
The Pivotal Role of NF-kB in the Pathogenesis and Therapeutics of Alzheimer’s Disease




Abstract
:1. Introduction
2. The NF-κB Family and Its General Role in Inflammation
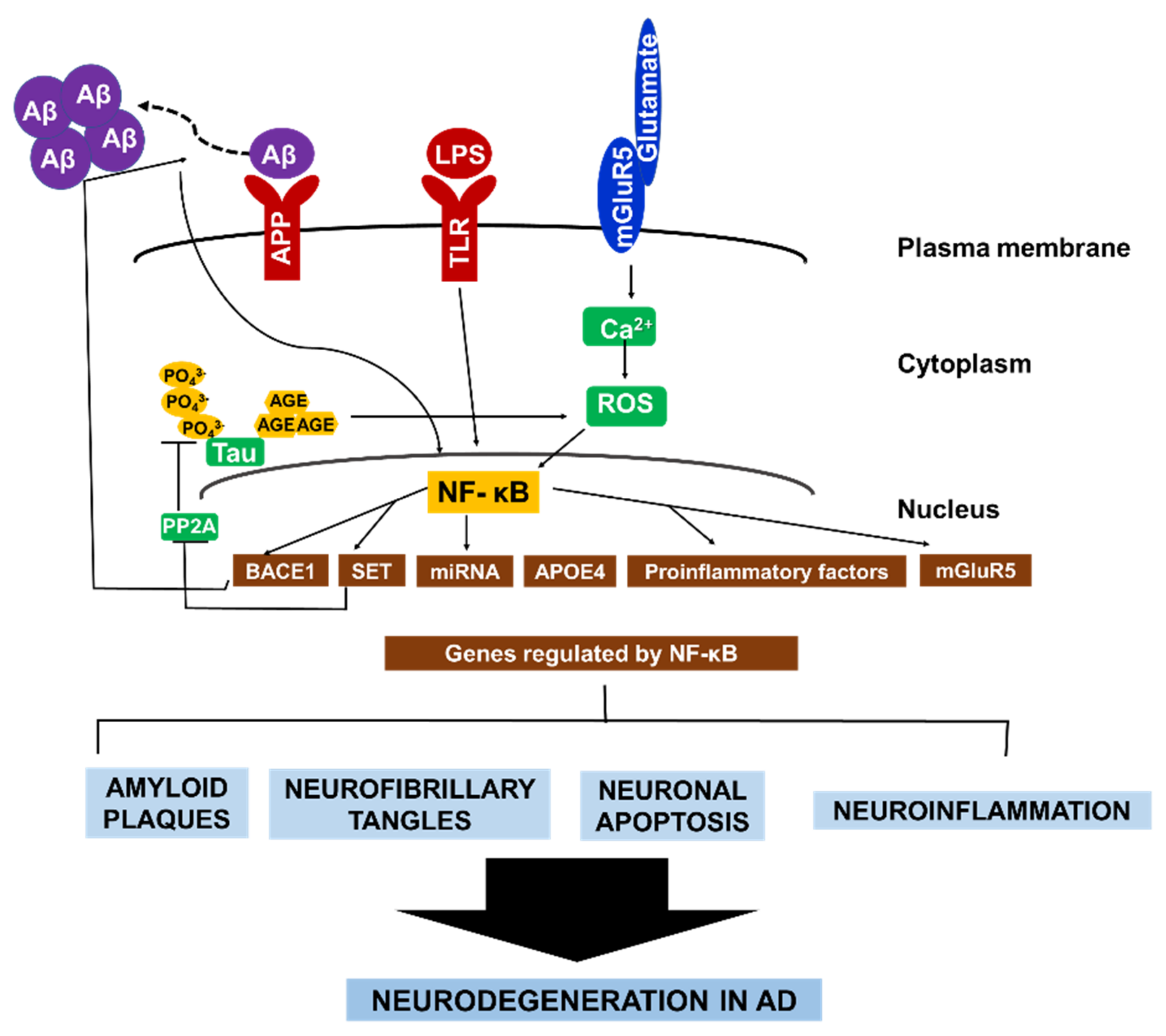
3. Role of NF-κB in AD
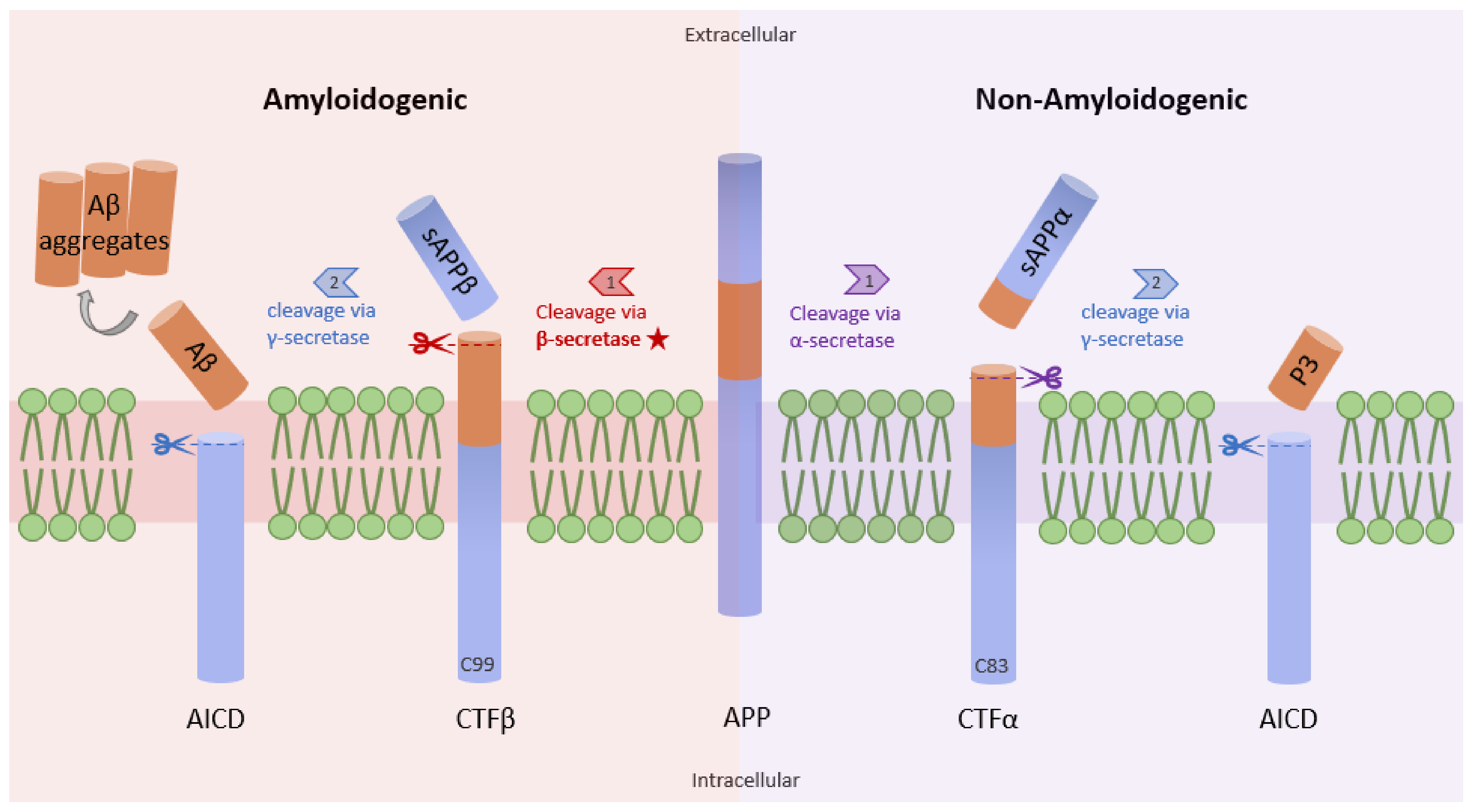
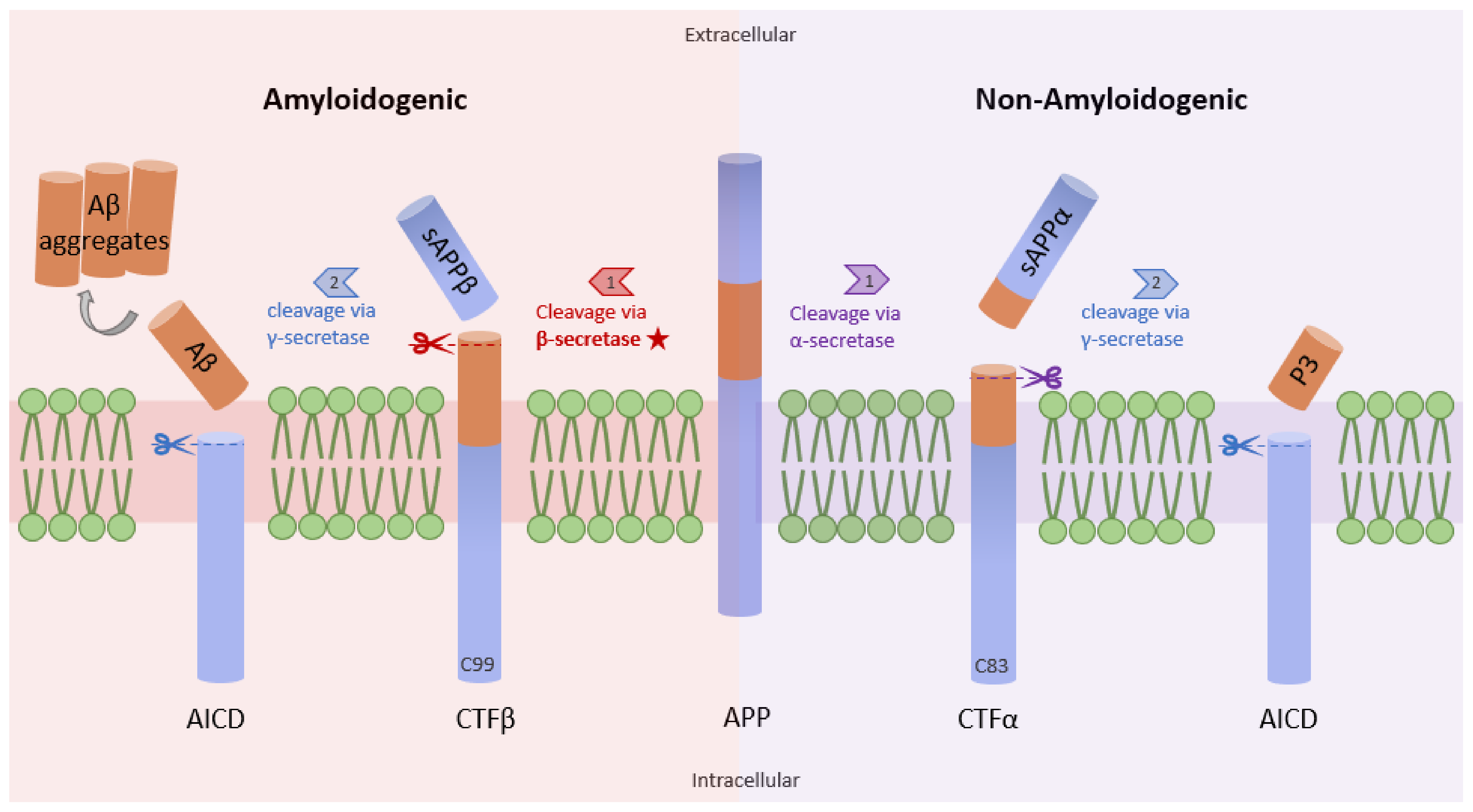
3.1. NF-κB and β-Secretase in AD
3.2. NF-κB in Reactive Microglia and Astrocytes
3.3. NF-κB and ApoE in AD
3.4. NF-κB and Glutamate in AD
3.5. NF-κB and miRNAs in AD
3.6. NF-κB and Tau Pathology in AD
4. Overview of Drugs That Interfere with NF-κB Signaling and Other AD Treatments
5. Conclusions and Perspective
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- NHS. Available online: https://www.nhs.uk/conditions/alzheimers-disease/ (accessed on 1 May 2022).
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef]
- Motolani, A.; Martin, M.; Sun, M.; Lu, T. NF-κB and Cancer Therapy Drugs. In Reference Module in Biomed Science; Elsevier: Amsterdam, The Netherlands, 2021. [Google Scholar]
- Sun, S.C. Non-canonical NF-κB signaling pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Chiarini, A.; Armato, U.; Hu, P.; Prà, I.D. Danger-Sensing/Patten Recognition Receptors and Neuroinflammation in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 9036. [Google Scholar] [CrossRef] [PubMed]
- Thawkar, B.S.; Kaur, G. Inhibitors of NF-κB and P2X7/NLRP3/Caspase 1 pathway in microglia: Novel therapeutic opportunities in neuroinflammation induced early-stage Alzheimer’s disease. J. Neuroimmun. 2019, 326, 62–74. [Google Scholar] [CrossRef]
- Ries, M.; Sastre, M. Mechanisms of Aβ Clearance and Degradation by Glial Cells. Front. Aging Neurosci. 2016, 8, 160. [Google Scholar] [CrossRef]
- Spires-Jones, T.L.; Hyman, B.T. The Intersection of Amyloid Beta and Tau at Synapses in Alzheimer’s Disease. Neuron 2014, 82, 756–771. [Google Scholar] [CrossRef]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimers Dement. 2016, 12, 719–732. [Google Scholar] [CrossRef]
- Jha, N.K.; Jha, S.K.; Kar, R.; Nand, P.; Swati, K.; Goswami, V.K. Nuclear factor κB as a therapeutic target for Alzheimer’s disease. J. Neurochem. 2019, 150, 113–137. [Google Scholar] [CrossRef]
- Sarnico, I.; Lanzillotta, A.; Benarese, M.; Alghisi, M.; Baiguera, C.; Battistin, L.; Spano, P.; Pizzi, M. NF-κB dimers in the regulation of neuronal survival. Int. Rev. Neurobiol. 2009, 85, 351–362. [Google Scholar]
- Srinivasan, M.; Lahiri, D.K. Significance of NF-κB as a pivotal therapeutic target in the neurodegenerative pathologies of Alzheimer’s disease and multiple sclerosis. Expert. Opin. Ther. Targets 2015, 19, 471–487. [Google Scholar] [CrossRef]
- Chen, C.H.; Zhou, W.; Liu, S.; Deng, Y.; Cai, F.; Tone, M.; Tone, Y.; Tong, Y.; Song, W. Increased NF-κB signalling upregulates BACE1 expression and its therapeutic potential in Alzheimer’s disease. Int. J. Neuropsychopharmacol. 2012, 15, 77–90. [Google Scholar] [CrossRef]
- Cole, S.L.; Vassar, R. The Alzheimer’s disease Beta-secretase enzyme, BACE1. Mol. Neurodegener. 2007, 2, 22. [Google Scholar] [CrossRef]
- Snow, W.M.; Albensi, B.C. Neuronal gene targets of NF-κB and their dysregulation in Alzheimer’s disease. Front. Mol. Neurosci. 2016, 9, 118. [Google Scholar] [CrossRef]
- Valerio, A.; Boroni, F.; Benarese, M.; Sarnico, I.; Ghisi, V.; Bresciani, L.G.; Ferrario, M.; Borsani, G.; Spano, P.; Pizzi, M. NF-κB pathway: A target for preventing β-amyloid (Aβ)-induced neuronal damage and Aβ42 production. Eur. J. Neurosci. 2006, 23, 1711–1720. [Google Scholar] [CrossRef]
- Behl, C.; Davis, J.B.; Lesley, F.; Schubert, D. Hydrogen peroxide mediates amyloid p protein toxicity. Cell 1994, 77, 817–827. [Google Scholar] [CrossRef]
- Lukiw, W.J. Bacteroides fragilis lipopolysaccharide and inflammatory signaling in Alzheimer’s Disease. Front. Microbiol. 2016, 7, 1544. [Google Scholar] [CrossRef]
- Zhan, X.; Stamova, B.; Sharp, F.R. Lipopolysaccharide associates with amyloid plaques, neurons and oligodendrocytes in Alzheimer’s disease brain: A review. Front. Aging Neurosci. 2018, 10, 42. [Google Scholar] [CrossRef]
- González-Reyes, R.E.; Nava-Mesa, M.O.; Vargas-Sánchez, K.; Ariza-Salamanca, D.; Mora-Muñoz, L. Involvement of Astrocytes in Alzheimer’s Disease from a Neuroinflammatory and Oxidative Stress Perspective. Front. Mol. Neurosci. 2017, 10, 427. [Google Scholar] [CrossRef]
- Kaur, D.; Sharma, V.; Deshmukh, R. Activation of microglia and astrocytes: A roadway to neuroinflammation and Alzheimer’s disease. Inflammopharmacology 2019, 27, 663–677. [Google Scholar] [CrossRef]
- Cirillo, C.; Capoccia, E.; Iuvone, T.; Cuomo, R.; Sarnelli, G.; Steardo, L.; Esposito, G. S100B Inhibitor Pentamidine Attenuates Reactive Gliosis and Reduces Neuronal Loss in a Mouse Model of Alzheimer’s Disease. BioMed. Res. Int. 2015, 2015, 508342. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Zhao, N.; Caulfield, T.R.; Liu, C.C.; Bu, G. Apolipoprotein E and Alzheimer disease: Pathobiology and targeting strategies. Nat. Rev. Neurol. 2019, 15, 501–518. [Google Scholar] [CrossRef]
- Du, Y.; Chen, X.; Wei, X.; Bales, K.R.; Berg, D.T.; Paul, S.M.; Farlow, M.R.; Maloney, B.; Ge, Y.W.; Lahiri, D.K. NF-κB mediates amyloid β peptide-stimulated activity of the human apolipoprotein E gene promoter in human astroglial cells. Mol. Brain Res. 2005, 136, 177–188. [Google Scholar] [CrossRef]
- Ophir, G.; Amariglio, N.; Jacob-Hirsch, J.; Elkon, R.; Rechavi, G.; Michaelson, D.M. Apolipoprotein E4 enhances brain inflammation by modulation of the NF-κB signaling cascade. Neurobiol. Dis. 2005, 20, 709–718. [Google Scholar] [CrossRef]
- Harkany, T.; Timmerman, W.; Laskay, G.; To, B.; Sasva Âri, M.; Ko, C.; Sebens, J.B.; Korf, J.; Nyakas, C.; Zara, M.; et al. b-Amyloid neurotoxicity is mediated by a glutamate-triggered excitotoxic cascade in rat nucleus basalis. Eur. J. Neurosci. 2000, 12, 2735–2745. [Google Scholar] [CrossRef]
- Mattson, M.P.; Cheng, B.; Davis, D.; Bryant, K.; Lieberburg, I.; Rydel, R.E. beta-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J. Neurosci. 1992, 12, 376–389. [Google Scholar] [CrossRef]
- Lim, D.; Iyer, A.; Ronco, V.; Grolla, A.A.; Canonico, P.L.; Aronica, E.; Genazzani, A.A. Amyloid beta deregulates astroglial mGluR5-mediated calcium signaling via calcineurin and NF-κB. Glia 2013, 61, 1134–1145. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Wertz, I.; O’Rourke, K.; Ultsch, M.; Seshagiri, S.; Eby, M.; Xiao, W.; Dixit, V.M. Bcl10 activates the NF-κB pathway through ubiquitination of NEMO. Nature 2004, 427, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J. NF-κB-regulated, proinflammatory miRNAs in Alzheimer’s disease. Alzheimer’s Res. Ther. 2012, 4, 47. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Bhattacharjee, S.; Jones, B.M.; Hill, J.; Dua, P.; Lukiw, W.J. Regulation of neurotropic signaling by the inducible, NF-κB-sensitive miRNA-125b in Alzheimer’s disease (AD) and in primary human neuronalglial (HNG) cells. Mol. Neurobiol. 2014, 50, 97–106. [Google Scholar] [CrossRef]
- Lukiw, W.J.; Alexandrov, P.N. Regulation of Complement Factor H (CFH) by Multiple miRNAs in Alzheimer’s Disease (AD) Brain. Mol. Neurobiol. 2012, 46, 11–19. [Google Scholar] [CrossRef]
- Zhao, Y.; Bhattacharjee, S.; Jones, B.M.; Dua, P.; Alexandrov, P.N.; Hill, J.M.; Lukiw, W.J. Regulation of TREM2 expression by an NF-κB-sensitive miRNA-34a. NeuroReport 2013, 24, 318–323. [Google Scholar] [CrossRef]
- Feng, Y.; Li, X.; Zhou, W.; Lou, D.; Huang, D.; Li, Y.; Kang, Y.; Xiang, Y.; Li, T.; Zhou, W.; et al. Regulation of SET gene expression by NF-κB. Mol. Neurobiol. 2017, 54, 4477–4485. [Google Scholar] [CrossRef]
- Yan, S.D.; Yan, S.F.; Chen, X.; Fu, J.; Chen, M.; Kuppusamy, P.; Smith, M.A.; Perry, G.; Godman, G.C.; Nawroth, P.; et al. Non-enzymatically glycated tau in Alzheimer’s disease induces neuronal oxidant stress resulting in cytokine gene expression and release of amyloid β-peptide. Nat. Med. 1995, 1, 693–699. [Google Scholar] [CrossRef]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and future treatments for Alzheimer’s disease. Ther. Adv. Neurol. Disord. 2013, 6, 19–33. [Google Scholar] [CrossRef]
- Wang, F.; Zou, Z.; Gong, Y.; Yuan, D.; Chen, X.; Sun, T. Regulation of Human Brain Microvascular Endothelial Cell Adhesion and Barrier Functions by Memantine. J. Mol. Neurosci. 2017, 62, 123–129. [Google Scholar] [CrossRef]
- Medeiros, R.; Kitazawa, M.; Passos, G.F.; Baglietto-Vargas, D.; Cheng, D.; Cribbs, D.H.; LaFerla, F.M. Aspirin-Triggered Lipoxin A4 Stimulates Alternative Activation of Microglia and Reduces Alzheimer Disease-Like Pathology in Mice. Am. J. Pathol. 2013, 182, 1780–1789. [Google Scholar] [CrossRef]
- Ryan, J.; Storey, E.; Murray, A.M.; Woods, R.L.; Wolfe, R.; Reid, C.M.; Nelson, M.R.; Chong, T.T.; Williamson, J.D.; Ward, S.A.; et al. Randomized placebo-controlled trial of the effects of aspirin on dementia and cognitive decline. Neurology 2020, 95, e320–e331. [Google Scholar] [CrossRef]
- Cai, Z.; Zhao, Y.; Yao, S.; Zhao, B. Increases in β-amyloid protein in the hippocampus caused by diabetic metabolic disorder are blocked by minocycline through inhibition of NF-κB pathway activation. Pharmacol. Rep. 2011, 63, 381–391. [Google Scholar] [CrossRef]
- Gagliardi, S.; Franco, V.; Sorrentino, S.; Zucca, S.; Pandini, C.; Rota, P.; Bernuzzi, S.; Costa, A.; Sinforiani, E.; Pansarasa, O.; et al. Curcumin and Novel Synthetic Analogs in Cell-Based Studies of Alzheimer’s Disease. Front. Pharmacol. 2018, 9, 1404. [Google Scholar] [CrossRef]
- Chen, J.; Zhou, Y.; Mueller-Steiner, S.; Chen, L.F.; Kwon, H.; Yi, S.; Mucke, L.; Gan, L. SIRT1 protects against microglia-dependent amyloid-beta toxicity through inhibiting NF-κB signaling. J. Biol. Chem. 2005, 280, 40364–40374. [Google Scholar] [CrossRef]
- Kong, F.; Jiang, X.; Wang, R.; Zhai, S.; Zhang, Y.; Wang, D. Forsythoside B attenuates memory impairment and neuroinflammation via inhibition on NF-κB signaling in Alzheimer’s disease. J. Neuroinflam. 2020, 17, 305. [Google Scholar] [CrossRef]
- Wang, C.; Fan, L.; Khawaja, R.R.; Liu, B.; Zhan, L.; Kodama, L.; Chin, M.; Li, Y.; Le, D.; Zhou, Y.; et al. Microglial NF-κB drives tau spreading and toxicity in a mouse model of tauopathy. Nat. Commun. 2022, 13, 1969. [Google Scholar] [CrossRef]
- Lindsay, A.; Hickman, D.; Srinivasan, M. A nuclear factor-kappa B inhibiting peptide suppresses innate immune receptors and gliosis in a transgenic mouse model of Alzheimer’s disease. Biomed. Pharmacother. 2021, 138, 111405. [Google Scholar] [CrossRef]
- Wei, H.; Wang, B.; Miyagi, M.; She, Y.; Gopalan, B.; Huang, D.B.; Ghosh, G.; Stark, G.R.; Lu, T. PRMT5 dimethylates R30 of the p65 subunit to activate NF-κB. Proc. Natl. Acad. Sci. USA 2013, 110, 13516–13521. [Google Scholar] [CrossRef]
- Prabhu, L.; Wei, H.; Chen, L.; Demir, Ö.; Sandusky, G.; Sun, E.; Wang, J.; Mo, J.; Zeng, L.; Fishel, M.; et al. Adapting AlphaLISA high throughput screen to discover a novel small-molecule inhibitor targeting protein arginine methyltransferase 5 in pancreatic and colorectal cancers. Oncotarget 2017, 20, 39963–39977. [Google Scholar] [CrossRef]
- Lu, T.; Jackson, M.W.; Singhi, A.D.; Kandel, E.S.; Yang, M.; Gudkov, A.V.; Stark, G.R. Validation-based insertional mutagenesis to identify the FBXL11 as a negative regulator of NF-κB. Proc. Natl. Acad. Sci. USA 2009, 106, 16339–16344. [Google Scholar] [CrossRef]
- Lu, T.; Jackson, M.W.; Wang, B.; Yang, M.; Chance, M.; Miyagi, M.; Gudkov, A.V.; Stark, G.R. Regulation of NF-κB by NSD1/FBXL11-dependent reversible lysine methylation of p65. Proc. Natl. Acad. Sci. USA 2010, 107, 46–51. [Google Scholar] [CrossRef]
- Lu, T.; Stark, G.R. NF-κB: Regulation by methylation. Cancer Res. 2015, 75, 3692–3695. [Google Scholar] [CrossRef] [PubMed]
- Quan, X.; Yue, W.; Luo, Y.; Cao, J.; Wang, H.; Wang, Y.; Lu, Z. The protein arginine methyltransferase PRMT5 regulates Aβ-induced toxicity in human cells and Caenorhabditis elegans models of Alzheimer’s disease. J. Neurochem. 2015, 134, 969–977. [Google Scholar] [CrossRef]
- Butchart, J.; Brook, L.; Hopkins, V.; Teeling, J.; Püntener, U.; Culliford, D.; Sharples, R.; Sharif, S.; McFarlane, B.; Raybould, R.; et al. Etanercept in Alzheimer disease: A randomized, placebo-controlled, double-blind, phase 2 trial. Neurology 2015, 84, 2161–2168. [Google Scholar] [CrossRef] [PubMed]
- Miao Yin, Y.; Du, J.; Wang, Z. Aβ metabolism and the role of APOE in Alzheimer’s disease. J. Alzheimer’s Dis. Par-Kinson 2016, 6, 285. [Google Scholar] [CrossRef]
- Hock, C.; Konietzko, U.; Streffer, J.R.; Tracy, J.; Signorell, A.; Mü Ller-Tillmanns, B.; Lemke, U.; Henke, K.; Moritz, E.; Garcia, E.; et al. Antibodies against beta-amyloid slow cognitive decline in Alzheimer’s disease. Neuron 2003, 38, 547–554. [Google Scholar] [CrossRef]
- Winblad, B.; Blum, K.I. Hints of a Therapeutic Vaccine for Alzheimer’s? Neuron 2003, 38, 517–518. [Google Scholar] [CrossRef]
- Lee, H.E.; Lim, D.; Lee, J.Y.; Lim, S.M.; Pae, A.N. Recent tau-targeted clinical strategies for the treatment of Alzheimer’s disease. Futur. Med. Chem. 2019, 11, 1845–1848. [Google Scholar] [CrossRef]
- Ostrowitzki, S.; Lasser, R.A.; Dorflinger, E.; Scheltens, P.; Barkhof, F.; Nikolcheva, T.; Ashford, E.; Retout, S.; Hofmann, C.; Delmar, P.; et al. A phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimer’s Res. Ther. 2017, 9, 95. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer disease and aducanumab: Adjusting our approach. Nat. Rev. Neurol. 2019, 15, 365–366. [Google Scholar] [CrossRef]
- Beshir, S.A.; Aadithsoorya, A.M.; Parveen, A.; Goh, S.S.L.; Hussain, N.; Menon, V.B. Aducanumab Therapy to Treat Alzheimer’s Disease: A Narrative Review. Int. J. Alzheimer’s Dis. 2022, 2022, 9343514. [Google Scholar] [CrossRef]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Moussa-Pacha, N.M.; Abdin, S.M.; Omar, H.A.; Alniss, H.; Al-Tel, T.H. BACE1 inhibitors: Current status and future directions in treating Alzheimer’s disease. Med. Res. Rev. 2020, 40, 339–384. [Google Scholar] [CrossRef]
- Reddy, A.; Reddy, P. Mitochondria-Targeted Molecules as Potential Drugs to Treat Patients with Alzheimer’s Disease. Prog. Mol. Biol. Transl. Sci. 2017, 146, 173–201. [Google Scholar] [CrossRef]
- Tong, C.K.B.; Wu, A.J.; Li, M.; Cheung, K.-H. Calcium signaling in Alzheimer’s disease & therapies. Biochim. Biophy. Acta-Mol. Cell Res. 2018, 1865, 1745–1760. [Google Scholar] [CrossRef]
- Liang, Z.Q.; Wang, X.; Li, L.Y.; Wang, Y.; Chen, R.W.; Chuang, D.M.; Chase, T.N.; Qin, Z.H. Nuclear factor-κB-dependent cyclin D1 induction and DNA replication associated with N-methyl-D-aspartate receptor-mediated apoptosis in rat striatum. J. Neurosci. Res. 2007, 85, 1295–1309. [Google Scholar] [CrossRef]
- Sarnico, I.; Boroni, F.; Benarese, M.; Alghisi, M.; Valerio, A.; Battistin, L.; Spano, P.; Pizzi, M. Targeting IKK2 by pharmacological inhibitor AS602868 prevents excitotoxic injury to neurons and oligodendrocytes. J. Neural. Transmiss. 2008, 115, 693–701. [Google Scholar] [CrossRef]
- Van Marum, R.J. Update on the use of memantine in Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2009, 5, 237–247. [Google Scholar] [CrossRef]
- O’Gorman, C.; Jones, A.; Tabuteau, H. P2-033: AXS-05 (Dextromethorphan/Bupropion): An Innovative Treatment in Clinical Development for Agitation Associated with Alzheimer’s Disease. Alzheimer’s Dement. 2006, 14, 679. [Google Scholar] [CrossRef]
- Cacabelos, R. Donepezil in Alzheimer’s disease: From conventional trials to pharmacogenetics. Neuropsychiatr. Dis. Treat. 2007, 3, 303–333. [Google Scholar]
- Zhang, Y.; Wang, Y.; Zhang, C.; Wang, Y.; Li, H.; Li, J.; Luo, Y.; Liu, D.; Bai, H. Pharmacological study of nimodipine plus donepezil in treating senile dementia. Int. J. Clin. Exp. Med. 2016, 9, 4497–4502. [Google Scholar]
- Birks, J.S.; Evans, J.G. Rivastigmine for Alzheimer’s disease. Cochrane Database Syst. Rev. 2015, 4, 1–98. [Google Scholar]
- Marcusson, J.; Bullock, R.; Gauthier, S.; Kurz, A.; Schwalen, S. Galantamine Demonstrates Efficacy and Safety in Elderly Patients with Alzheimer Disease. Alzheimer Dis. Assoc. Disord. 2003, 17, S86–S91. [Google Scholar] [CrossRef]
- Cao, J.; Hou, J.; Ping, J.; Cai, D. Advances in developing novel therapeutic strategies for Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 64. [Google Scholar] [CrossRef]
- Lopez Lopez, C.; Tariot, P.N.; Caputo, A.; Langbaum, J.B.; Liu, F.; Riviere, M.E.; Langlois, C.; Rouzade-Dominguez, M.L.; Zalesak, M.; Hendrix, S.; et al. The Alzheimer’s prevention initiative generation program: Study design of two randomized controlled trials for individuals at risk for clinical onset of Alzheimer’s disease. Alzheimer’s Dement Transl. Res. Clin. Interv. 2019, 5, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Lynch, S.Y.; Kaplow, J.; Zhao, J.; Dhadda, S.; Luthman, J.; Albala, B. P4-389: Elenbecestat, E2609, a bace inhibitor: Results from a phase-2 study in subjects with mild cognitive impairment and mild-to-moderate dementia due to Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, P1623. [Google Scholar] [CrossRef]
- Cramer, P.E.; Cirrito, J.R.; Wesson, D.W.; Lee, C.Y.D.; Karlo, J.C.; Zinn, A.E.; Casali, B.T.; Restivo, J.L.; Goebel, W.D.; James, M.J.; et al. ApoE-Directed Therapeutics Rapidly Clear β-Amyloid and Reverse Deficits in AD Mouse Models. Science 2012, 335, 1503–1506. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Breitbart, A.; Sun, Y.; Mehta, P.D.; Boutajangout, A.; Scholtzova, H.; Wisniewski, T. Blocking the apolipoprotein E/amyloid β interaction in triple transgenic mice ameliorates Alzheimer’s disease related amyloid β and tau pathology. J. Neurochem. 2014, 128, 577–591. [Google Scholar] [CrossRef]
- Alam, R.; Driver, D.; Wu, S.; Lozano, E.; Key, S.L.; Hole, J.T.; Hayashi, M.L.; Lu, J. Preclinical characterization of an antibody [LY3303560] targeting aggregated tau. Alzheimer’s Dement. 2017, 13, P592–P593. [Google Scholar] [CrossRef]
- He, W.; Wang, C.; Chen, Y.; He, Y.; Cai, Z. Berberine attenuates cognitive impairment and ameliorates tau hyperphosphorylation by limiting the self-perpetuating pathogenic cycle between NF-κB signaling, oxidative stress and neuroinflammation. Pharm. Rep. 2017, 69, 1341–1348. [Google Scholar] [CrossRef]
- Serenó, L.; Coma, M.; Rodríguez, M.; Sánchez-Ferrer, P.; Sánchez, M.; Gich, I.; Agulló, J.; Pérez, M.; Avila, J.; Guardia-Laguarta, C.; et al. A novel GSK-3β inhibitor reduces Alzheimer’s pathology and rescues neuronal loss in vivo. Neurobiol. Dis. 2009, 35, 359–367. [Google Scholar] [CrossRef]
- Morich, F.J.; Bieber, F.; Lewis, J.M.; Kaiser, L.; Cutler, N.R.; Escobar, J.I.; Willmer, J.; Petersen, R.C.; Reisbergs, B. Nimodipine in the treatment of probable Alzheimer’s disease results of two multicentre trials. Clin. Use Clin. Drug Investig. 1996, 11, 185–195. [Google Scholar] [CrossRef]
- Popović, N.; Morales-Delgado, N.; Vidal Mena, D.; Alonso, A.; Pascual Martínez, M.; Caballero Bleda, M.; Popović, M. Verapamil and Alzheimer’s disease: Past, present, and future. Front. Pharmacol. 2020, 11, 562. [Google Scholar] [CrossRef]
- Green, K.N.; Khashwji, H.; Estrada, T.; Laferla, F.M. ST101 induces a novel 17 kDa APP cleavage that precludes Aβ generation in vivo. Ann. Neurol. 2011, 69, 831–844. [Google Scholar] [CrossRef]
- Kon, N.; Satoh, A.; Miyoshi, N. A small-molecule DS44170716 inhibits Ca2+-induced mitochondrial permeability transition. Sci. Rep. 2017, 7, 3864. [Google Scholar] [CrossRef]
- Manczak, M.; Mao, P.; Calkins, M.J.; Cornea, A.; Reddy, A.P.; Murphy, M.P.; Szeto, H.H.; Park, B.; Reddy, P.H. Mitochondria-targeted antioxidants protect against amyloid-β toxicity in Alzheimer’s disease neurons. J. Alzheimer’s Dis. 2010, 20, S609–S631. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Target Protein/Pathway | Drug | Mechanism of Action | Inhibition of NF-κB Signaling | Stage of Development | References/Clinical trial ID |
|---|---|---|---|---|---|
| NF-κB | Etanercept (Enbrel™) | Inhibits TNF-α activity, and consequently NF-κB signaling | Yes | Phase 2 clinical trial | [52] ClinicalTrials.gov Identifier: NCT01068353 |
| NSAIDs | Inhibits NF-κB signaling and other inflammatory pathways. | Yes | Preclinical | [13] | |
| SN50 | Blocks NF-κB nuclear translocation | Yes | Preclinical | [64] | |
| AS62868 | Inhibits IKKβ | Yes | Preclinical | [13,65] | |
| Curcumin and curcuminoids | Decreases NF-κB and BACE1 expression | Yes | Preclinical | [41] | |
| Resveratrol | Deacetylation of lysine 310 on p65 | Yes | Preclinical | [42] | |
| Forsythoside B | Decreases phosphorylation of IKKα/β, IκBα, and p65 at serine 536 | Yes | Preclinical | [43] | |
| TPCA-1 | Inhibits IKKβ | Yes | Preclinical | [44] | |
| Glucocorticoid induced leucine zipper (GILZ) analogs | Bind to p65 transactivation domain | Yes | Preclinical | [45] | |
| NMDAR | Memantine | Antagonizes NMDA receptor | Yes | FDA Approved | [66] |
| AXS-05 | Antagonizes NMDAR, nicotinic receptor, serotonin and norepinephrine transporters, and agonizes sigma-1 receptor. | Unknown | Phase 2/3 clinical trial | [67] ClinicalTrials.gov Identifier: NCT03226522 | |
| Cholinergic system | Donepezil | Inhibits acetylcholinesterase | Unknown | FDA approved | [68,69] |
| Rivastigmine | Inhibits acetylcholinesterase | Unknown | FDA approved | [70] | |
| Galantamine | Allosterically potentiates nicotinic receptor activity and inhibits acetylcholinesterase | Unknown | FDA approved | [71] | |
| Amyloid-β | ALZT-OP1 (cromolyn+ ibuprofen) | Prevents Aβ aggregation and neuroinflammation | Unknown | Phase 3 clinical trial | [72] ClinicalTrials.gov Identifier: NCT02547818 |
| CAD 106 | Binds to Aβ to elicit immune response | Unknown | Phase 2/3 clinical trial | [72] ClinicalTrials.gov Identifier: NCT02565511 | |
| CNP520 | Inhibits BACE1 | Unknown | Phase 2/3 clinical trial | [73] ClinicalTrials.gov Identifier: NCT02565511 | |
| E2609 (Elenbecestat) | BACE inhibitor | Unknown | Phase 3 clinical trial | [74] ClinicalTrials.gov Identifier: NCT02956486 | |
| Solanezumab, gantenerumab | Aβ monoclonal antibodies | Unknown | Phase 2/3 clinical trial | ClinicalTrials.gov Identifier: NCT01760005 | |
| APOE | Bexarotene | Binds to Retinoid X receptor (RXR) agonist to increase expression of APOE which facilitates Aβ clearance. | Unknown | Preclinical | [23,75] |
| PH002 | Corrects the structure of the APOE4 protein associated with neuropathology in AD | Unknown | Preclinical | [76] | |
| Aβ12–28P | Binds to APOE4 to prevent Aβ binding, inhibiting Aβ fibril formation. | Unknown | Preclinical | [76] | |
| Tau | LMTX(TRx0237) | Inhibits aggregation of hyperphosphorylated tau | Unknown | Phase 3 clinical trial | ClinicalTrials.gov Identifier: NCT03446001 |
| BIIB080(IONIS MAPTRx) | Inhibits the translation of tau mRNA | Unknown | Phase 2 clinical trial | [56] ClinicalTrials.gov Identifier: NCT03186989 | |
| LY3303560 (Zagotenemab) | Monoclonal antibody to tau aggregates | Unknown | Phase 2 | [77] ClinicalTrials.gov Identifier: NCT03518073 | |
| Berberine | Inhibits tau phosphorylation and NF-κB signaling | Yes | Preclinical | [78] | |
| NP12 | Inhibits GSK-3β to reduce tau phosphorylation | Unknown | Preclinical | [79] | |
| Calcium signaling | Nimodepine | Inhibits L-type Voltage-gated calcium channel (VGCC) | Unknown | Preclinical as single agent. Phase 1 clinical trial in combination with donepezil | [69,80] |
| Verapamil | Blocks L-, N-, R- and T-type VGCC in 3xTg AD mice | Unknown | Preclinical | [81] | |
| ST101 | Inhibits T-type VGCC in 3xTg AD mice | Unknown | Preclinical | [82] | |
| Mitochondrial proteins | DS44170716 | Inhibits mitochondrial permeability transition which mediates cell death | Unknown | Preclinical | [83] |
| Mito Q, SS31, resveratrol | Targets multiple mitochondrial protein to decrease Aβ induced toxicity and oxidative stress. | Unknown | Preclinical | [84] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, E.; Motolani, A.; Campos, L.; Lu, T. The Pivotal Role of NF-kB in the Pathogenesis and Therapeutics of Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 8972. https://doi.org/10.3390/ijms23168972
Sun E, Motolani A, Campos L, Lu T. The Pivotal Role of NF-kB in the Pathogenesis and Therapeutics of Alzheimer’s Disease. International Journal of Molecular Sciences. 2022; 23(16):8972. https://doi.org/10.3390/ijms23168972
Chicago/Turabian StyleSun, Emily, Aishat Motolani, Leonardo Campos, and Tao Lu. 2022. "The Pivotal Role of NF-kB in the Pathogenesis and Therapeutics of Alzheimer’s Disease" International Journal of Molecular Sciences 23, no. 16: 8972. https://doi.org/10.3390/ijms23168972
APA StyleSun, E., Motolani, A., Campos, L., & Lu, T. (2022). The Pivotal Role of NF-kB in the Pathogenesis and Therapeutics of Alzheimer’s Disease. International Journal of Molecular Sciences, 23(16), 8972. https://doi.org/10.3390/ijms23168972