
Epigenetic Regulation of Optic Nerve Development, Protection, and Repair

 , ,
, ,
Abstract
1. Introduction
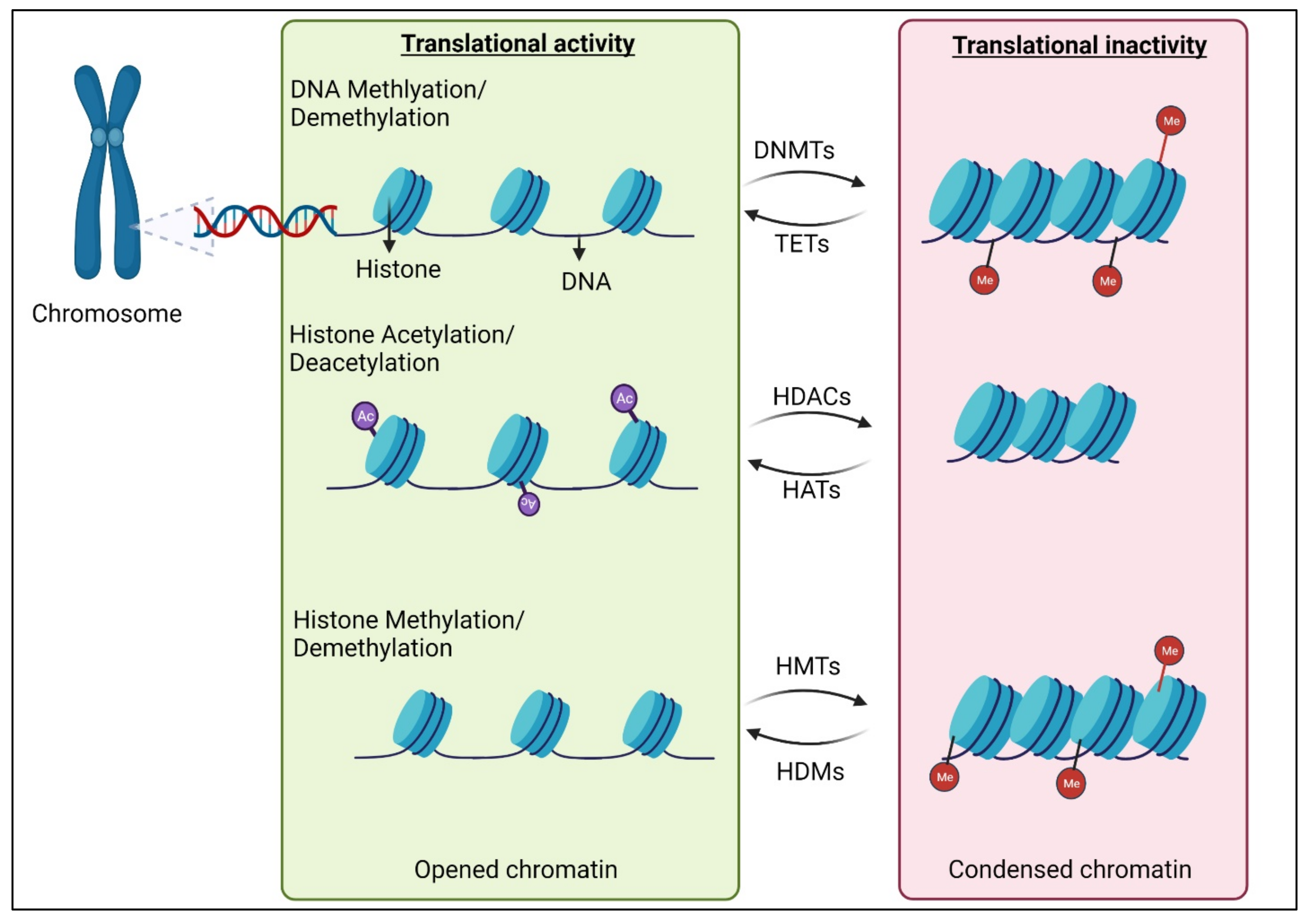
2. Epigenetic Modifications of DNAs and Histones
3. Epigenetics in Optic Nerve and Retinal Development
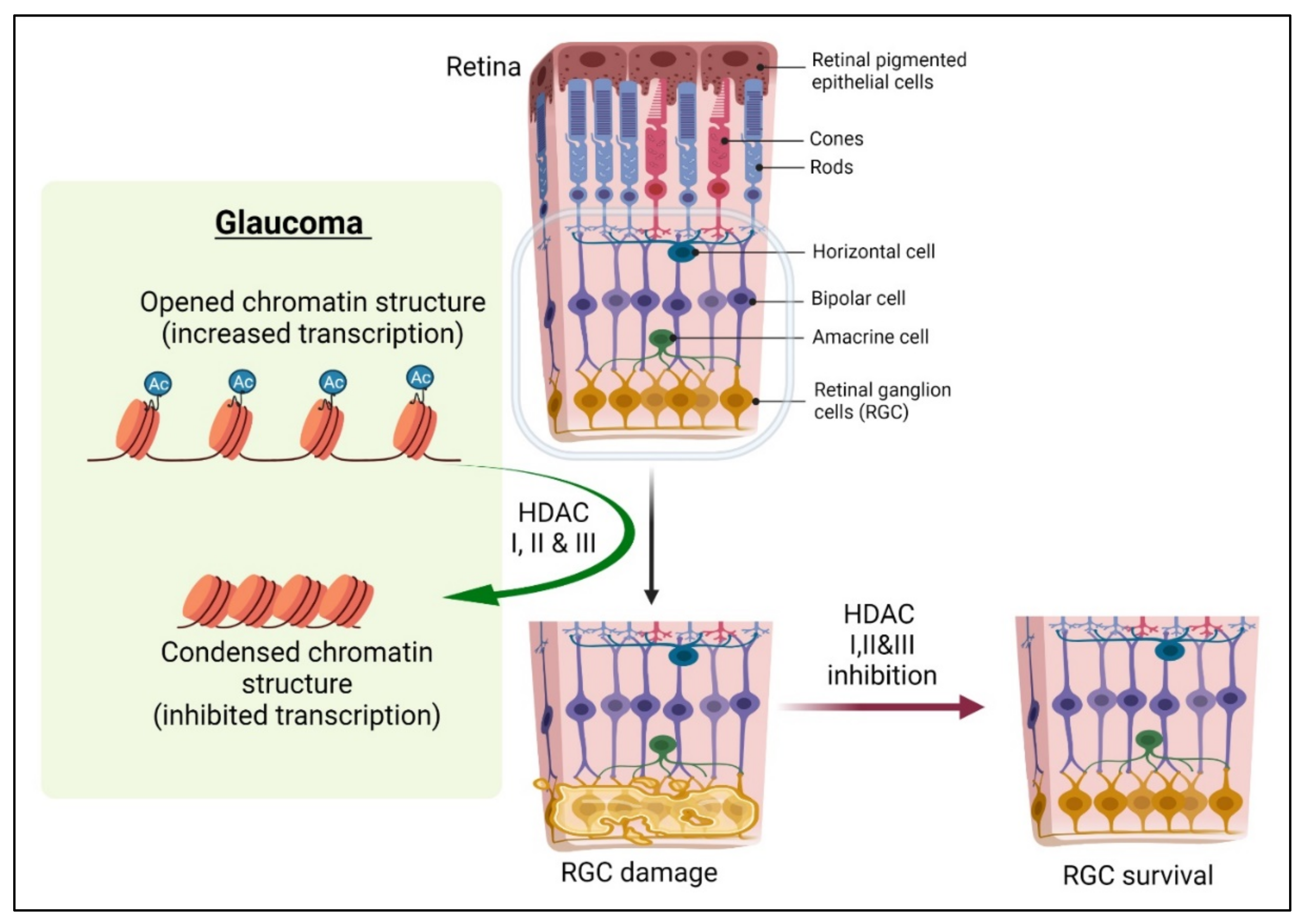
4. DNA Modification in Optic Nerve Repair and Protection
5. Histone Modifications in Optic Nerve Repair and Protection
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Waddington, C.H. The epigenotype. Int. J. Epidemiol. 1942, 1, 18–20. [Google Scholar] [CrossRef] [PubMed]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Pelzel, H.R.; Nickells, R.W. A role for epigenetic changes in the development of retinal neurodegenerative conditions. J. Ocul. Biol. Dis. Inform. 2011, 4, 104–110. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pennington, K.L.; DeAngelis, M.M. Epigenetic mechanisms of the aging human retina. J. Exp. Neurosci. 2015, 9, 51–79. [Google Scholar] [CrossRef]
- Yohannan, J.; Boland, M.V. The evolving role of the relationship between optic nerve structure and function in glaucoma. Ophthalmology 2017, 124, S66–S70. [Google Scholar] [CrossRef]
- Fawcett, J.W. The struggle to make CNS axons regenerate: Why has it been so difficult? Neurochem. Res. 2020, 45, 144–158. [Google Scholar] [CrossRef]
- Yun, M.H. Changes in regenerative capacity through lifespan. Int. J. Mol. Sci. 2015, 16, 25392–25432. [Google Scholar] [CrossRef]
- Gauthier, A.C.; Liu, J. Epigenetics and signaling pathways in glaucoma. BioMed Res. Int. 2017, 2017, 1–12. [Google Scholar] [CrossRef]
- Schmitt, H.M.; Schlamp, C.L.; Nickells, R.W. Role of HDACs in optic nerve damage-induced nuclear atrophy of retinal ganglion cells. Neurosci. Lett. 2016, 625, 11–15. [Google Scholar] [CrossRef]
- Mancino, R.; Cesareo, M.; Martucci, A.; Di Carlo, E.; Ciuffoletti, E.; Giannini, C.; Morrone, L.A.; Nucci, C.; Garaci, F. Neurodegenerative process linking the eye and the brain. Curr. Med. Chem. 2019, 26, 3754–3763. [Google Scholar] [CrossRef]
- Ashok, A.; Singh, N.; Chaudhary, S.; Bellamkonda, V.; Kritikos, A.E.; Wise, A.S.; Rana, N.; McDonald, D.; Ayyagari, R. Retinal degeneration and Alzheimer’s disease: An evolving link. Int. J. Mol. Sci. 2020, 21, 7290. [Google Scholar] [CrossRef]
- Lu, Y.; Brommer, B.; Tian, X.; Krishnan, A.; Meer, M.; Wang, C.; Vera, D.L.; Zeng, Q.; Yu, D.; Bonkowski, M.S. Reprogramming to recover youthful epigenetic information and restore vision. Nature 2020, 588, 124–129. [Google Scholar] [CrossRef]
- Pita-Thomas, W.; Gonçalves, T.M.; Kumar, A.; Zhao, G.; Cavalli, V. Genome-wide chromatin accessibility analyses provide a map for enhancing optic nerve regeneration. Sci. Rep. 2021, 11, 1–17. [Google Scholar] [CrossRef]
- Yao, B.; Christian, K.M.; He, C.; Jin, P.; Ming, G.-l.; Song, H. Epigenetic mechanisms in neurogenesis. Nat. Rev. Neurosci. 2016, 17, 537–549. [Google Scholar] [CrossRef]
- VandenBosch, L.S.; Reh, T.A. Seminars in cell & developmental bioloy. In Epigenetics in Neuronal Regeneration; Elsevier: Amsterdam, The Netherlands, 2020; pp. 63–73. [Google Scholar]
- Liu, W.; Wu, G.; Xiong, F.; Chen, Y. Advances in the DNA methylation hydroxylase TET1. Biomark. Res. 2021, 9, 1–12. [Google Scholar] [CrossRef]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef]
- D’Oto, A.; Tian, Q.W.; Davidoff, A.M.; Yang, J. Histone demethylases and their roles in cancer epigenetics. J. Med. Oncol. Ther. 2016, 1, 34–40. [Google Scholar] [CrossRef]
- Lopez, N.; Clark, A.F.; Tovar-Vidales, T. Epigenetic regulation of optic nerve head fibrosis in glaucoma. Investig. Ophthalmol. Vis. Sci. 2019, 60, 5668. [Google Scholar]
- Ramaiah, M.J.; Tangutur, A.D.; Manyam, R.R. Epigenetic modulation and understanding of HDAC inhibitors in cancer therapy. Life Sci. 2021, 277, 119504. [Google Scholar] [CrossRef]
- Tiwari, S.; Dharmarajan, S.; Shivanna, M.; Otteson, D.C.; Belecky-Adams, T.L. Histone deacetylase expression patterns in developing murine optic nerve. BMC Dev. Biol. 2014, 14, 30. [Google Scholar] [CrossRef]
- Rao, R.C.; Tchedre, K.T.; Malik, M.T.A.; Coleman, N.; Fang, Y.; Marquez, V.E.; Chen, D.F. Dynamic patterns of histone lysine methylation in the developing retina. Investig. Ophthalmol. Vis. Sci. 2010, 51, 6784–6792. [Google Scholar] [CrossRef]
- Gaub, P.; Joshi, Y.; Wuttke, A.; Naumann, U.; Schnichels, S.; Heiduschka, P.; Di Giovanni, S. The histone acetyltransferase p300 promotes intrinsic axonal regeneration. Brain 2011, 134, 2134–2148. [Google Scholar] [CrossRef]
- Schmitt, H.M.; Pelzel, H.R.; Schlamp, C.L.; Nickells, R.W. Histone deacetylase 3 (HDAC3) plays an important role in retinal ganglion cell death after acute optic nerve injury. Mol. Neurodegener. 2014, 9, 1–15. [Google Scholar] [CrossRef]
- Wang, X.-W.; Yang, S.-G.; Hu, M.-W.; Wang, R.-Y.; Zhang, C.; Kosanam, A.R.; Ochuba, A.J.; Jiang, J.-J.; Luo, X.; Qian, J. Histone methyltransferase Ezh2 coordinates mammalian axon regeneration via epigenetic regulation of key regenerative pathways. BioRxiv 2022. [Google Scholar] [CrossRef]
- Bradley, M.C.; Markenscoff-Papadimitriou, E.; Duffié, R.; Lomvardas, S. Dnmt3a regulates global gene expression in olfactory sensory neurons and enables odorant-induced transcription. Neuron 2014, 83, 823–838. [Google Scholar]
- Hamidi, T.; Singh, A.K.; Chen, T. Genetic alterations of DNA methylation machinery in human diseases. Epigenomics 2015, 7, 247–265. [Google Scholar] [CrossRef]
- Angileri, K.M.; Gross, J.M. Dnmt1 function is required to maintain retinal stem cells within the ciliary marginal zone of the zebrafish eye. Sci. Rep. 2020, 10, 11293. [Google Scholar] [CrossRef]
- Zhu, X.; Li, D.; Du, Y.; He, W.; Lu, Y. DNA hypermethylation-mediated downregulation of antioxidant genes contributes to the early onset of cataracts in highly myopic eyes. Redox Biol. 2018, 19, 179–189. [Google Scholar] [CrossRef]
- Rai, K.; Chidester, S.; Zavala, C.V.; Manos, E.J.; James, S.R.; Karpf, A.R.; Jones, D.A.; Cairns, B.R. Dnmt2 functions in the cytoplasm to promote liver, brain, and retina development in zebrafish. Genes Dev. 2007, 21, 261–266. [Google Scholar] [CrossRef]
- Singh, R.K.; Mallela, R.K.; Hayes, A.; Dunham, N.R.; Hedden, M.E.; Enke, R.A.; Fariss, R.N.; Sternberg, H.; West, M.D.; Nasonkin, I.O. Dnmt1, Dnmt3a and Dnmt3b cooperate in photoreceptor and outer plexiform layer development in the mammalian retina. Exp. Eye Res. 2017, 159, 132–146. [Google Scholar] [CrossRef]
- Moyon, S.; Frawley, R.; Marechal, D.; Huang, D.; Marshall-Phelps, K.L.; Kegel, L.; Bøstrand, S.M.; Sadowski, B.; Jiang, Y.H.; Lyons, D.A.; et al. TET1-mediated DNA hydroxymethylation regulates adult remyelination in mice. Nat. Commun. 2021, 12, 3359. [Google Scholar] [CrossRef]
- Bergles, D.E.; Richardson, W.D. Oligodendrocyte development and plasticity. Cold Spring Harb. Perspect. Biol. 2015, 8, a020453. [Google Scholar] [CrossRef]
- Emery, B.; Lu, Q.R. Transcriptional and epigenetic regulation of oligodendrocyte development and myelination in the central nervous system. Cold Spring Harb. Perspect. Biol. 2015, 7, a020461. [Google Scholar] [CrossRef]
- Liu, J.; Magri, L.; Zhang, F.; Marsh, N.O.; Albrecht, S.; Huynh, J.L.; Kaur, J.; Kuhlmann, T.; Zhang, W.; Slesinger, P.A. Chromatin landscape defined by repressive histone methylation during oligodendrocyte differentiation. J. Neurosci. 2015, 35, 352–365. [Google Scholar] [CrossRef]
- Moyon, S.; Huynh, J.L.; Dutta, D.; Zhang, F.; Ma, D.; Yoo, S.; Lawrence, R.; Wegner, M.; John, G.R.; Emery, B.; et al. Functional characterization of DNA methylation in the oligodendrocyte lineage. Cell Rep. 2016, 15, 748–760. [Google Scholar] [CrossRef]
- Li, E.; Bestor, T.H.; Jaenisch, R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 1992, 69, 915–926. [Google Scholar] [CrossRef]
- Jackson-Grusby, L.; Beard, C.; Possemato, R.; Tudor, M.; Fambrough, D.; Csankovszki, G.; Dausman, J.; Lee, P.; Wilson, C.; Lander, E. Loss of genomic methylation causes p53-dependent apoptosis and epigenetic deregulation. Nat. Genet. 2001, 27, 31–39. [Google Scholar] [CrossRef]
- Unterberger, A.; Andrews, S.D.; Weaver, I.C.; Szyf, M. DNA methyltransferase 1 knockdown activates a replication stress checkpoint. Mol. Cell. Biol. 2006, 26, 7575–7586. [Google Scholar] [CrossRef]
- Moyon, S.; Ma, D.; Huynh, J.L.; Coutts, D.J.; Zhao, C.; Casaccia, P.; Franklin, R.J. Efficient remyelination requires DNA methylation. Eneuro 2017, 4. [Google Scholar] [CrossRef]
- MacArthur, I.C.; Dawlaty, M.M. TET enzymes and 5-hydroxymethylcytosine in neural progenitor cell biology and neurodevelopment. Front. Cell Dev. Biol. 2021, 9, 645335. [Google Scholar] [CrossRef]
- Zhang, W.; Xia, W.; Wang, Q.; Towers, A.J.; Chen, J.; Gao, R.; Zhang, Y.; Yen, C.-a.; Lee, A.Y.; Li, Y. Isoform switch of TET1 regulates DNA demethylation and mouse development. Mol. Cell 2016, 64, 1062–1073. [Google Scholar] [CrossRef]
- Seritrakul, P.; Gross, J.M. Tet-mediated DNA hydroxymethylation regulates retinal neurogenesis by modulating cell-extrinsic signaling pathways. PLoS Genet. 2017, 13, e1006987. [Google Scholar] [CrossRef]
- Vishweswaraiah, S.; Swierkowska, J.; Ratnamala, U.; Mishra, N.K.; Guda, C.; Chettiar, S.S.; Johar, K.R.; Mrugacz, M.; Karolak, J.A.; Gajecka, M.; et al. Epigenetically dysregulated genes and pathways implicated in the pathogenesis of non-syndromic high myopia. Sci. Rep. 2019, 9, 4145. [Google Scholar] [CrossRef]
- Calió, M.L.; Henriques, E.; Siena, A.; Bertoncini, C.R.A.; Gil-Mohapel, J.; Rosenstock, T.R. Mitochondrial dysfunction, neurogenesis, and epigenetics: Putative implications for amyotrophic lateral sclerosis neurodegeneration and treatment. Front. Neurosci. 2020, 14, 679. [Google Scholar] [CrossRef]
- Schmitt, H.M.; Schlamp, C.L.; Nickells, R.W. Targeting HDAC3 activity with RGFP966 protects against retinal ganglion cell nuclear atrophy and apoptosis after optic nerve injury. J. Ocul. Pharmacol. Ther. 2018, 34, 260–273. [Google Scholar] [CrossRef]
- Li, P.; Ma, Y.; Yu, C.; Wu, S.; Wang, K.; Yi, H.; Liang, W. Autophagy and aging: Roles in skeletal muscle, eye, brain and hepatic tissue. Front. Cell Dev. Biol. 2021, 9, 752962. [Google Scholar] [CrossRef]
- Li, J.; Zhao, L.; Urabe, G.; Fu, Y.; Guo, L.-W. Epigenetic intervention with a BET inhibitor ameliorates acute retinal ganglion cell death in mice. Mol. Vis. 2017, 23, 149. [Google Scholar]
- Wiese, M.; Bannister, A.J. Two genomes, one cell: Mitochondrial-nuclear coordination via epigenetic pathways. Mol. Metab. 2020, 38, 100942. [Google Scholar] [CrossRef]
- Xie, K.; Ngo, S.; Rong, J.; Sheppard, A. Modulation of mitochondrial respiration underpins neuronal differentiation enhanced by lutein. Neural Regen. Res. 2019, 14, 87. [Google Scholar] [CrossRef]
- Faraco, G.; Pittelli, M.; Cavone, L.; Fossati, S.; Porcu, M.; Mascagni, P.; Fossati, G.; Moroni, F.; Chiarugi, A. Histone deacetylase (HDAC) inhibitors reduce the glial inflammatory response in vitro and in vivo. Neurobiol. Dis. 2009, 36, 269–279. [Google Scholar] [CrossRef]
- Suh, H.-S.; Choi, S.; Khattar, P.; Choi, N.; Lee, S.C. Histone deacetylase inhibitors suppress the expression of inflammatory and innate immune response genes in human microglia and astrocytes. J. Neuroimmune Pharmacol. 2010, 5, 521–532. [Google Scholar] [CrossRef]
- Kanski, R.; Sneeboer, M.A.; van Bodegraven, E.J.; Sluijs, J.A.; Kropff, W.; Vermunt, M.W.; Creyghton, M.P.; De Filippis, L.; Vescovi, A.; Aronica, E. Histone acetylation in astrocytes suppresses GFAP and stimulates a reorganization of the intermediate filament network. J. Cell Sci. 2014, 127, 4368–4380. [Google Scholar] [CrossRef]
- Xuan, A.; Long, D.; Li, J.; Ji, W.; Hong, L.; Zhang, M.; Zhang, W. Neuroprotective effects of valproic acid following transient global ischemia in rats. Life Sci. 2012, 90, 463–468. [Google Scholar] [CrossRef]
- Buczek-Thomas, J.A.; Hsia, E.; Rich, C.B.; Foster, J.A.; Nugent, M.A. Inhibition of histone acetyltransferase by glycosaminoglycans. J. Cell. Biochem. 2008, 105, 108–120. [Google Scholar] [CrossRef]
- Kuboyama, T.; Wahane, S.; Huang, Y.; Zhou, X.; Wong, J.K.; Koemeter-Cox, A.; Martini, M.; Friedel, R.H.; Zou, H. HDAC3 inhibition ameliorates spinal cord injury by immunomodulation. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Skowronska-Krawczyk, D.; Ballivet, M.; Dynlacht, B.; Matter, J.-M. Highly specific interactions between bHLH transcription factors and chromatin during retina development. Development 2004, 131, 4447–4454. [Google Scholar] [CrossRef]
- Li, Y.; Du, Z.; Xie, X.; Zhang, Y.; Liu, H.; Zhou, Z.; Zhao, J.; Lee, R.S.; Xiao, Y.; Ivanoviski, S.; et al. Epigenetic changes caused by diabetes and their potential role in the development of periodontitis. J. Diabetes Investig. 2021, 12, 1326–1335. [Google Scholar] [CrossRef]
- Reverdatto, S.; Prasad, A.; Belrose, J.L.; Zhang, X.; Sammons, M.A.; Gibbs, K.M.; Szaro, B.G. Developmental and injury-induced changes in DNA methylation in regenerative versus non-regenerative regions of the vertebrate central nervous system. BMC Genom. 2022, 23, 2. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; Sen, S.; Weeks, R.J.; Eccles, M.R.; Chatterjee, A. Promoter DNA hypermethylation and paradoxical gene activation. Trends Cancer 2020, 6, 392–406. [Google Scholar] [CrossRef] [PubMed]
- Takasawa, K.; Arai, Y.; Yamazaki-Inoue, M.; Toyoda, M.; Akutsu, H.; Umezawa, A.; Nishino, K. DNA hypermethylation enhanced telomerase reverse transcriptase expression in human-induced pluripotent stem cells. Hum. Cell 2018, 31, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Struebing, F.L.; Wang, J.; Li, Y.; King, R.; Mistretta, O.C.; English, A.W.; Geisert, E.E. Differential expression of Sox11 and Bdnf mRNA isoforms in the injured and regenerating nervous systems. Front. Mol. Neurosci. 2017, 10, 354. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, J.; Liao, Y.; Jin, Y.; Yu, X.; Li, H.; Yang, Q.; Li, X.; Chen, R.; Wu, D. DNMT1-mediated DNA methylation targets CDKN2B to promote the repair of retinal ganglion cells in streptozotocin-induced mongolian gerbils during diabetic retinopathy. Comput. Math. Methods Med. 2022, 2022, 1–9. [Google Scholar] [CrossRef]
- Pelzel, H.R.; Schlamp, C.L.; Nickells, R.W. Histone H4 deacetylation plays a critical role in early gene silencing during neuronal apoptosis. BMC Neurosci. 2010, 11, 1–20. [Google Scholar] [CrossRef]
- Cho, Y.; Cavalli, V. HDAC5 is a novel injury-regulated tubulin deacetylase controlling axon regeneration. EMBO J. 2012, 31, 3063–3078. [Google Scholar] [CrossRef]
- Rivieccio, M.A.; Brochier, C.; Willis, D.E.; Walker, B.A.; D’Annibale, M.A.; McLaughlin, K.; Siddiq, A.; Kozikowski, A.P.; Jaffrey, S.R.; Twiss, J.L.; et al. HDAC6 is a target for protection and regeneration following injury in the nervous system. Proc. Natl. Acad. Sci. USA 2009, 106, 19599–19604. [Google Scholar] [CrossRef]
- Gaub, P.; Tedeschi, A.; Puttagunta, R.; Nguyen, T.; Schmandke, A.; Di Giovanni, S. HDAC inhibition promotes neuronal outgrowth and counteracts growth cone collapse through CBP/p300 and P/CAF-dependent p53 acetylation. Cell Death Differ. 2010, 17, 1392–1408. [Google Scholar] [CrossRef]
- Lebrun-Julien, F.; Suter, U. Combined HDAC1 and HDAC2 depletion promotes retinal ganglion cell survival after injury through reduction of p53 target gene expression. ASN Neuro 2015, 7, 1759091415593066. [Google Scholar] [CrossRef]
- Zou, S.; Tian, C.; Ge, S.; Hu, B. Neurogenesis of retinal ganglion cells is not essential to visual functional recovery after optic nerve injury in adult zebrafish. PLoS ONE 2013, 8, e57280. [Google Scholar] [CrossRef]
- Siebzehnrubl, F.A.; Buslei, R.; Eyupoglu, I.Y.; Seufert, S.; Hahnen, E.; Blumcke, I. Histone deacetylase inhibitors increase neuronal differentiation in adult forebrain precursor cells. Exp. Brain Res. 2007, 176, 672–678. [Google Scholar] [CrossRef]
- Dincman, T.A.; Beare, J.E.; Ohri, S.S.; Gallo, V.; Hetman, M.; Whittemore, S.R. Histone deacetylase inhibition is cytotoxic to oligodendrocyte precursor cells in vitro and in vivo. Int. J. Dev. Neurosci. 2016, 54, 53–61. [Google Scholar] [CrossRef]
- Huang, X.; Wu, D.-Y.; Chen, G.; Manji, H.; Chen, D.F. Support of retinal ganglion cell survival and axon regeneration by lithium through a Bcl-2-dependent mechanism. Investig. Ophthalmol. Vis. Sci. 2003, 44, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Biermann, J.; Boyle, J.; Pielen, A.; Lagrèze, W.A. Histone deacetylase inhibitors sodium butyrate and valproic acid delay spontaneous cell death in purified rat retinal ganglion cells. Mol. Vis. 2011, 17, 395–403. [Google Scholar] [PubMed]
- Phiel, C.J.; Zhang, F.; Huang, E.Y.; Guenther, M.G.; Lazar, M.A.; Klein, P.S. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J. Biol. Chem. 2001, 276, 36734–36741. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, S.A.H.; Guzman, W.; Singh, S.; Mehrotra, S.; Husain, S. Changes in class I and IIb HDACs by δ-opioid in chronic rat glaucoma model. Investig. Ophthalmol. Vis. Sci. 2020, 61, 4. [Google Scholar] [CrossRef]
- Luo, J.; Su, F.; Chen, D.; Shiloh, A.; Gu, W. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature 2000, 408, 377–381. [Google Scholar] [CrossRef]
- Vaziri, H.; Dessain, S.K.; Eaton, E.N.; Imai, S.-I.; Frye, R.A.; Pandita, T.K.; Guarente, L.; Weinberg, R.A. hSIR2SIRT1 functions as an NAD-dependent p53 deacetylase. Cell 2001, 107, 149–159. [Google Scholar] [CrossRef]
- Mrakovcic, M.; Kleinheinz, J.; Fröhlich, L.F. p53 at the crossroads between different types of HDAC inhibitor-mediated cancer cell death. Int. J. Mol. Sci. 2019, 20, 2415. [Google Scholar] [CrossRef]
- Shi, J.; Vakoc, C.R. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol. Cell 2014, 54, 728–736. [Google Scholar] [CrossRef]
- Gupta, R.; Saha, P.; Sen, T.; Sen, N. An augmentation in histone dimethylation at lysine nine residues elicits vision impairment following traumatic brain injury. Free. Radic. Biol. Med. 2019, 134, 630–643. [Google Scholar] [CrossRef]
- Chase, K.; Feiner, B.; Ramaker, M.; Hu, E.; Rosen, C.; Sharma, R. Examining the effects of the histone methyltransferase inhibitor BIX-01294 on histone modifications and gene expression in both a clinical population and mouse models. PLoS ONE 2019, 14, e0216463. [Google Scholar] [CrossRef]
- Cheng, L.; Wong, L.J.; Yan, N.; Han, R.C.; Yu, H.; Guo, C.; Batsuuri, K.; Zinzuwadia, A.; Guan, R.; Cho, K.-S. Ezh2 does not mediate retinal ganglion cell homeostasis or their susceptibility to injury. PLoS ONE 2018, 13, e0191853. [Google Scholar] [CrossRef]
- Yan, N.; Cheng, L.; Cho, K.; Malik, M.T.A.; Xiao, L.; Guo, C.; Yu, H.; Zhu, R.; Rao, R.C.; Chen, D.F. Postnatal onset of retinal degeneration by loss of embryonic Ezh2 repression of Six1. Sci. Rep. 2016, 6, 33887. [Google Scholar] [CrossRef]
- Xiao, L.; Hou, C.; Cheng, L.; Zheng, S.; Zhao, L.; Yan, N. DZNep protects against retinal ganglion cell death in an NMDA-induced mouse model of retinal degeneration. Exp. Eye Res. 2021, 212, 108785. [Google Scholar] [CrossRef]
- Tsutsumi, T.; Iwao, K.; Hayashi, H.; Kirihara, T.; Kawaji, T.; Inoue, T.; Hino, S.; Nakao, M.; Tanihara, H. Potential neuroprotective effects of an LSD1 inhibitor in retinal ganglion cells via p38 mapk activity. Investig. Ophthalmol. Vis. Sci. 2016, 57, 6461–6473. [Google Scholar] [CrossRef]
- Chung, D.; Shum, A.; Caraveo, G. GAP-43 and BASP1 in axon regeneration: Implications for the treatment of neurodegenerative diseases. Front. Cell Dev. Biol. 2020, 8, 567537. [Google Scholar] [CrossRef]
- Latremoliere, A.; Cheng, L.; DeLisle, M.; Wu, C.; Chew, S.; Hutchinson, E.B.; Sheridan, A.; Alexandre, C.; Latremoliere, F.; Sheu, S.-H. Neuronal-specific TUBB3 is not required for normal neuronal function but is essential for timely axon regeneration. Cell Rep. 2018, 24, 1865–1879. [Google Scholar] [CrossRef]
- Smith, P.D.; Sun, F.; Park, K.K.; Cai, B.; Wang, C.; Kuwako, K.; Martinez-Carrasco, I.; Connolly, L.; He, Z. SOCS3 Deletion Promotes Optic Nerve Regeneration In Vivo. Neuron 2009, 64, 617–623. [Google Scholar] [CrossRef]
- Lindborg, J.A.; Tran, N.M.; Chenette, D.M.; DeLuca, K.; Foli, Y.; Kannan, R.; Sekine, Y.; Wang, X.; Wollan, M.; Kim, I.-J.; et al. Optic nerve regeneration screen identifies multiple genes restricting adult neural repair. Cell Reports 2021, 34, 108777. [Google Scholar] [CrossRef]
- Park, K.K.; Liu, K.; Hu, Y.; Smith, P.D.; Wang, C.; Cai, B.; Xu, B.; Connolly, L.; Kramvis, I.; Sahin, M. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science 2008, 322, 963–966. [Google Scholar] [CrossRef]
- Wang, Y.H.; Huang, M.L. Organogenesis and tumorigenesis: Insight from the JAK/STAT pathway in the Drosophila eye. Dev. Dyn. 2010, 239, 2522–2533. [Google Scholar] [CrossRef]
- Madsen, A.S.; Kristensen, H.M.; Lanz, G.; Olsen, C.A. The effect of various zinc binding groups on inhibition of histone deacetylases 1–11. ChemMedChem 2014, 9, 614–626. [Google Scholar] [CrossRef]
- Younes, A.; Oki, Y.; Bociek, R.G.; Kuruvilla, J.; Fanale, M.; Neelapu, S.; Copeland, A.; Buglio, D.; Galal, A.; Besterman, J. Mocetinostat for relapsed classical Hodgkin’s lymphoma: An open-label, single-arm, phase 2 trial. Lancet Oncol. 2011, 12, 1222–1228. [Google Scholar] [CrossRef]
- Weng, Y.-L.; An, R.; Cassin, J.; Joseph, J.; Mi, R.; Wang, C.; Zhong, C.; Jin, S.-G.; Pfeifer, G.P.; Bellacosa, A. An intrinsic epigenetic barrier for functional axon regeneration. Neuron 2017, 94, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Xu, Y.; Huang, X.; Ye, D.; Han, M.; Wang, H.-L. Regulatory roles of histone deacetylases 1 and 2 in Pb-induced neurotoxicity. Toxicol. Sci. 2018, 162, 688–701. [Google Scholar] [CrossRef] [PubMed]
- Douvaras, P.; Rusielewicz, T.; Kim, K.H.; Haines, J.D.; Casaccia, P.; Fossati, V. Epigenetic modulation of human induced pluripotent stem cell differentiation to oligodendrocytes. Int. J. Mol. Sci. 2016, 17, 614. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Zhang, X.; Liang, F.; Cao, Y.; Li, Z.; Qu, W.; Zhang, J.; Bi, Y.; Sun, C.; Zhang, J.; et al. Tet1 regulates astrocyte development and cognition of mice through modulating GluA1. Front. Cell Dev. Biol. 2021, 9, 644375. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Cell Type | Epigenetic Player | Experimental Model | Effect | References |
|---|---|---|---|---|
| RGCs (Retinal ganglion cells) | TET1-dependent deletion of PTEN | Optic nerve crush model | Optic nerve regeneration | [94] |
| Class I HDACs and HDAC 2 & 3 upregulation and HDAC3 nuclear localization in RGCs | Optic nerve crush model | Optic nerve degeneration/RGC apoptosis | [64] | |
| Increased G9a expression and H3K9Me2 activity in the retina (RGC) and optic nerve | Traumatic brain injury (TBI) model | TBI causes apoptosis and oxidative stress in the retina (RGC) and optic nerve | [80] | |
| Inhibition of retinal HDAC activity (post valproic acid treatment) |
| Neuroprotection and histone hyperacetylation | [73,74] | |
| Inhibition of HDAC3 activity (RGFP966 activity) | Optic nerve crush model | RGC survival and repression of the apoptotic gene in RGCs post optic nerve injury | [24,64] | |
| Double knock out of HDAC1&2 | Optic nerve axotomy | Anti-apoptosis and neuroprotection effect | [68] | |
| 3-deazaneplanocin (DZNep) inhibits Ezh2 inhibition using 3-deazaneplanocin (DZNep) -reduces the trimethylation of histone 3 lysine 27 (H3K27me3) or activity | Retinal/RGC damage caused by intravitreal injection of N-methyl-D-aspartate (NMDA) | Prevent cell death and inner nuclear layer thinning induced by NMDA and improved visual function | [84] | |
| Increased Histone H3K9 acetylation using Trichostatin A (TSA) | Lead-induced neurotoxicity | Promotes neurite outgrowth and branching, neuroprotection, neuronal differentiation, and neurite branching | [95] | |
| Intravitreal JQ1 (BET inhibitor) administration | RGC damage induced by NMDA excitotoxicity | Sustained RGC number and gene expression and decreased TUNEL-positive cells in the ganglion cell layer | [48] | |
| Promotion of p38 MAPKc activity and intravitreal administration of tranylcypromine (lysine-specific demethylase 1 (LSD1) inhibitor) | NMDA-induced excitotoxicity | Enhanced RGC survival | [85] | |
| OSK-mediated vision restoration is TET1/2 dependent ectopic expression of Oct4 (also known as Pou5f1), Sox2, and Klf4 genes (OSK) in RGC | Optic nerve crush model | Axon regeneration | [12] | |
| OPCs (Oligodendrocyte progenitor cells) | Increased levels of H3K27me3 from NSCs (neural stem cells) to immature OL and significantly decreased levels of histone acetylation (i.e., H3K9ac) at the early stages of OPC differentiation associated with increasing levels of H3K9me3 during OPC maturation | Human pluripotent stem cell culture | Differentiation of OPCs into OLs | [96] |
| DNMT1 downregulation in oligodendrocytes |
|
| [38,39] | |
| Myelin | Downregulation of TET1 |
|
| [32] |
| [97] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ashok, A.; Pooranawattanakul, S.; Tai, W.L.; Cho, K.-S.; Utheim, T.P.; Cestari, D.M.; Chen, D.F. Epigenetic Regulation of Optic Nerve Development, Protection, and Repair. Int. J. Mol. Sci. 2022, 23, 8927. https://doi.org/10.3390/ijms23168927
Ashok A, Pooranawattanakul S, Tai WL, Cho K-S, Utheim TP, Cestari DM, Chen DF. Epigenetic Regulation of Optic Nerve Development, Protection, and Repair. International Journal of Molecular Sciences. 2022; 23(16):8927. https://doi.org/10.3390/ijms23168927
Chicago/Turabian StyleAshok, Ajay, Sarita Pooranawattanakul, Wai Lydia Tai, Kin-Sang Cho, Tor P. Utheim, Dean M. Cestari, and Dong Feng Chen. 2022. "Epigenetic Regulation of Optic Nerve Development, Protection, and Repair" International Journal of Molecular Sciences 23, no. 16: 8927. https://doi.org/10.3390/ijms23168927
APA StyleAshok, A., Pooranawattanakul, S., Tai, W. L., Cho, K.-S., Utheim, T. P., Cestari, D. M., & Chen, D. F. (2022). Epigenetic Regulation of Optic Nerve Development, Protection, and Repair. International Journal of Molecular Sciences, 23(16), 8927. https://doi.org/10.3390/ijms23168927