/AMPK/mTOR pathways in human glioma U251 cells. J. Cell. Physiol. 2018, 233, 2434–2443. [Google Scholar] [CrossRef]
- Lin, S.S.; Bassik, M.C.; Suh, H.; Nishino, M.; Arroyo, J.D.; Hahn, W.C.; Korsmeyer, S.J.; Roberts, T.M. PP2A regulates BCL-2 phosphorylation and proteasome-mediated degradation at the endoplasmic reticulum. Int. J. Biol. Chem. 2006, 281, 23003–23012. [Google Scholar] [CrossRef]
- Vinayanuwattikun, C.; Prakhongcheep, O.; Tungsukruthai, S.; Petsri, K.; Thirasastr, P.; Leelayuwatanakul, N.; Chanvorachote, P. Feasibility technique of low-passage in vitro drug sensitivity testing of malignant pleural effusion from advanced-stage non-small cell lung cancer for prediction of clinical outcome. Anticancer Res. 2019, 39, 6981–6988. [Google Scholar] [CrossRef]
- Zhang, C.; Jia, X.; Bao, J.; Chen, S.; Wang, K.; Zhang, Y.; Li, P.; Wan, J.B.; Su, H.; Wang, Y.; et al. Polyphyllin VII induces apoptosis in HepG2 cells through ROS-mediated mitochondrial dysfunction and MAPK pathways. BMC Complement. Altern. Med. 2016, 16, 58. [Google Scholar] [CrossRef]
- Zhang, S.; Lu, Y.; Li, H.; Ji, Y.; Fang, F.; Tang, H.; Qio, P. A steroidal saponin form Paris vietnamensis (Takht.) reverses temozolomide resistance in glioblastoma cells via inducing apoptosis through ROS/PI3K/Akt pathway. Biosci. Trends 2020, 14, 123–133. [Google Scholar] [CrossRef]
- Chen, C.R.; Zhang, J.; Wu, K.W.; Liu, P.Y.; Wang, S.J.; Chen, D.Y.; Ji, Z.N. Gracillin induces apoptosis in HL60 human leukemic cell line via oxidative stress and cell cycle arrest of G1. Pharmazie 2015, 70, 199–204. [Google Scholar] [PubMed]
- Wang, Z.; Cheng, Y.; Wang, N.; Wang, D.M.; Li, Y.W.; Han, F.; Shen, J.G.; Yang, D.P.; Guan, X.Y.; Chen, J.P. Dioscin induces cancer cell apoptosis through elevated oxidative stress mediated by downregulation of peroxiredoxins. Cancer Biol. Ther. 2012, 13, 138–147. [Google Scholar]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Hu, X.; Liu, Y.; Dong, S.; Wen, Z.; He, W.; Zhang, S.; Huang, Q.; Shi, M. ROS signaling under metabolic stress: Cross-talk between AMPK and AKT pathway. Mol. Cancer. 2017, 16, 79. [Google Scholar] [CrossRef]
- Kim, J.; Yang, G.; Kim, Y.; Kim, J.; Ha, J. AMPK activators: Mechanisms of action and physiological activities. Exp. Mol. Med. 2016, 48, e224. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Ji, J.; Yan, X.H. Cross-talk between AMPK and mTOR in regulating energy balance. Crit. Rev. Food Sci. Nutr. 2012, 52, 373–381. [Google Scholar] [CrossRef]
- Zhou, H.; Shang, C.; Wang, M.; Shen, T.; Kong, L.; Yu, C.; Ye, Z.; Luo, Y.; Liu, L.; Li, Y.; et al. Ciclopirox Olamine inhibits mTORC1 signaling by activation of AMPK. Biochem. Pharmacol. 2016, 116, 39–50. [Google Scholar] [CrossRef]
- Han, G.; Gong, H.; Wang, Y.; Guo, S.; Liu, K. AMPK/mTOR-mediated inhibition of survivin partly contributes to metformin-induced apoptosis in human gastric cancer cell. Cancer Biol. Ther. 2015, 16, 77–87. [Google Scholar] [CrossRef]
- Kang, M.H.; Reynolds, C.P. Bcl-2 inhibitors: Targeting mitochondrial apoptotic pathways in cancer therapy. Clin. Cancer Res. 2009, 15, 1126–1132. [Google Scholar] [CrossRef]
- Kutuk, O.; Letai, A. Regulation of Bcl-2 family proteins by posttranslational modifications. Curr. Mol. Med. 2008, 8, 102–118. [Google Scholar]
- Pongrakhananon, V.; Nimmannit, U.; Luanpitpong, S.; Rojanasakul, Y.; Chanvorachote, P. Curcumin sensitizes non-small cell lung cancer cell anoikis through reactive oxygen species-mediated Bcl-2 downregulation. Apoptosis 2010, 15, 574–585. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.X.; Kong, C.Z.; Wang, L.H.; Li, J.Y.; Liu, X.K.; Xu, B.; Xu, C.L.; Sun, Y.H. Ursolic acid overcomes Bcl-2-mediated resistance to apoptosis in prostate cancer cells involving activation of JNK-induced Bcl-2 phosphorylation and degradation. J. Cell Biochem. 2010, 109, 764–773. [Google Scholar] [CrossRef]
- Chen, H.; Li, Y.; Li, Y.; Chen, Z.; Xie, L.; Li, W.; Zhu, Y.; Xue, H.; Koeffler, H.P.; Wu, W.; et al. PARK2 promotes mitochondrial pathway of apoptosis and antimicrotubule drugs chemosensitivity via degradation of phospho-BCL-2. Theranostics 2020, 10, 9984–10000. [Google Scholar] [CrossRef]
- Guo, H.; Ding, H.; Yan, Y.; Chen, Q.; Zhang, J.; Chen, B.; Cao, J. Intermittent hypoxia-induced autophagy via AMPK/mTOR signaling pathway attenuates endothelial apoptosis and dysfunction in vitro. Sleep Breath 2021, 25, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Miyake, N.; Chikumi, H.; Takata, M.; Nakamoto, M.; Igishi, T.; Shimizu, E. Rapamycin induces p53 -independent apoptosis through the mitochondrial pathway in non-small cell lung cancer cells. Oncol. Rep. 2012, 28, 848–854. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhai, B.; Gygi, S.P.; Goldberg, A.L. mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. Proc. Natl. Acad. Sci. USA 2015, 112, 15790–15797. [Google Scholar] [CrossRef]
- Li, L.; Tan, H.; Yang, H.; Li, F.; He, X.; Gu, Z.; Zhao, M.; Su, L. Reactive oxygen species mediate heat stress-induced apoptosis via ERK dephosphorylation and Bcl-2 ubiquitination in human umbilical vein endothelial cells. Oncotarget 2017, 8, 12902–12916. [Google Scholar] [CrossRef]
- Liu, Y.B.; Gao, X.; Deeb, D.; Arbab, A.S.; Gautum, S.C. Pristimerin induces apoptosis in prostate cancer cells by down-regulating Bcl-2 through ROS-dependent ubiquitin-proteasomal degradation pathway. J. Carcinog. Mutagen. 2013, Suppl. 6, 005. [Google Scholar]
- Sun, J.; Feng, Y.; Wang, Y.; Ji, Q.; Cai, G.; Shi, L.; Wang, Y.; Huang, Y.; Zhang, J.; Li, Q. α-hederin induces autophagic cell death in colorectal cancer cells through reactive oxygen species dependent AMPK/mTOR signaling pathway activation. Int. J. Oncol. 2019, 54, 1601–1612. [Google Scholar] [CrossRef]
- Zhou, X.; Chen, Y.; Wang, F.; Wu, H.; Zhang, Y.; Liu, J.; Cai, Y.; Huang, S.; He, N.; Hu, Z.; et al. Artesunate induces autophagy dependent apoptosis through upregulating ROS and activating AMPK-mTOR-ULK1 axis in human bladder cancer cells. Chem.-Biol. Interact 2020, 331, 109273. [Google Scholar] [CrossRef]
- Guo, H.; Ding, H.; Tang, X.; Liang, M.; Li, S.; Zhang, J.; Cao, J. Quercetin induces pro-apoptotic autophagy via SIRT1/AMPK signaling pathway in human lung cancer cell lines A549 and H1299 in vitro. Thorac. Cancer 2021, 12, 1415–1422. [Google Scholar] [CrossRef] [PubMed]
- Kodack, D.P.; Farago, A.F.; Dastur, A.; Held, M.A.; Dardaei, L.; Friboulet, L.; von Flotow, F.; Damon, L.J.; Lee, D.; Parks, M.; et al. Primary patient-derived cancer cells and their potential for personalized cancer patient care. Cell Rep. 2017, 21, 3298–3309. [Google Scholar] [CrossRef]
- Petsri, K.; Yokoya, M.; Tungsukruthai, S.; Rungrotmongkol, T.; Nutho, B.; Vinayanuwattikun, C.; Saito, N.; Takehiro, M.; Sato, R.; Chanvorachote, P. Structure-activity relationships and molecular docking analysis of Mcl-1 targeting renieramycin T analogues in patient-derived lung cancer cells. Cancer 2020, 12, 875. [Google Scholar] [CrossRef] [PubMed]
- Sriratanasak, N.; Petsri, K.; Laobuthee, A.; Wattanathana, W.; Vinayanuwattikun, C.; Luanpitpong, S.; Chanvorachote, P. Novel c-Myc -targeting compound N, N -Bis (5-Ethyl-2-Hydroxybenzyl) methylamine for mediated c-Myc ubiquitin-proteasomal degradation in lung cancer cells. Mol. Pharmacol. 2020, 98, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.H.; Li, H.; Jia, Y.; Mak, P.I.; Martins, R.; Liu, Y.; Vong, C.M.; Wong, H.C.; Wong, P.K.; Wang, H.; et al. Drug screening of cancer cell lines and human primary tumors using droplet microfluidics. Sci. Rep. 2017, 7, 9109. [Google Scholar] [CrossRef] [PubMed]
- Nyga, A.; Cheema, U.; Loizidou, M. 3D tumour models: Novel in vitro approaches to cancer studies. J. Cell Commun. Signal. 2011, 5, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Vinci, M.; Gowan, S.; Boxall, F.; Patterson, L.; Zimmermann, M.; Court, W.; Lomas, C.; Mendiola, M.; Hardisson, D.; Eccles, S.A. Advances in establishment and analysis of three-dimensional tumor spheroid-based functional assays for target validation and drug evaluation. BMC Biol. 2012, 10, 29. [Google Scholar] [CrossRef]
- Mikhail, A.S.; Eetezadi, S.; Allen, C. Multicellular tumor spheroids for evaluation of cytotoxicity and tumor growth inhibitory effects of nanomedicines in vitro: A comparison of docetaxel- loaded block copolymer micelles and taxotere®. PLoS ONE 2013, 8, e62630. [Google Scholar] [CrossRef]
- Kasper, S.H.; Morell-perez, C.; Wyche, T.P.; Sana, T.R.; Lieberman, L.A.; Hett, E.C. Colorectal cancer-associated anaerobic bacteria proliferate in tumor spheroids and alter the microenvironment. Sci. Rep. 2020, 10, 5321. [Google Scholar] [CrossRef]
- Singh, K.; Gangrade, A.; Jana, A.; Mandal, B.B.; Das, N.J.A.O. Design, synthesis, characterization, and antiproliferative activity of organoplatinum compounds bearing a 1, 2, 3-triazole ring. ACS Omega 2019, 4, 835. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Witayateeraporn, W.; Nguyen, H.M.; Ho, D.V.; Nguyen, H.T.; Chanvorachote, P.; Vinayanuwattikun, C.; Pongrakhananon, V. Aspiletrein A Induces Apoptosis Cell Death via Increasing Reactive Oxygen Species Generation and AMPK Activation in Non-Small-Cell Lung Cancer Cells. Int. J. Mol. Sci. 2022, 23, 9258. https://doi.org/10.3390/ijms23169258
Witayateeraporn W, Nguyen HM, Ho DV, Nguyen HT, Chanvorachote P, Vinayanuwattikun C, Pongrakhananon V. Aspiletrein A Induces Apoptosis Cell Death via Increasing Reactive Oxygen Species Generation and AMPK Activation in Non-Small-Cell Lung Cancer Cells. International Journal of Molecular Sciences. 2022; 23(16):9258. https://doi.org/10.3390/ijms23169258
Chicago/Turabian StyleWitayateeraporn, Wasita, Hien Minh Nguyen, Duc Viet Ho, Hoai Thi Nguyen, Pithi Chanvorachote, Chanida Vinayanuwattikun, and Varisa Pongrakhananon. 2022. "Aspiletrein A Induces Apoptosis Cell Death via Increasing Reactive Oxygen Species Generation and AMPK Activation in Non-Small-Cell Lung Cancer Cells" International Journal of Molecular Sciences 23, no. 16: 9258. https://doi.org/10.3390/ijms23169258
APA StyleWitayateeraporn, W., Nguyen, H. M., Ho, D. V., Nguyen, H. T., Chanvorachote, P., Vinayanuwattikun, C., & Pongrakhananon, V. (2022). Aspiletrein A Induces Apoptosis Cell Death via Increasing Reactive Oxygen Species Generation and AMPK Activation in Non-Small-Cell Lung Cancer Cells. International Journal of Molecular Sciences, 23(16), 9258. https://doi.org/10.3390/ijms23169258