
The IRE1α–XBP1s Arm of the Unfolded Protein Response Activates N-Glycosylation to Remodel the Subepithelial Basement Membrane in Paramyxovirus Infection


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
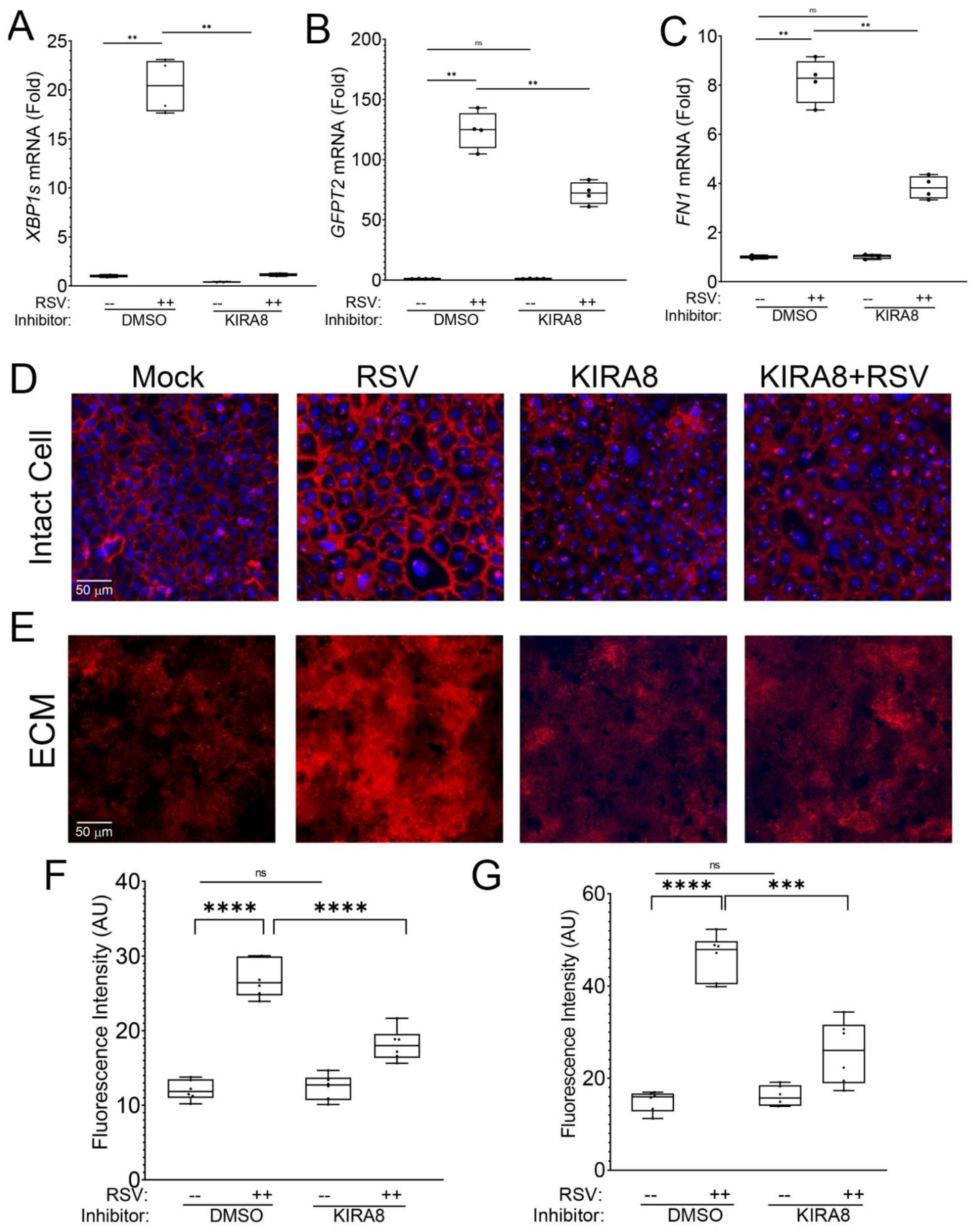
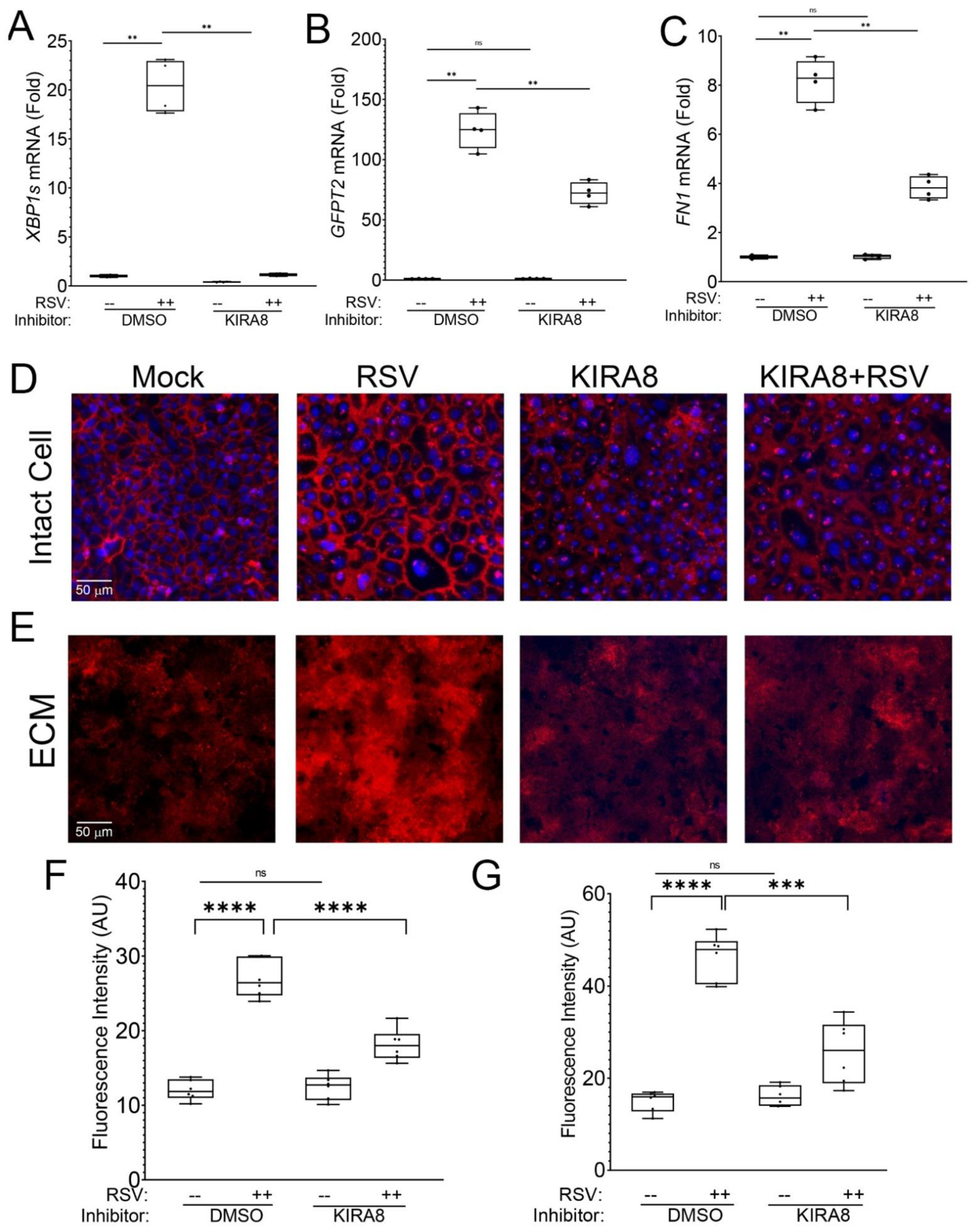
2.1. RSV Infection Remodels the Epithelial Basement Membrane
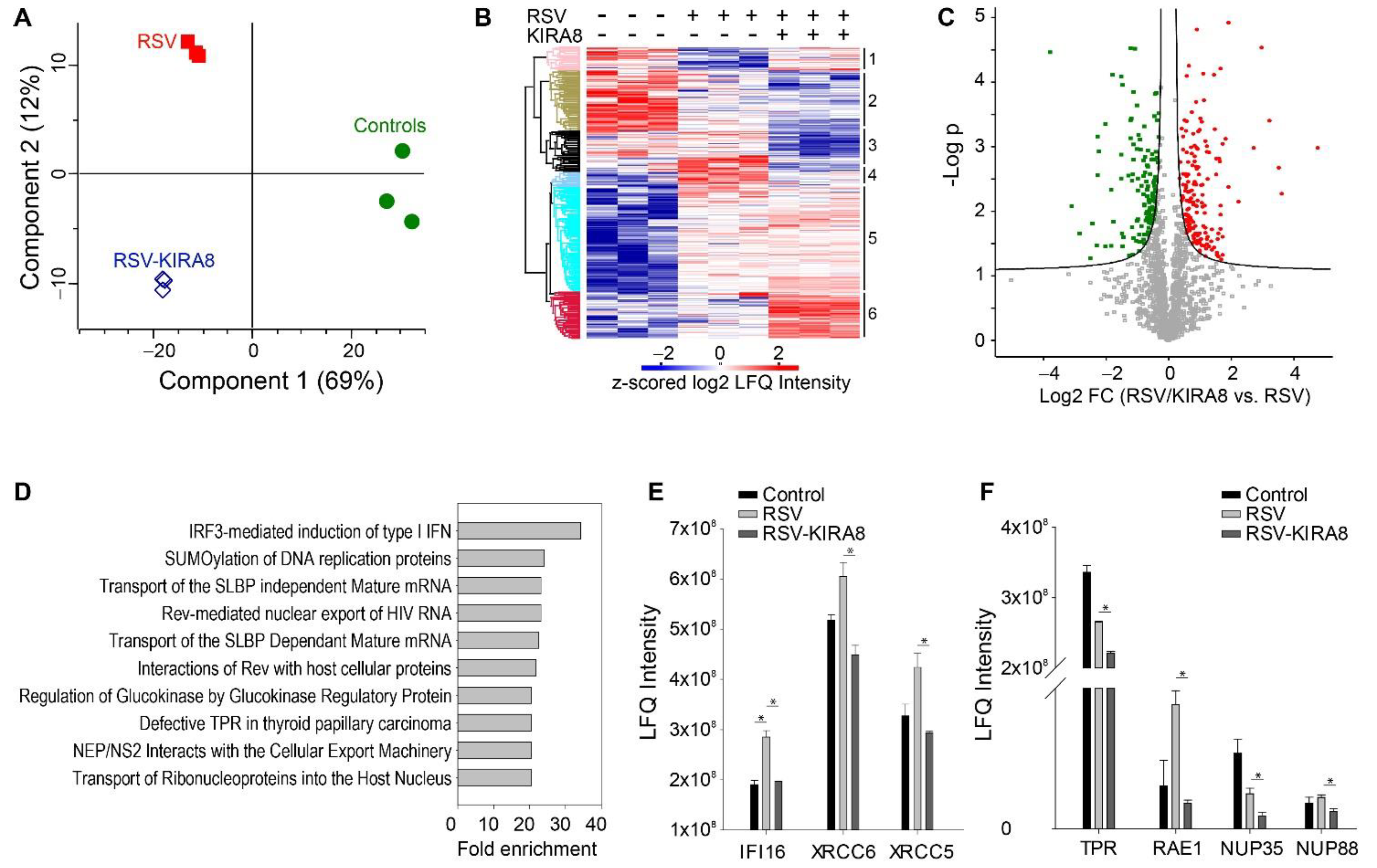
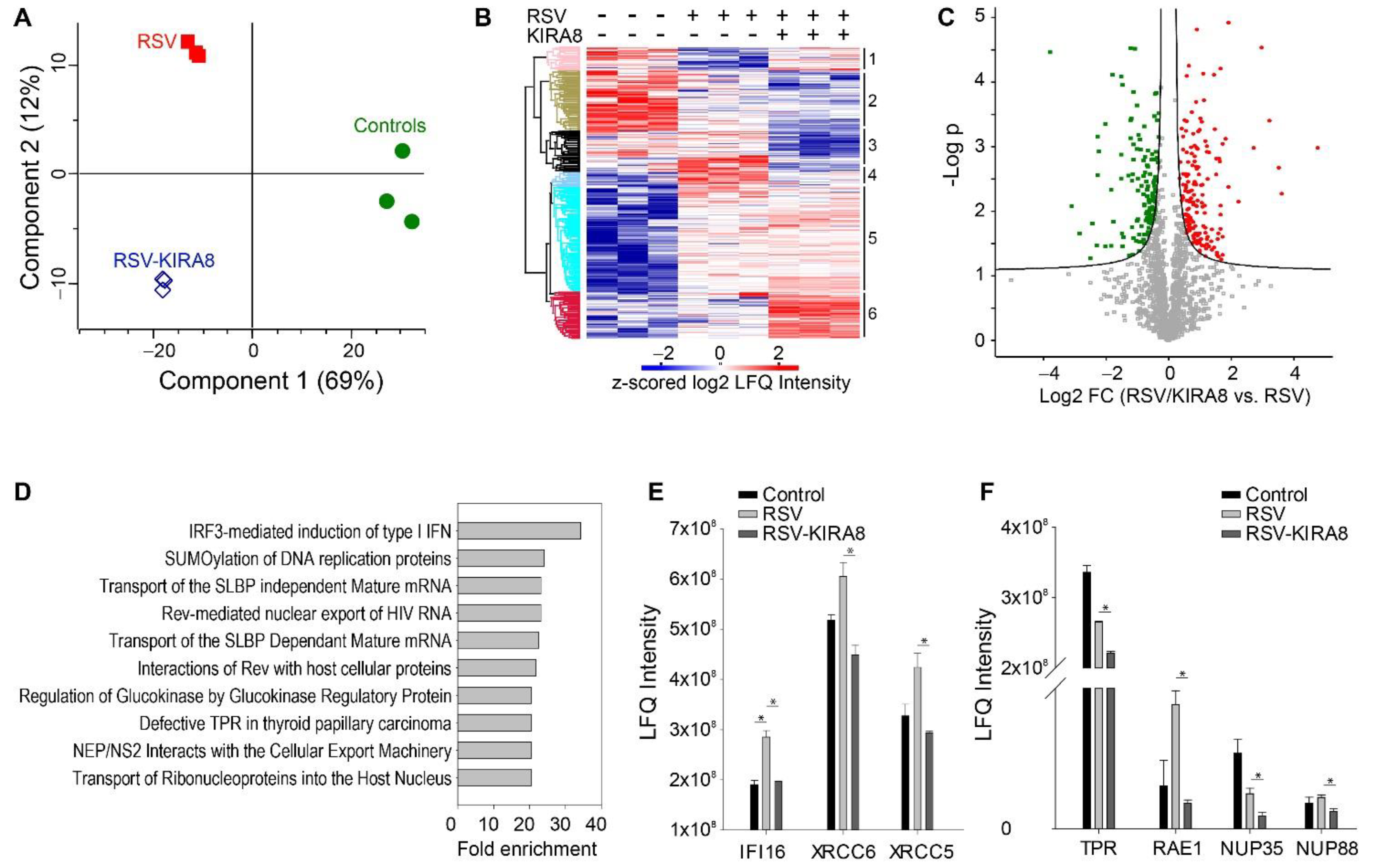
2.2. Proteomics Analysis of the Effect of the IRE1α–XBP1 Arm of UPR on RSV-Induced Host Response
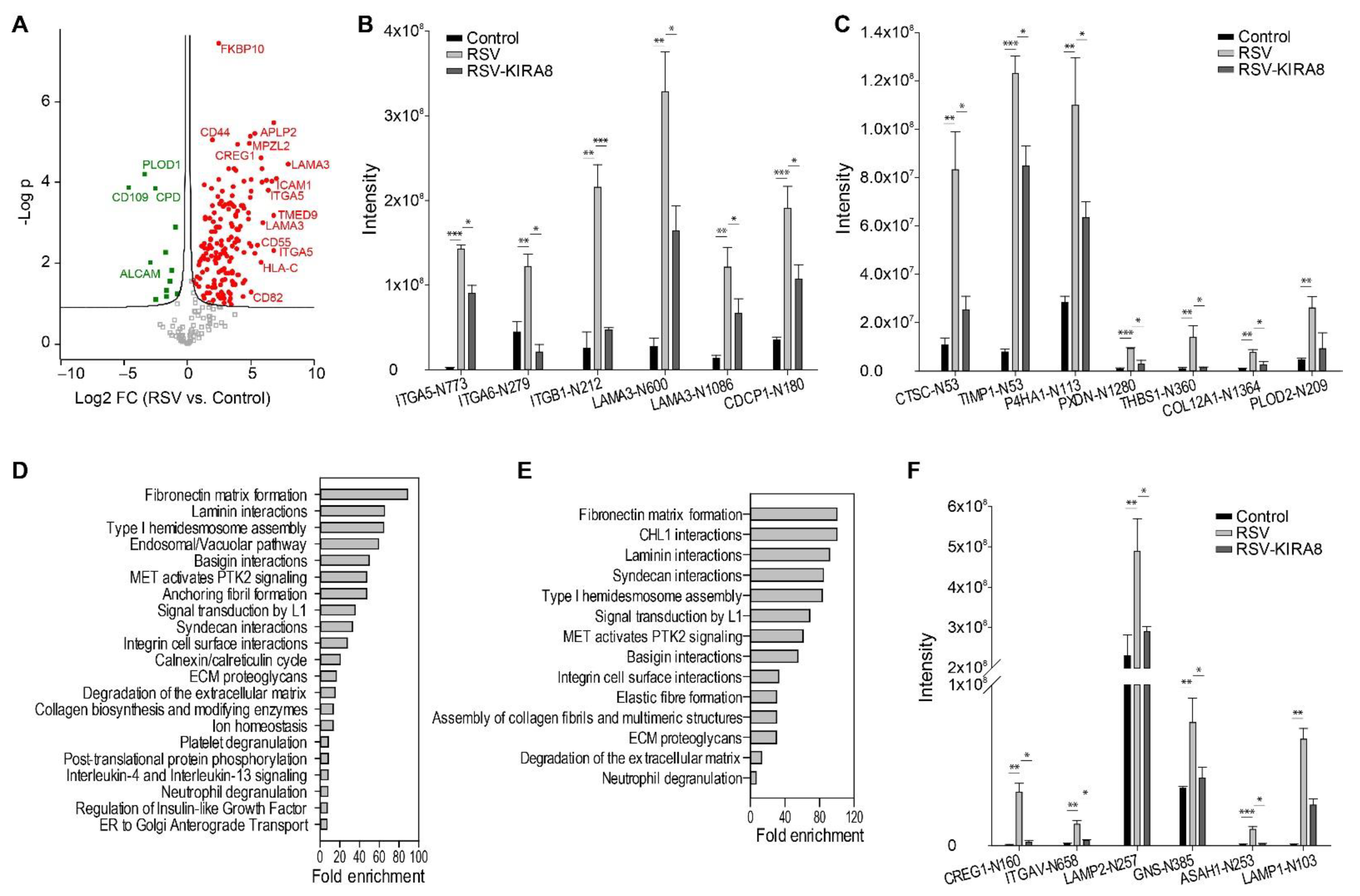
2.3. IRE1α–XBP1 Arm of UPR Regulates N-Glycosylation in RSV-Induced hSAECs
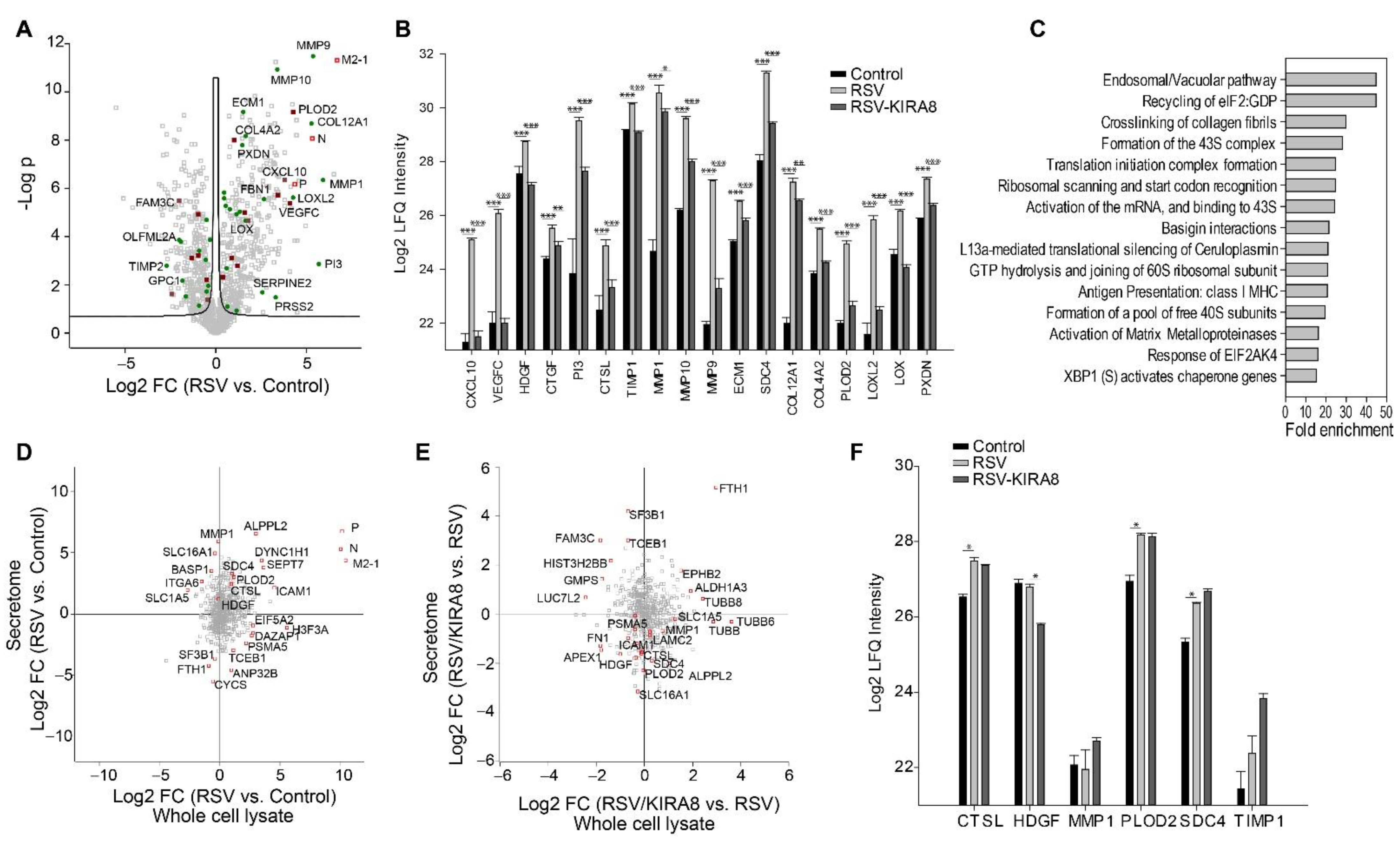
2.4. IRE1α–XBP1 Arm of UPR Regulates RSV-Induced Secretome
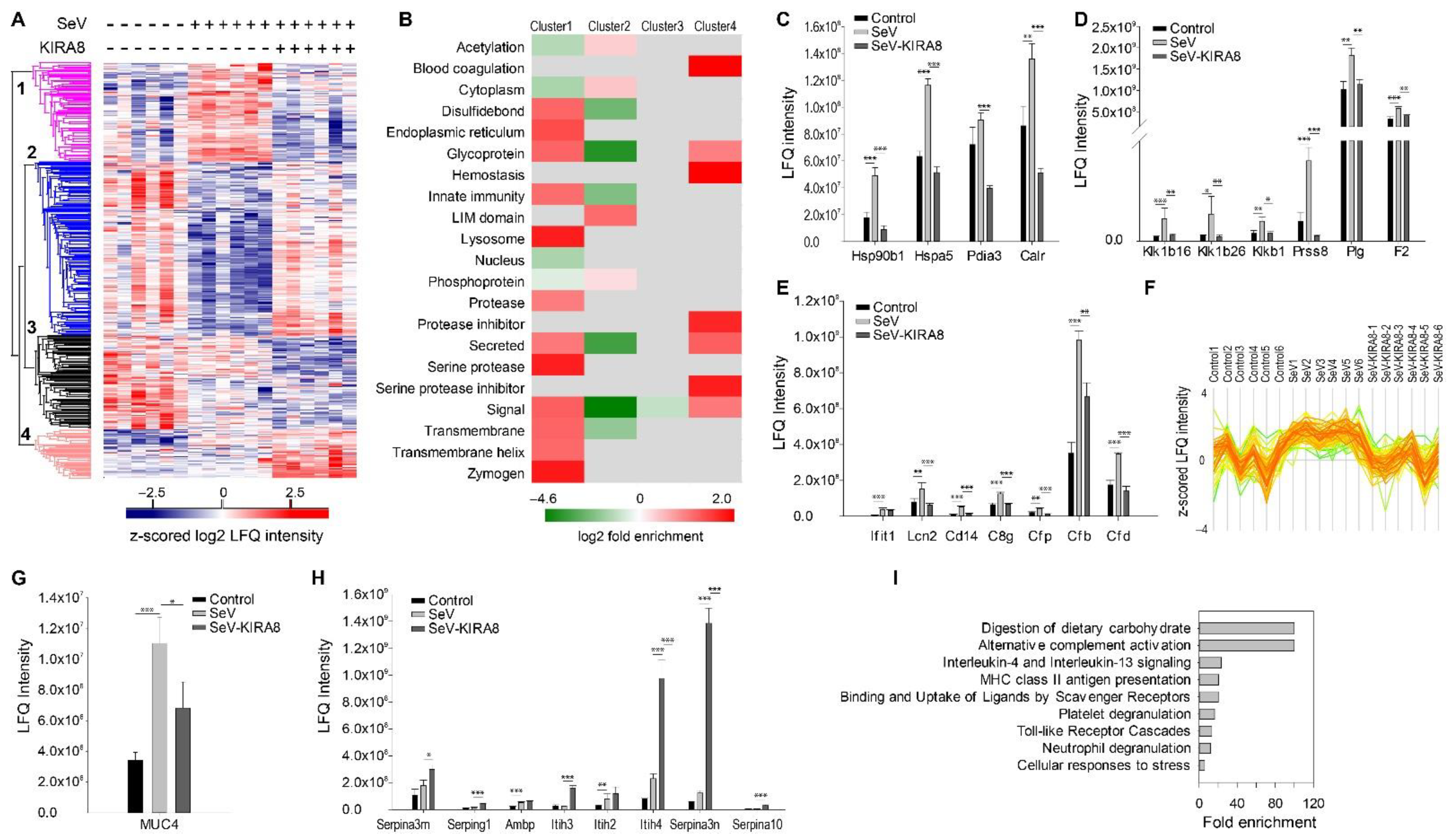
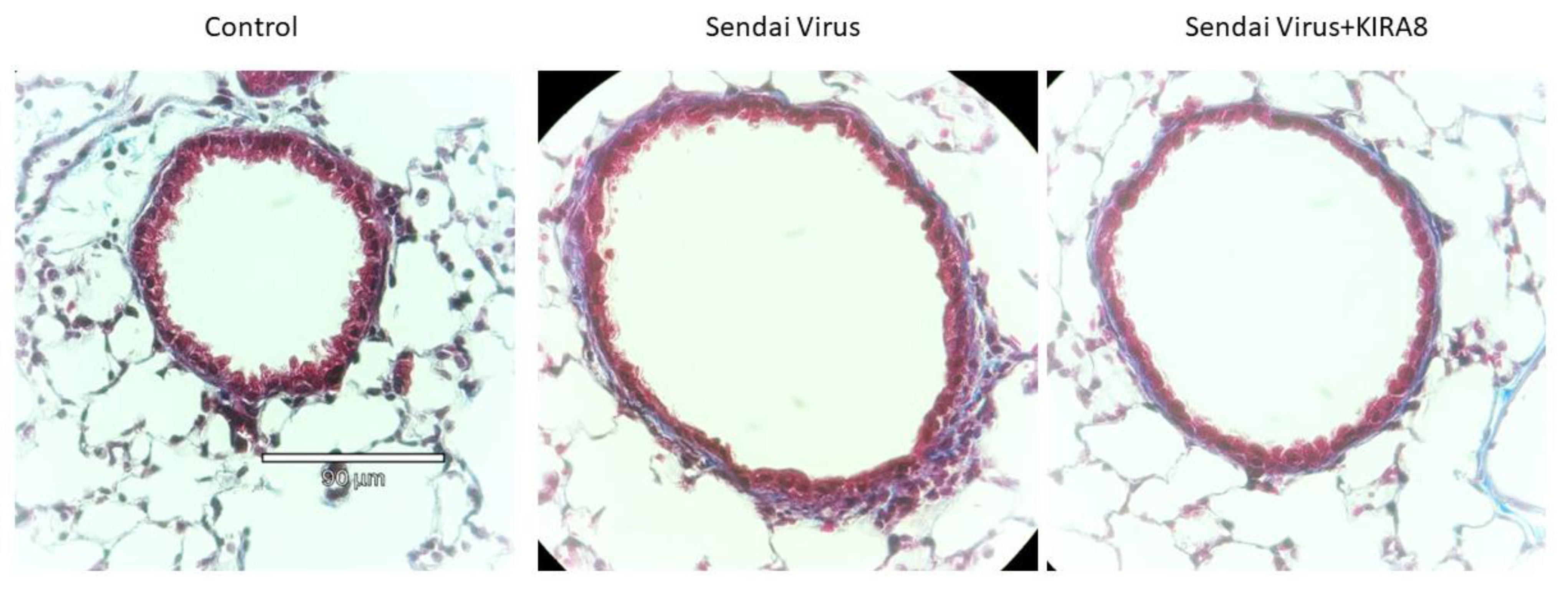
2.5. IRE1α–XBP1 Arm of UPR Regulates N-Glycoprotein Secretion In Vivo
3. Discussion
3.1. RSV-Induced Remodeling of the Basal Lamina in Chronic Airway Disease
3.2. IRE1α–XBP1 Arm of the UPR Regulates Antiviral Response
3.3. IRE1α–XBP1 Arm of the UPR Regulates N-Glycosylation in Response to RSV Infection
3.4. IRE1α–XBP1 Arm of the UPR Regulates RSV Secretome
3.5. IRE1α–XBP1 Arm of UPR Regulates ECM and Mediators of Innate Immunity In Vivo
4. Materials and Methods
4.1. Human Small Airway Epithelial Cell (hSAEC) Culture and Treatment
4.2. Immunofluorescence of ECM Deposition
4.3. Protein Extraction and Trypsin Digestion
4.4. Enrichment of N-Glycosylation
4.5. NanoLC-MS/MS Analysis
4.6. Proteomics Data Analysis and Statistical Analysis
4.7. RNA Isolation and qRT-PCR
4.8. Murine Respirovirus (Sendai Virus (SeV)) Infection
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Borchers, A.T.; Chang, C.; Gershwin, M.E.; Gershwin, L.J. Respiratory syncytial virus—A comprehensive review. Clin. Rev. Allergy Immunol. 2013, 45, 331–379. [Google Scholar] [CrossRef] [PubMed]
- Stockman, L.J.; Curns, A.T.; Anderson, L.J.; Fischer-Langley, G. Respiratory syncytial virus-associated hospitalizations among infants and young children in the United States, 1997–2006. Pediatric Infect. Dis. J. 2012, 31, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; McAllister, D.A.; O’Brien, K.L.; Simoes, E.A.F.; Madhi, S.A.; Gessner, B.D.; Polack, F.P.; Balsells, E.; Acacio, S.; Aguayo, C.; et al. Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in young children in 2015: A systematic review and modelling study. Lancet 2017, 390, 946–958. [Google Scholar] [CrossRef]
- Zhang, Y.; Luxon, B.A.; Casola, A.; Garofalo, R.P.; Jamaluddin, M.; Brasier, A.R. Expression of respiratory syncytial virus-induced chemokine gene networks in lower airway epithelial cells revealed by cDNA microarrays. J. Virol. 2001, 75, 9044–9058. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Zhang, Y.; Luxon, B.A.; Garofalo, R.P.; Casola, A.; Sinha, M.; Brasier, A.R. Identification of NF-kappaB-dependent gene networks in respiratory syncytial virus-infected cells. J. Virol. 2002, 76, 6800–6814. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Jamaluddin, M.; Li, K.; Garofalo, R.P.; Casola, A.; Brasier, A.R. Retinoic Acid-Inducible Gene I Mediates Early Antiviral Response and Toll-Like Receptor 3 Expression in Respiratory Syncytial Virus-Infected Airway Epithelial Cells. J. Virol. 2007, 81, 1401–1411. [Google Scholar] [CrossRef] [PubMed]
- Hosakote, Y.M.; Brasier, A.R.; Casola, A.; Garofalo, R.P.; Kurosky, A. Respiratory Syncytial Virus Infection Triggers Epithelial HMGB1 Release as a Damage-Associated Molecular Pattern Promoting a Monocytic Inflammatory Response. J. Virol. 2016, 90, 9618–9631. [Google Scholar] [CrossRef]
- Johnson, J.E.; Gonzales, R.A.; Olson, S.J.; Wright, P.F.; Graham, B.S. The histopathology of fatal untreated human respiratory syncytial virus infection. Mod. Pathol. Off. J. USA Can. Acad. Pathol. Inc. 2007, 20, 108–119. [Google Scholar] [CrossRef]
- Fauroux, B.; Simões, E.A.F.; Checchia, P.A.; Paes, B.; Figueras-Aloy, J.; Manzoni, P.; Bont, L.; Carbonell-Estrany, X. The Burden and Long-term Respiratory Morbidity Associated with Respiratory Syncytial Virus Infection in Early Childhood. Infect. Dis. Ther. 2017, 6, 173–197. [Google Scholar] [CrossRef]
- Sigurs, N.; Aljassim, F.; Kjellman, B.; Robinson, P.D.; Sigurbergsson, F.; Bjarnason, R.; Gustafsson, P.M. Asthma and allergy patterns over 18 years after severe RSV bronchiolitis in the first year of life. Thorax 2010, 65, 1045–1052. [Google Scholar] [CrossRef]
- Zomer-Kooijker, K.; van der Ent, C.K.; Ermers, M.J.; Uiterwaal, C.S.; Rovers, M.M.; Bont, L.J. Increased risk of wheeze and decreased lung function after respiratory syncytial virus infection. PLoS ONE 2014, 9, e87162. [Google Scholar] [CrossRef]
- Schuurhof, A.; Bont, L.; Hodemaekers, H.M.; de Klerk, A.; de Groot, H.; Hofland, R.W.; van de Pol, A.C.; Kimpen, J.L.L.; Janssen, R. Proteins involved in extracellular matrix dynamics are associated with respiratory syncytial virus disease severity. Eur. Respir. J. 2012, 39, 1475–1481. [Google Scholar] [CrossRef] [PubMed]
- Kong, M.Y.; Clancy, J.P.; Peng, N.; Li, Y.; Szul, T.J.; Xu, X.; Oster, R.; Sullender, W.; Ambalavanan, N.; Blalock, J.E.; et al. Pulmonary matrix metalloproteinase-9 activity in mechanically ventilated children with respiratory syncytial virus. Eur. Respir. J. 2014, 43, 1086–1096. [Google Scholar] [CrossRef] [PubMed]
- Kellar, G.G.; Reeves, S.R.; Barrow, K.A.; Debley, J.S.; Wight, T.N.; Ziegler, S.F. Juvenile, but Not Adult, Mice Display Increased Myeloid Recruitment and Extracellular Matrix Remodeling during Respiratory Syncytial Virus Infection. J. Immunol. 2020, 205, 3050–3057. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Qiao, D.; Dong, C.; Mann, M.; Garofalo, R.P.; Keles, S.; Brasier, A.R. The SWI/SNF-Related, Matrix Associated, Actin-Dependent Regulator of Chromatin A4 Core Complex Represses Respiratory Syncytial Virus-Induced Syncytia Formation and Subepithelial Myofibroblast Transition. Front. Immunol. 2021, 12, 633654. [Google Scholar] [CrossRef]
- Xu, X.; Qiao, D.; Mann, M.; Garofalo, R.P.; Brasier, A.R. Respiratory Syncytial Virus Infection Induces Chromatin Remodeling to Activate Growth Factor and Extracellular Matrix Secretion Pathways. Viruses 2020, 12, 804. [Google Scholar] [CrossRef]
- Qiao, D.; Skibba, M.; Xu, X.; Garofalo, R.P.; Zhao, Y.; Brasier, A.R. Paramyxovirus replication induces the hexosamine biosynthetic pathway and mesenchymal transition via the IRE1alpha-XBP1s arm of the unfolded protein response. Am. J. Physiol. Lung Cell. Mol. Physiol. 2021, 321, L576–L594. [Google Scholar] [CrossRef]
- Brasier, A.R.; Qiao, D.; Zhao, Y. The Hexosamine Biosynthetic Pathway Links Innate Inflammation With Epithelial-Mesenchymal Plasticity in Airway Remodeling. Front. Pharmacol. 2021, 12, 808735. [Google Scholar] [CrossRef]
- Harrington, P.E.; Biswas, K.; Malwitz, D.; Tasker, A.S.; Mohr, C.; Andrews, K.L.; Dellamaggiore, K.; Kendall, R.; Beckmann, H.; Jaeckel, P.; et al. Unfolded Protein Response in Cancer: IRE1alpha Inhibition by Selective Kinase Ligands Does Not Impair Tumor Cell Viability. ACS Med. Chem. Lett. 2015, 6, 68–72. [Google Scholar] [CrossRef]
- Halper, J.; Kjaer, M. Basic components of connective tissues and extracellular matrix: Elastin, fibrillin, fibulins, fibrinogen, fibronectin, laminin, tenascins and thrombospondins. Adv. Exp. Med. Biol. 2014, 802, 31–47. [Google Scholar] [CrossRef]
- Neumann, G.; Hughes, M.T.; Kawaoka, Y. Influenza a virus NS2 protein mediates vRNP nuclear export through NES-independent interaction with hCRM1. EMBO J. 2000, 19, 6751–6758. [Google Scholar] [CrossRef]
- Faisca, P.; Desmecht, D. Sendai virus, the mouse parainfluenza type 1: A longstanding pathogen that remains up-to-date. Res. Vet. Sci. 2007, 82, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.B. Respiratory syncytial virus and parainfluenza virus. N. Engl. J. Med. 2001, 344, 1917–1928. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.B.; Weinberg, G.A.; Iwane, M.K.; Blumkin, A.K.; Edwards, K.M.; Staat, M.A.; Auinger, P.; Griffin, M.R.; Poehling, K.A.; Erdman, D.; et al. The burden of respiratory syncytial virus infection in young children. N. Engl. J. Med. 2009, 360, 588–598. [Google Scholar] [CrossRef] [PubMed]
- Sigurs, N.; Gustafsson, P.M.; Bjarnason, R.; Lundberg, F.; Schmidt, S.; Sigurbergsson, F.; Kjellman, B. Severe respiratory syncytial virus bronchiolitis in infancy and asthma and allergy at age 13. Am. J. Respir. Crit. Care Med. 2005, 171, 137–141. [Google Scholar] [CrossRef]
- Tian, B.; Yang, J.; Zhao, Y.; Ivanciuc, T.; Sun, H.; Wakamiya, M.; Garofalo, R.P.; Brasier, A.R. Central Role of the NF-kappaB Pathway in the Scgb1a1-Expressing Epithelium in Mediating Respiratory Syncytial Virus-Induced Airway Inflammation. J. Virol. 2018, 92, e00441-18. [Google Scholar] [CrossRef]
- Zhao, Y.; Jamaluddin, M.; Zhang, Y.; Sun, H.; Ivanciuc, T.; Garofalo, R.P.; Brasier, A.R. Systematic Analysis of Cell-Type Differences in the Epithelial Secretome Reveals Insights into the Pathogenesis of Respiratory Syncytial Virus-Induced Lower Respiratory Tract Infections. J. Immunol. 2017, 198, 3345–3364. [Google Scholar] [CrossRef]
- Cutz, E.; Levison, H.; Cooper, D.M. Ultrastructure of airways in children with asthma. Histopathology 1978, 2, 407–421. [Google Scholar] [CrossRef]
- Boulet, L.-P.; Laviolette, M.; Turcotte, H.; Cartier, A.; Dugas, M.; Malo, J.-L.; Boutet, M. Bronchial Subepithelial Fibrosis Correlates With Airway Responsiveness to Methacholine. CHEST 1997, 112, 45–52. [Google Scholar] [CrossRef]
- Mathew, C.; Tamir, S.; Tripp, R.A.; Ghildyal, R. Reversible disruption of XPO1-mediated nuclear export inhibits respiratory syncytial virus (RSV) replication. Sci. Rep. 2021, 11, 19223. [Google Scholar] [CrossRef]
- Jorquera, P.A.; Mathew, C.; Pickens, J.; Williams, C.; Luczo, J.M.; Tamir, S.; Ghildyal, R.; Tripp, R.A. Verdinexor (KPT-335), a Selective Inhibitor of Nuclear Export, Reduces Respiratory Syncytial Virus Replication In Vitro. J. Virol. 2019, 93, e01684-18. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Xie, Y.; Munoz-Moreno, R.; Wang, J.; Zhang, L.; Esparza, M.; Garcia-Sastre, A.; Fontoura, B.M.A.; Ren, Y. Structural basis for influenza virus NS1 protein block of mRNA nuclear export. Nat. Microbiol. 2019, 4, 1671–1679. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Miorin, L.; Makio, T.; Dehghan, I.; Gao, S.; Xie, Y.; Zhong, H.; Esparza, M.; Kehrer, T.; Kumar, A.; et al. Nsp1 protein of SARS-CoV-2 disrupts the mRNA export machinery to inhibit host gene expression. Sci. Adv. 2021, 7, eabe7386. [Google Scholar] [CrossRef] [PubMed]
- Satterly, N.; Tsai, P.L.; van Deursen, J.; Nussenzveig, D.R.; Wang, Y.; Faria, P.A.; Levay, A.; Levy, D.E.; Fontoura, B.M. Influenza virus targets the mRNA export machinery and the nuclear pore complex. Proc. Natl. Acad. Sci. USA 2007, 104, 1853–1858. [Google Scholar] [CrossRef]
- von Kobbe, C.; van Deursen, J.M.; Rodrigues, J.P.; Sitterlin, D.; Bachi, A.; Wu, X.; Wilm, M.; Carmo-Fonseca, M.; Izaurralde, E. Vesicular stomatitis virus matrix protein inhibits host cell gene expression by targeting the nucleoporin Nup98. Mol. Cell. 2000, 6, 1243–1252. [Google Scholar] [CrossRef]
- Xu, X.; Mann, M.; Qiao, D.; Brasier, A.R. Alternative mRNA Processing of Innate Response Pathways in Respiratory Syncytial Virus (RSV) Infection. Viruses 2021, 13, 218. [Google Scholar] [CrossRef]
- Taniguchi, T.; Ogasawara, K.; Takaoka, A.; Tanaka, N. IRF family of transcription factors as regulators of host defense. Annu. Rev. Immunol. 2001, 19, 623–655. [Google Scholar] [CrossRef]
- Barnes, B.; Lubyova, B.; Pitha, P.M. On the role of IRF in host defense. J. Interferon Cytokine Res. 2002, 22, 59–71. [Google Scholar] [CrossRef]
- Tian, B.; Zhao, Y.; Kalita, M.; Edeh, C.B.; Paessler, S.; Casola, A.; Teng, M.N.; Garofalo, R.P.; Brasier, A.R. CDK9-dependent transcriptional elongation in the innate interferon-stimulated gene response to respiratory syncytial virus infection in airway epithelial cells. J. Virol. 2013, 87, 7075–7092. [Google Scholar] [CrossRef]
- Ferguson, B.J.; Mansur, D.S.; Peters, N.E.; Ren, H.; Smith, G.L. DNA-PK is a DNA sensor for IRF-3-dependent innate immunity. eLife 2012, 1, e00047. [Google Scholar] [CrossRef]
- Sui, H.; Zhou, M.; Imamichi, H.; Jiao, X.; Sherman, B.T.; Lane, H.C.; Imamichi, T. STING is an essential mediator of the Ku70-mediated production of IFN-lambda1 in response to exogenous DNA. Sci. Signal. 2017, 10, eaah5054. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Jamaluddin, M.; Zhang, Y.; Widen, S.G.; Sun, H.; Brasier, A.R.; Zhao, Y. Type II Epithelial-Mesenchymal Transition Upregulates Protein N-Glycosylation To Maintain Proteostasis and Extracellular Matrix Production. J. Proteome Res. 2019, 18, 3447–3460. [Google Scholar] [CrossRef] [PubMed]
- Collins, P.L.; Graham, B.S. Viral and host factors in human respiratory syncytial virus pathogenesis. J. Virol. 2008, 82, 2040–2055. [Google Scholar] [CrossRef] [PubMed]
- Harburger, D.S.; Calderwood, D.A. Integrin signalling at a glance. J. Cell. Sci. 2009, 122, 159–163. [Google Scholar] [CrossRef]
- Hang, Q.; Isaji, T.; Hou, S.; Wang, Y.; Fukuda, T.; Gu, J. A Key Regulator of Cell Adhesion: Identification and Characterization of Important N-Glycosylation Sites on Integrin alpha5 for Cell Migration. Mol. Cell. Biol. 2017, 37, e00558-16. [Google Scholar] [CrossRef]
- Hang, Q.; Isaji, T.; Hou, S.; Zhou, Y.; Fukuda, T.; Gu, J. N-Glycosylation of integrin alpha5 acts as a switch for EGFR-mediated complex formation of integrin alpha5beta1 to alpha6beta4. Sci. Rep. 2016, 6, 33507. [Google Scholar] [CrossRef]
- Laubli, H.; Borsig, L. Altered Cell Adhesion and Glycosylation Promote Cancer Immune Suppression and Metastasis. Front. Immunol. 2019, 10, 2120. [Google Scholar] [CrossRef]
- Nicolaou, N.; Margadant, C.; Kevelam, S.H.; Lilien, M.R.; Oosterveld, M.J.; Kreft, M.; van Eerde, A.M.; Pfundt, R.; Terhal, P.A.; van der Zwaag, B.; et al. Gain of glycosylation in integrin alpha3 causes lung disease and nephrotic syndrome. J. Clin. Investig. 2012, 122, 4375–4387. [Google Scholar] [CrossRef]
- Hollander, N.; Haimovich, J. Altered N-Linked Glycosylation in Follicular Lymphoma and Chronic Lymphocytic Leukemia: Involvement in Pathogenesis and Potential Therapeutic Targeting. Front. Immunol. 2017, 8, 912. [Google Scholar] [CrossRef]
- Huang, X.; Ye, Q.; Chen, M.; Li, A.; Mi, W.; Fang, Y.; Zaytseva, Y.Y.; O’Connor, K.L.; Vander Kooi, C.W.; Liu, S.; et al. N-glycosylation-defective splice variants of neuropilin-1 promote metastasis by activating endosomal signals. Nat. Commun. 2019, 10, 3708. [Google Scholar] [CrossRef]
- Maki, J.M.; Sormunen, R.; Lippo, S.; Kaarteenaho-Wiik, R.; Soininen, R.; Myllyharju, J. Lysyl oxidase is essential for normal development and function of the respiratory system and for the integrity of elastic and collagen fibers in various tissues. Am. J. Pathol. 2005, 167, 927–936. [Google Scholar] [CrossRef]
- Kim, Y.M.; Kim, E.C.; Kim, Y. The human lysyl oxidase-like 2 protein functions as an amine oxidase toward collagen and elastin. Mol. Biol. Rep. 2011, 38, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Xu, R. Roles of PLODs in Collagen Synthesis and Cancer Progression. Front. Cell. Dev. Biol. 2018, 6, 66. [Google Scholar] [CrossRef] [PubMed]
- van der Slot, A.J.; Zuurmond, A.M.; Bardoel, A.F.; Wijmenga, C.; Pruijs, H.E.; Sillence, D.O.; Brinckmann, J.; Abraham, D.J.; Black, C.M.; Verzijl, N.; et al. Identification of PLOD2 as telopeptide lysyl hydroxylase, an important enzyme in fibrosis. J. Biol. Chem. 2003, 278, 40967–40972. [Google Scholar] [CrossRef] [PubMed]
- Peterfi, Z.; Donko, A.; Orient, A.; Sum, A.; Prokai, A.; Molnar, B.; Vereb, Z.; Rajnavolgyi, E.; Kovacs, K.J.; Muller, V.; et al. Peroxidasin is secreted and incorporated into the extracellular matrix of myofibroblasts and fibrotic kidney. Am. J. Pathol. 2009, 175, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Peterfi, Z.; Geiszt, M. Peroxidasins: Novel players in tissue genesis. Trends Biochem. Sci. 2014, 39, 305–307. [Google Scholar] [CrossRef] [PubMed]
- Taggart, C.; Mall, M.A.; Lalmanach, G.; Cataldo, D.; Ludwig, A.; Janciauskiene, S.; Heath, N.; Meiners, S.; Overall, C.M.; Schultz, C.; et al. Protean proteases: At the cutting edge of lung diseases. Eur. Respir. J. 2017, 49, 1501200. [Google Scholar] [CrossRef]
- McKelvey, M.C.; Brown, R.; Ryan, S.; Mall, M.A.; Weldon, S.; Taggart, C.C. Proteases, Mucus, and Mucosal Immunity in Chronic Lung Disease. Int. J. Mol. Sci. 2021, 22, 5018. [Google Scholar] [CrossRef]
- Pfeffer, P.E.; Corrigan, C.J. An Imbalance between Proteases and Endogenous Protease Inhibitors in Eosinophilic Airway Disease. Am. J. Respir. Crit. Care Med. 2017, 195, 707–708. [Google Scholar] [CrossRef]
- Mulinari Turin de Oliveira, N.; Fernandes da Silva Figueiredo, I.; Cristine Malaquias da Silva, L.; Sauruk da Silva, K.; Regis Bueno, L.; Barbosa da Luz, B.; Rita Corso, C.; de Paula Werner, M.F.; Soares Fernandes, E.; Maria-Ferreira, D. Tissue Proteases and Immune Responses: Influencing Factors of COVID-19 Severity and Mortality. Pathogens 2020, 9, 817. [Google Scholar] [CrossRef]
- Ramu, S.; Akbarshahi, H.; Mogren, S.; Berlin, F.; Cerps, S.; Menzel, M.; Hvidtfeldt, M.; Porsbjerg, C.; Uller, L.; Andersson, C.K. Direct effects of mast cell proteases, tryptase and chymase, on bronchial epithelial integrity proteins and anti-viral responses. BMC Immunol. 2021, 22, 35. [Google Scholar] [CrossRef] [PubMed]
- Pejler, G. The emerging role of mast cell proteases in asthma. Eur. Respir. J. 2019, 54, 1900685. [Google Scholar] [CrossRef] [PubMed]
- McKelvey, M.C.; Weldon, S.; McAuley, D.F.; Mall, M.A.; Taggart, C.C. Targeting Proteases in Cystic Fibrosis Lung Disease. Paradigms, Progress, and Potential. Am. J. Respir. Crit. Care Med. 2020, 201, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Guay, C.; Laviolette, M.; Tremblay, G.M. Targeting serine proteases in asthma. Curr. Top. Med. Chem. 2006, 6, 393–402. [Google Scholar] [CrossRef]
- Ramirez, R.D.; Sheridan, S.; Girard, L.; Sato, M.; Kim, Y.; Pollack, J.; Peyton, M.; Zou, Y.; Kurie, J.M.; Dimaio, J.M.; et al. Immortalization of human bronchial epithelial cells in the absence of viral oncoproteins. Cancer Res. 2004, 64, 9027–9034. [Google Scholar] [CrossRef]
- Kalita, M.; Tian, B.; Gao, B.; Choudhary, S.; Wood, T.G.; Carmical, J.R.; Boldogh, I.; Mitra, S.; Minna, J.D.; Brasier, A.R. Systems Approaches to Modeling Chronic Mucosal Inflammation. BioMed Res. Int. 2013, 2013, 505864. [Google Scholar] [CrossRef]
- Garofalo, R.; Sabry, M.; Jamaluddin, M.; Yu, R.K.; Casola, A.; Ogra, P.L.; Brasier, A.R. Transcriptional activation of the interleukin-8 gene by respiratory syncytial virus infection in alveolar epithelial cells: Nuclear translocation of the RelA transcription factor as a mechanism producing airway mucosal inflammation. J. Virol. 1996, 70, 8773–8781. [Google Scholar] [CrossRef]
- Thamsen, M.; Ghosh, R.; Auyeung, V.C.; Brumwell, A.; Chapman, H.A.; Backes, B.J.; Perara, G. Small molecule inhibition of IRE1α kinase/RNase has anti-fibrotic effects in the lung. PLoS ONE 2019, 14, e0209824. [Google Scholar] [CrossRef]
- Zhao, Y.; Tian, B.; Sadygov, R.G.; Zhang, Y.; Brasier, A.R. Integrative proteomic analysis reveals reprograming tumor necrosis factor signaling in epithelial mesenchymal transition. J. Proteom. 2016, 148, 126–138. [Google Scholar] [CrossRef]
- Wisniewski, J.R.; Zielinska, D.F.; Mann, M. Comparison of ultrafiltration units for proteomic and N-glycoproteomic analysis by the filter-aided sample preparation method. Anal. Biochem. 2011, 410, 307–309. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. Available online: https://www.nature.com/articles/nbt.1511 (accessed on 21 June 2022). [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, P.; Livstone, M.S.; Lewis, S.E.; Thomas, P.D. Phylogenetic-based propagation of functional annotations within the Gene Ontology consortium. Brief. Bioinform. 2011, 12, 449–462. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Y.; Qiao, D.; Skibba, M.; Brasier, A.R. The IRE1α–XBP1s Arm of the Unfolded Protein Response Activates N-Glycosylation to Remodel the Subepithelial Basement Membrane in Paramyxovirus Infection. Int. J. Mol. Sci. 2022, 23, 9000. https://doi.org/10.3390/ijms23169000
Zhao Y, Qiao D, Skibba M, Brasier AR. The IRE1α–XBP1s Arm of the Unfolded Protein Response Activates N-Glycosylation to Remodel the Subepithelial Basement Membrane in Paramyxovirus Infection. International Journal of Molecular Sciences. 2022; 23(16):9000. https://doi.org/10.3390/ijms23169000
Chicago/Turabian StyleZhao, Yingxin, Dianhua Qiao, Melissa Skibba, and Allan R. Brasier. 2022. "The IRE1α–XBP1s Arm of the Unfolded Protein Response Activates N-Glycosylation to Remodel the Subepithelial Basement Membrane in Paramyxovirus Infection" International Journal of Molecular Sciences 23, no. 16: 9000. https://doi.org/10.3390/ijms23169000
APA StyleZhao, Y., Qiao, D., Skibba, M., & Brasier, A. R. (2022). The IRE1α–XBP1s Arm of the Unfolded Protein Response Activates N-Glycosylation to Remodel the Subepithelial Basement Membrane in Paramyxovirus Infection. International Journal of Molecular Sciences, 23(16), 9000. https://doi.org/10.3390/ijms23169000