
Alterations of Mitochondrial Structure in Methamphetamine Toxicity

 ,
,  ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
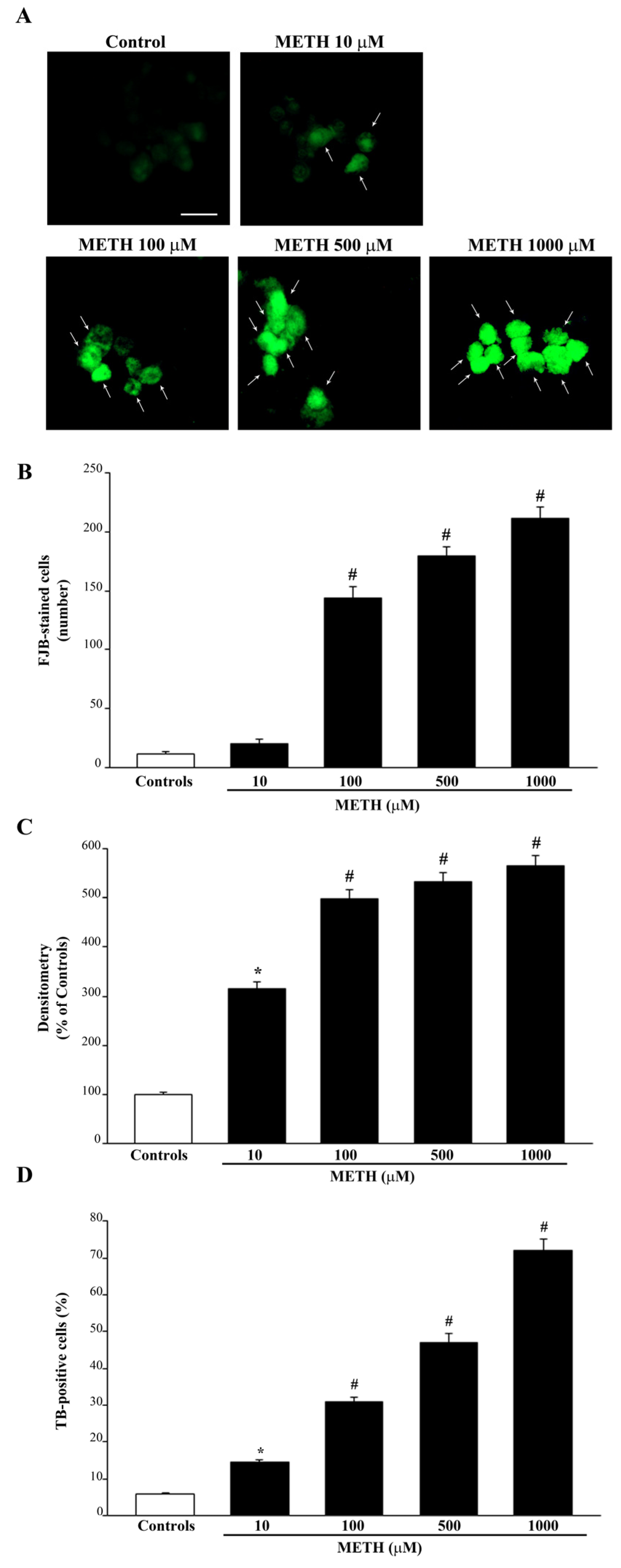
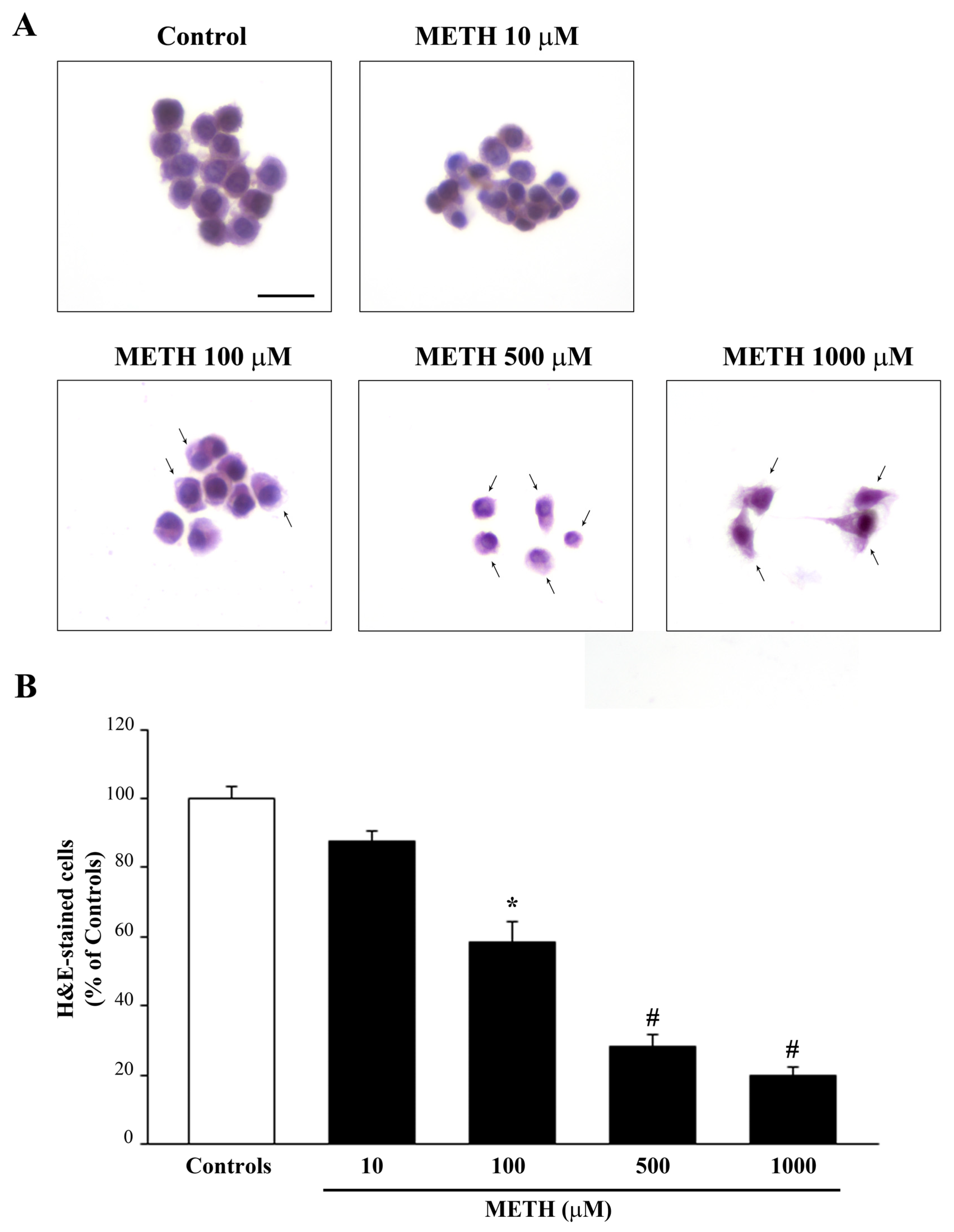
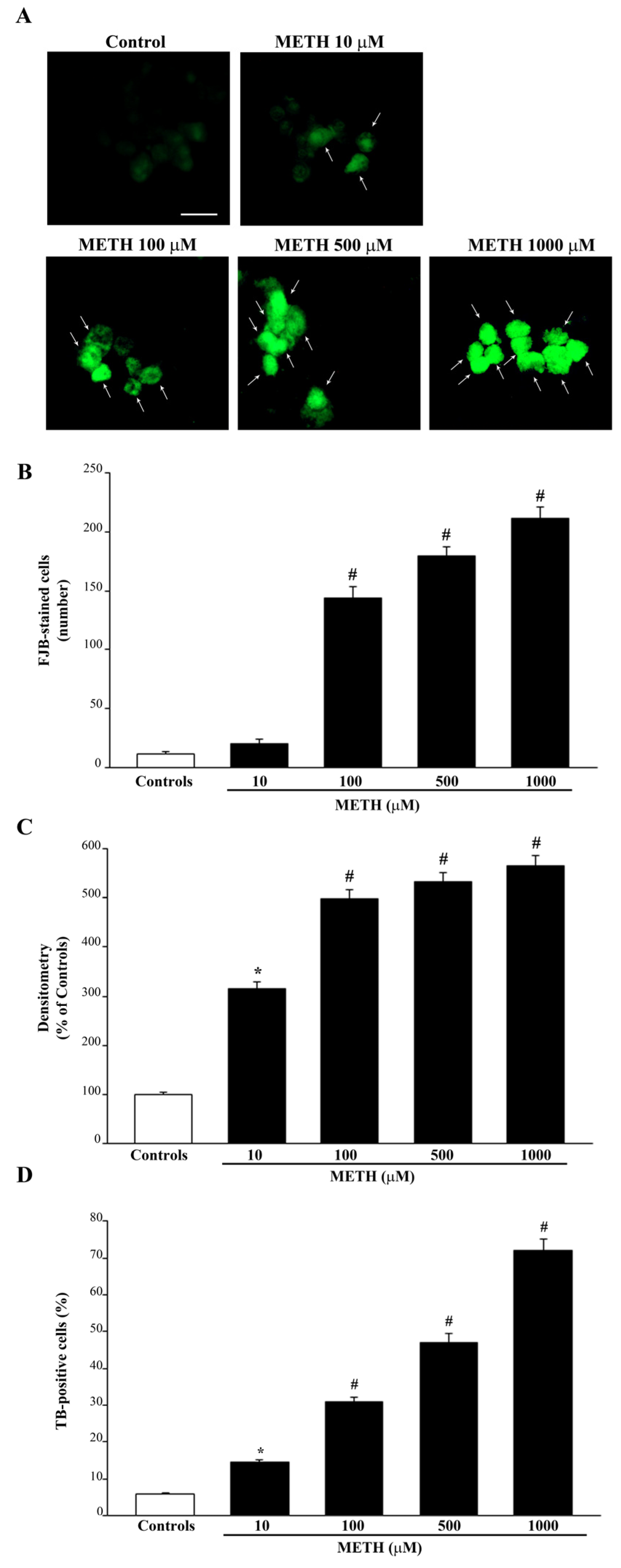
2.1. Dose-Dependent Effects of METh on Cell Survival and Cell Degeneration
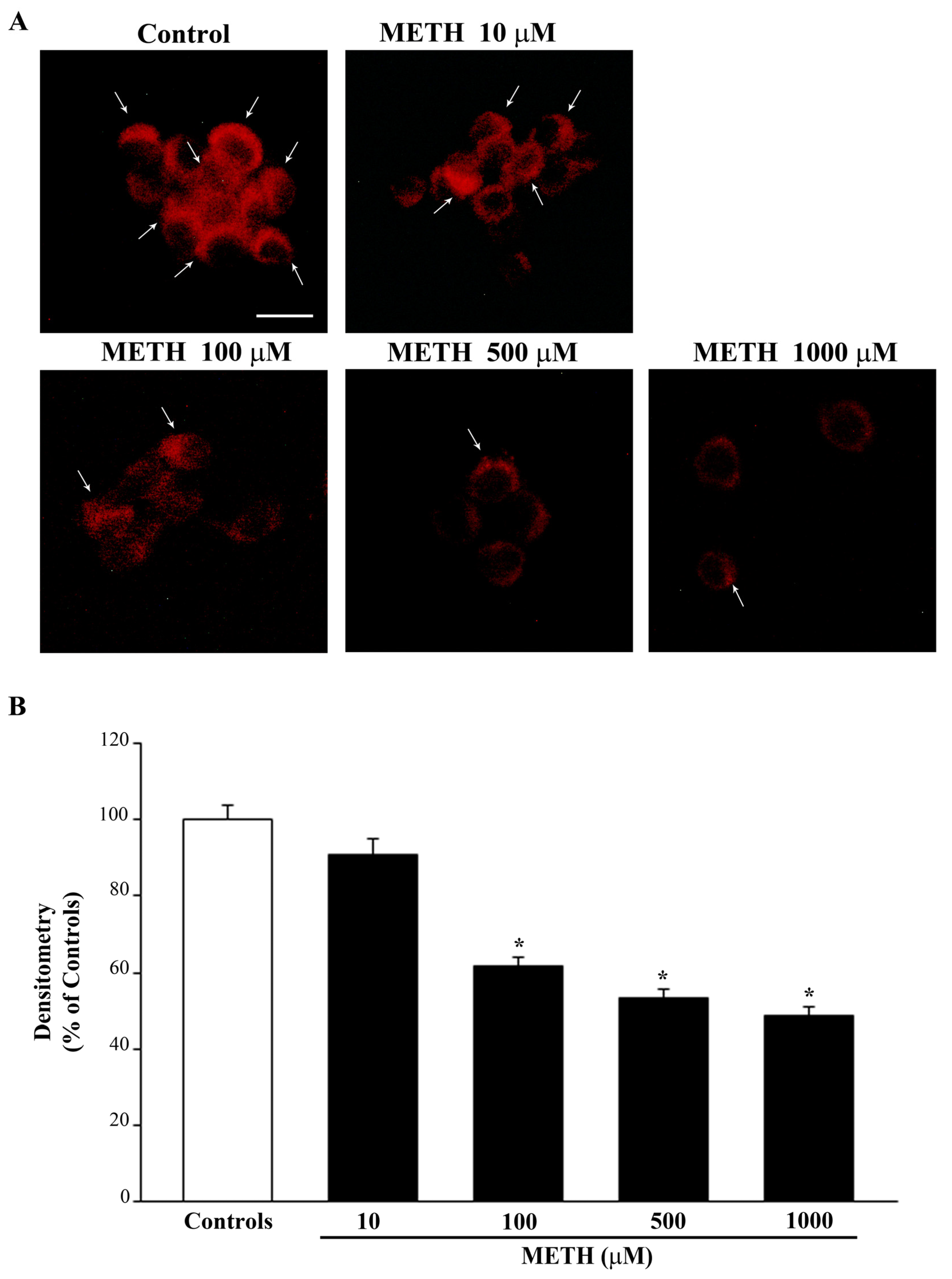
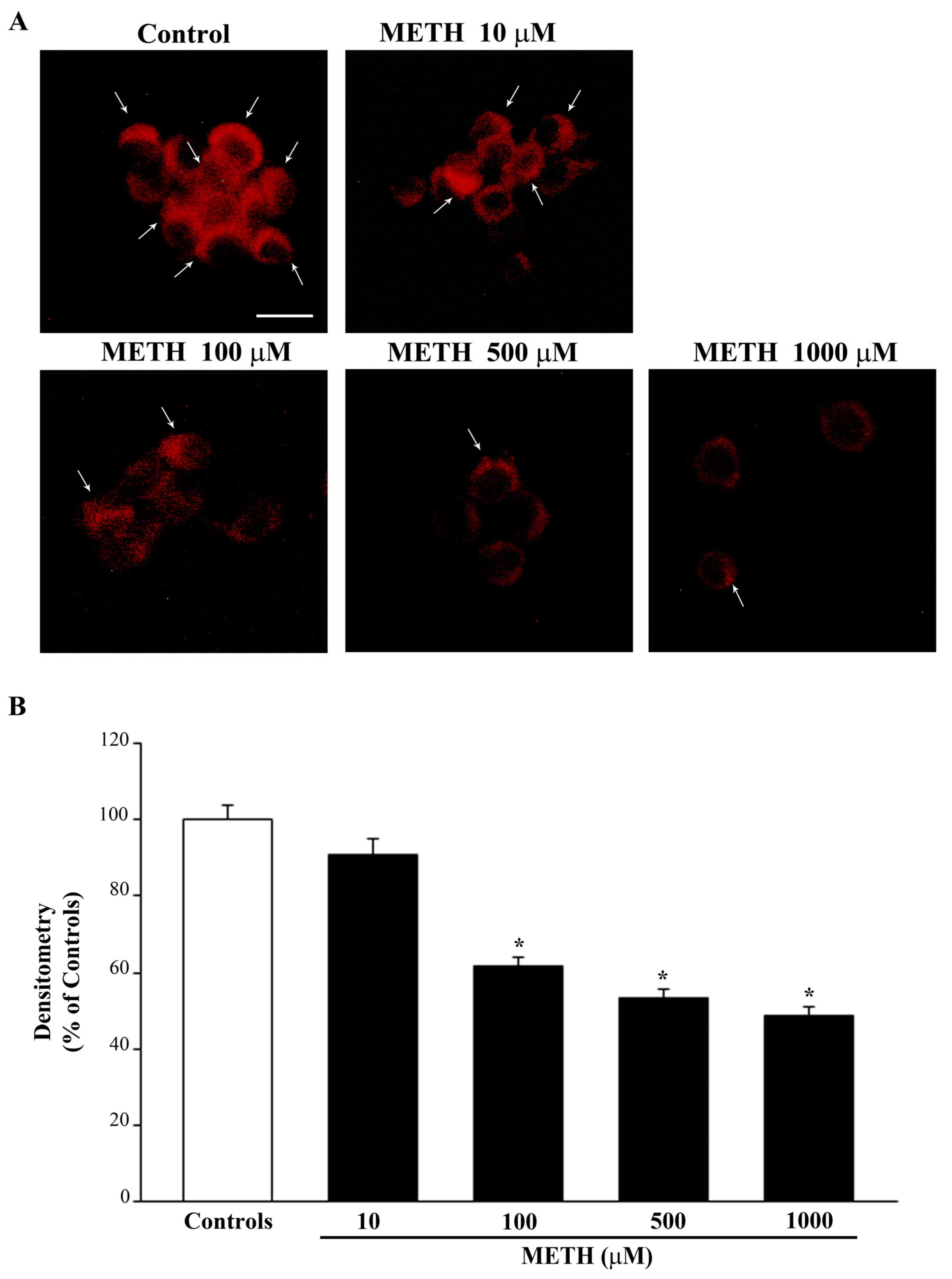
2.2. Dose-Dependent Effects of METH on the Amount of Total Mitochondria (Mitotracker Green, MTR-G)
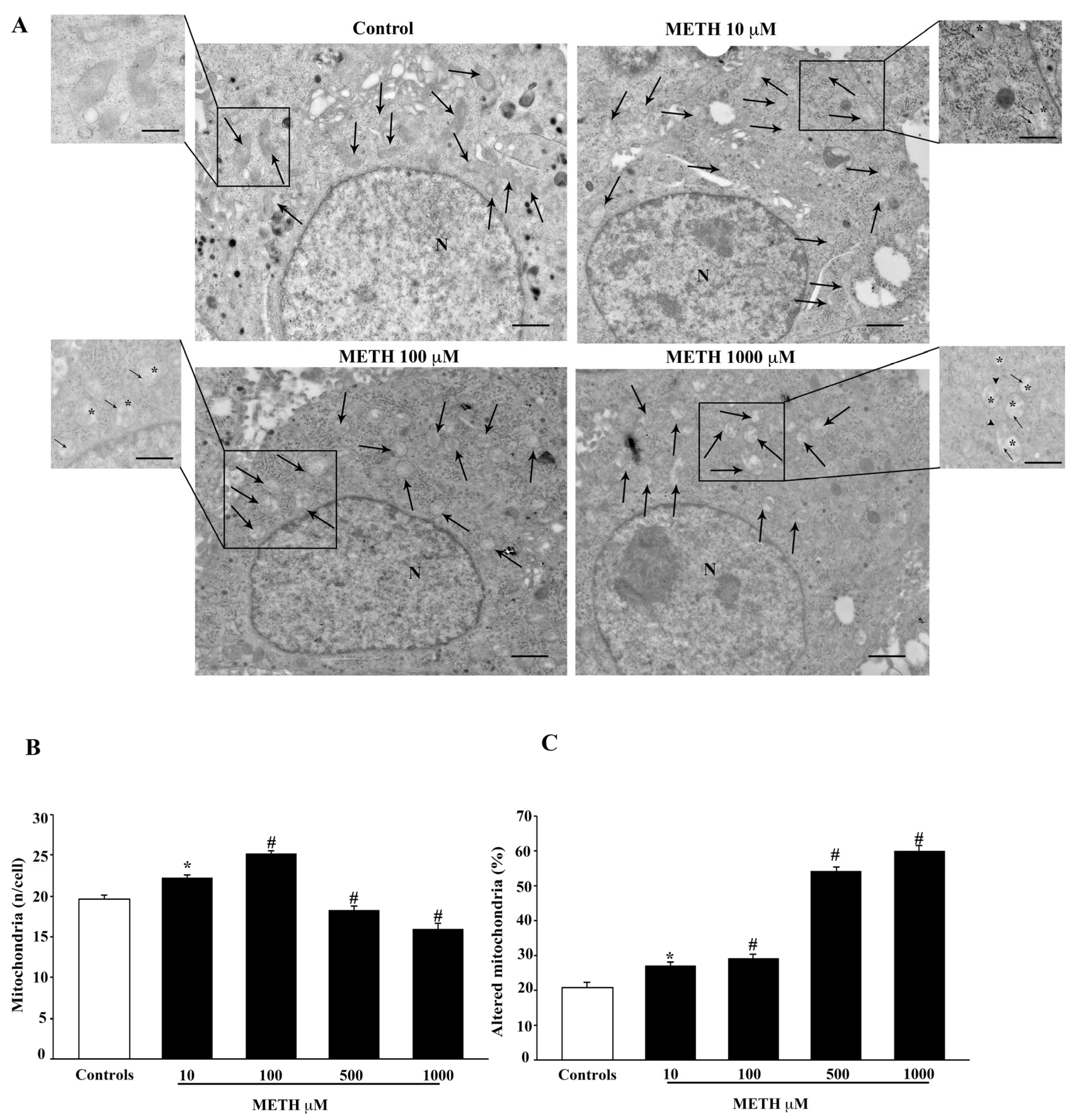
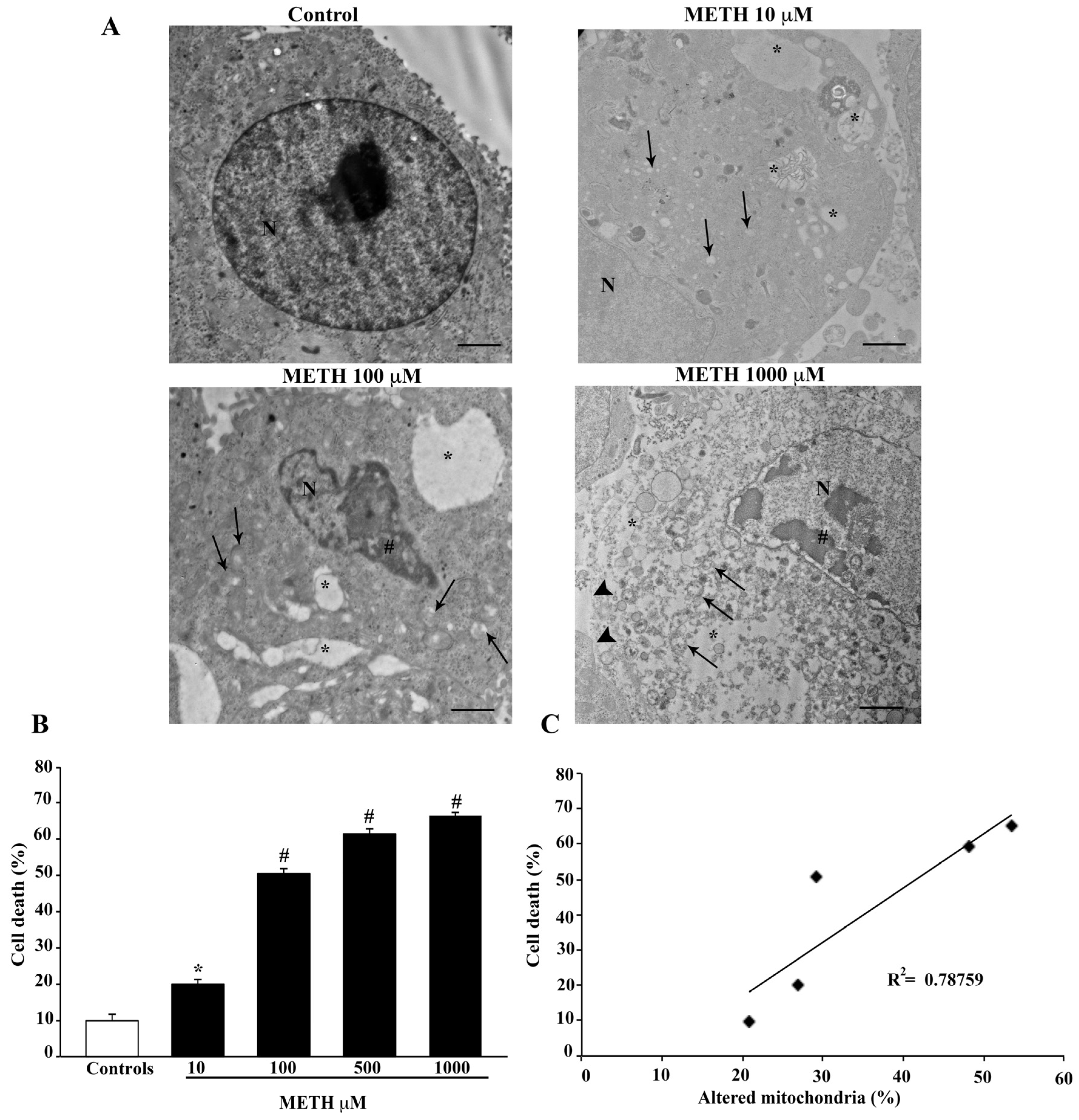
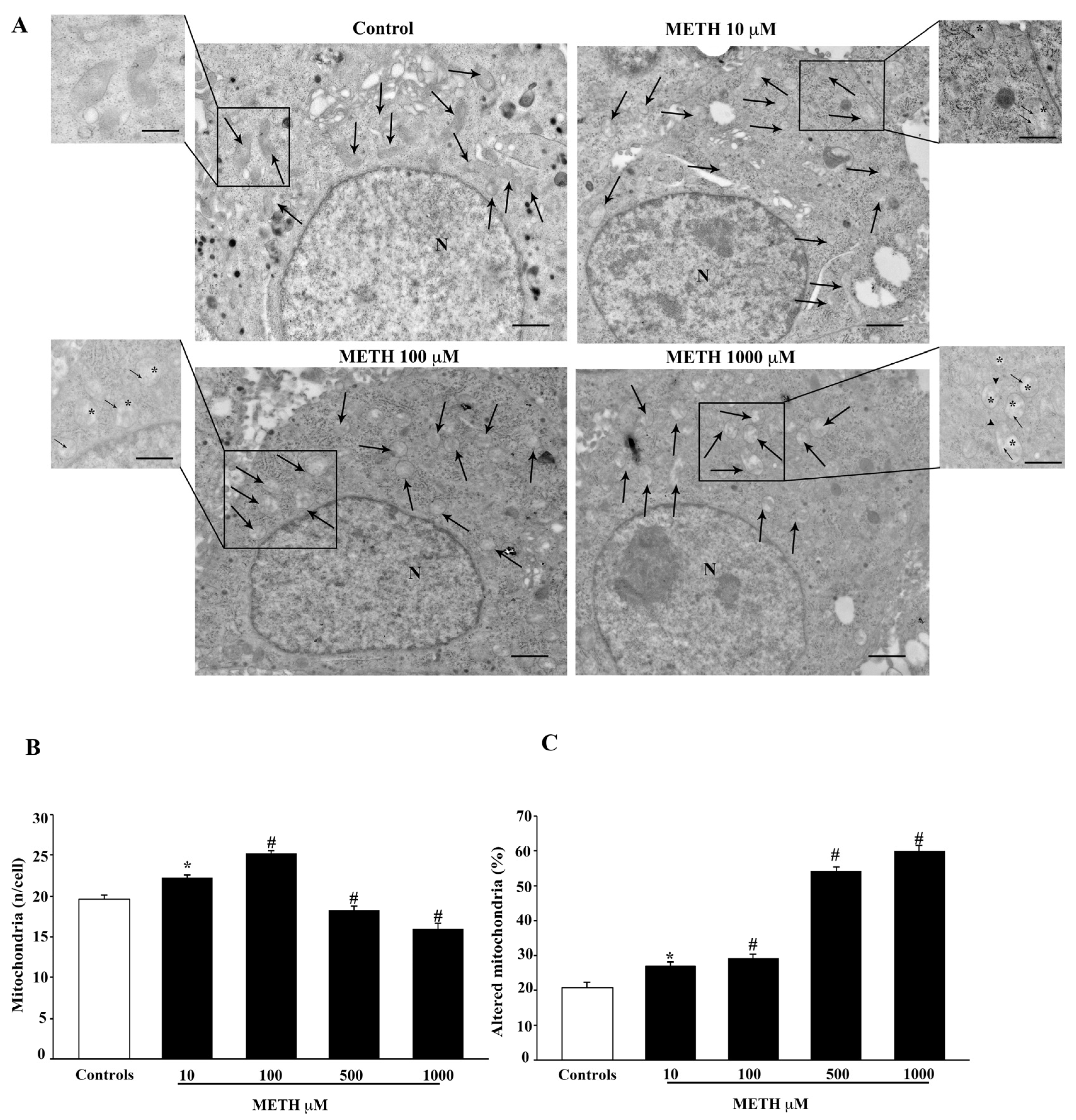
2.3. Dose-Dependent Effects of METH on TEM-Detected Cell Death Correlates with Mitochondral Alterations
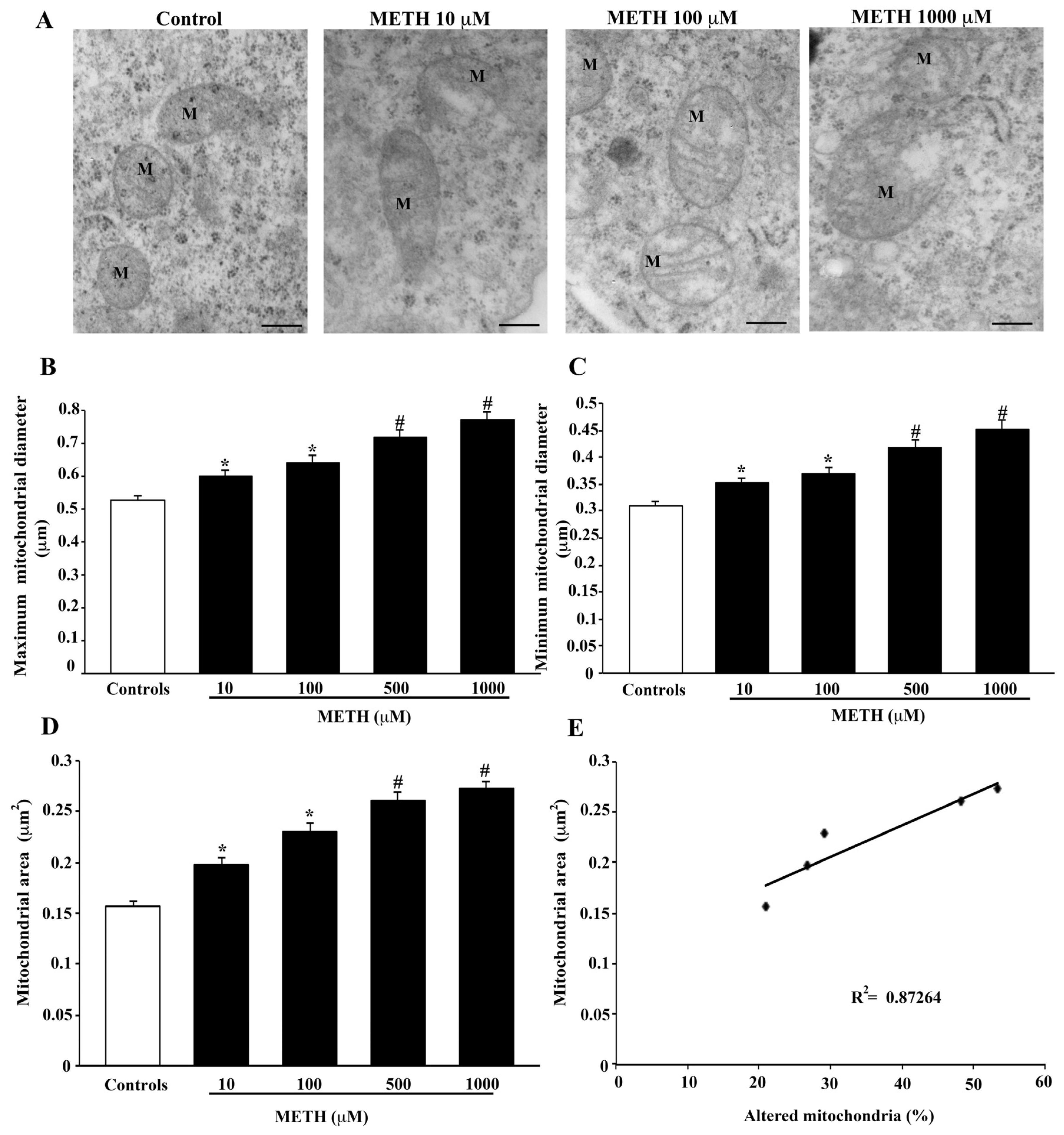
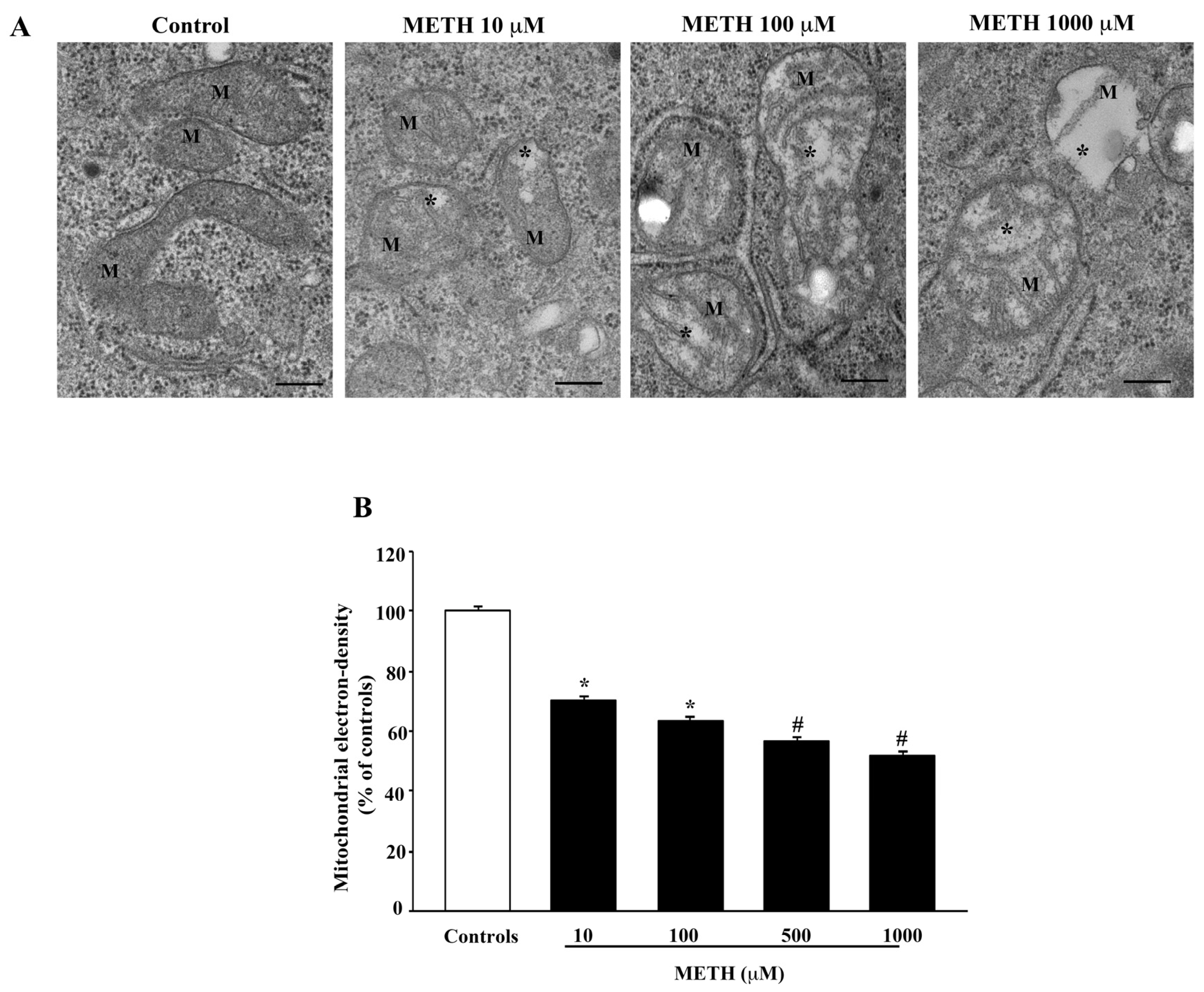
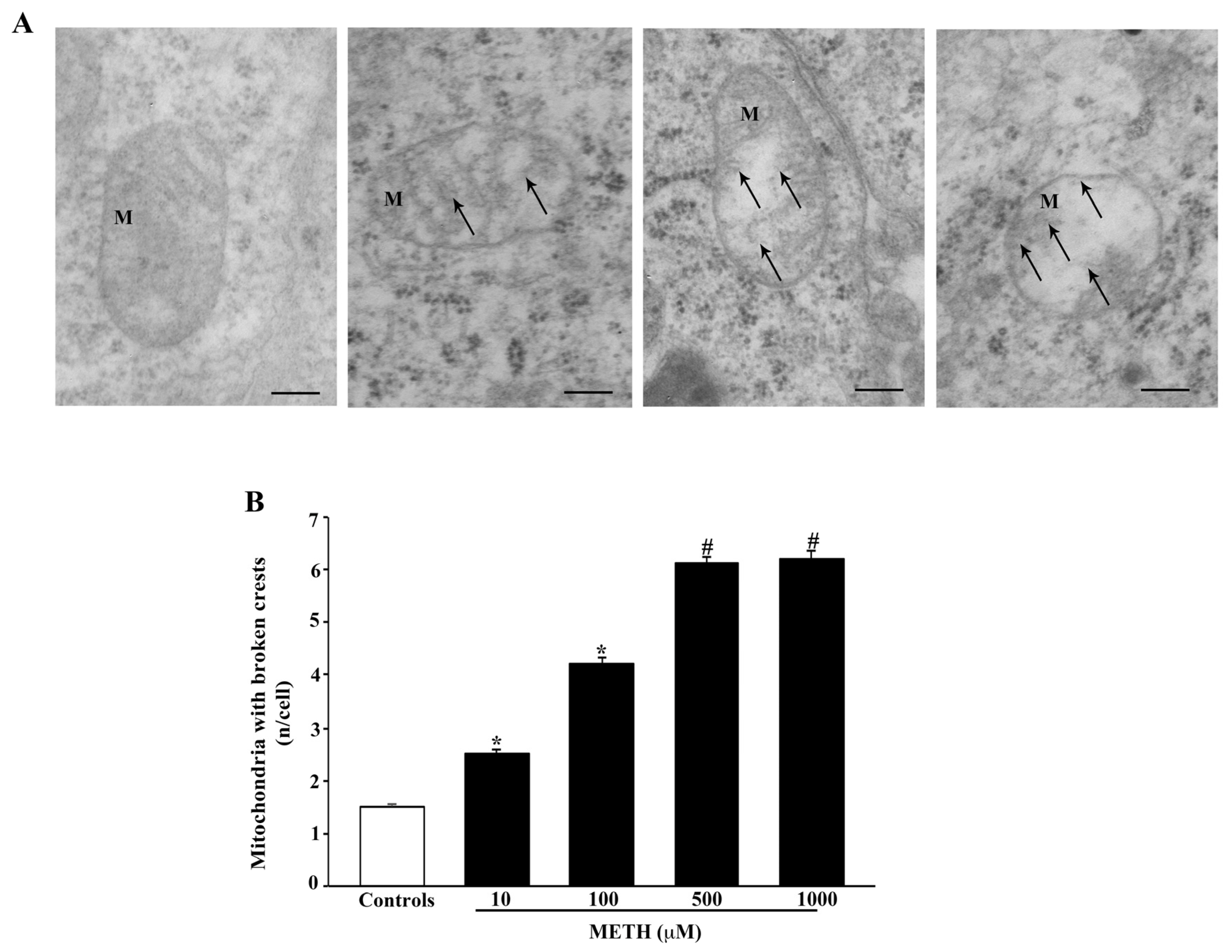
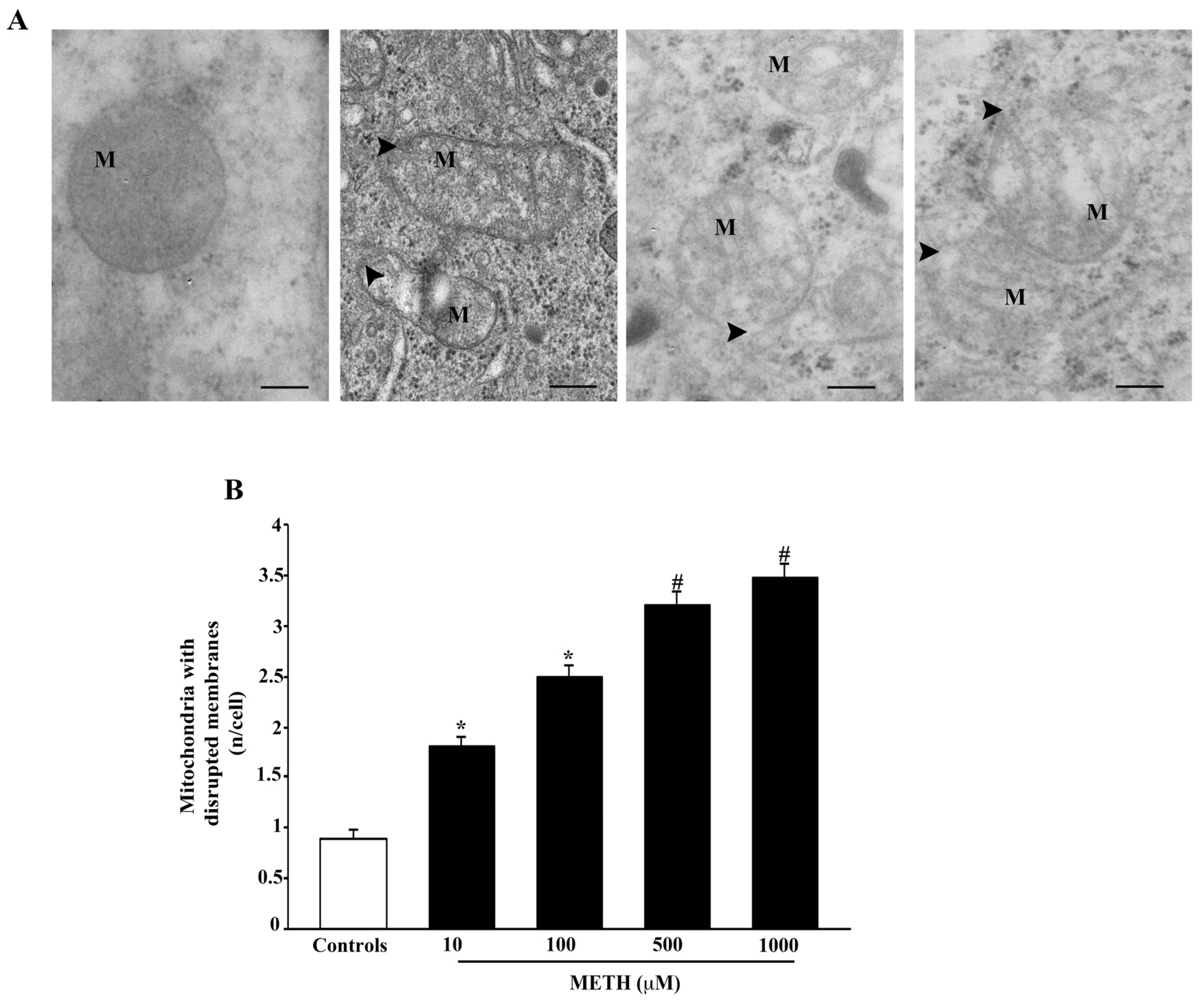
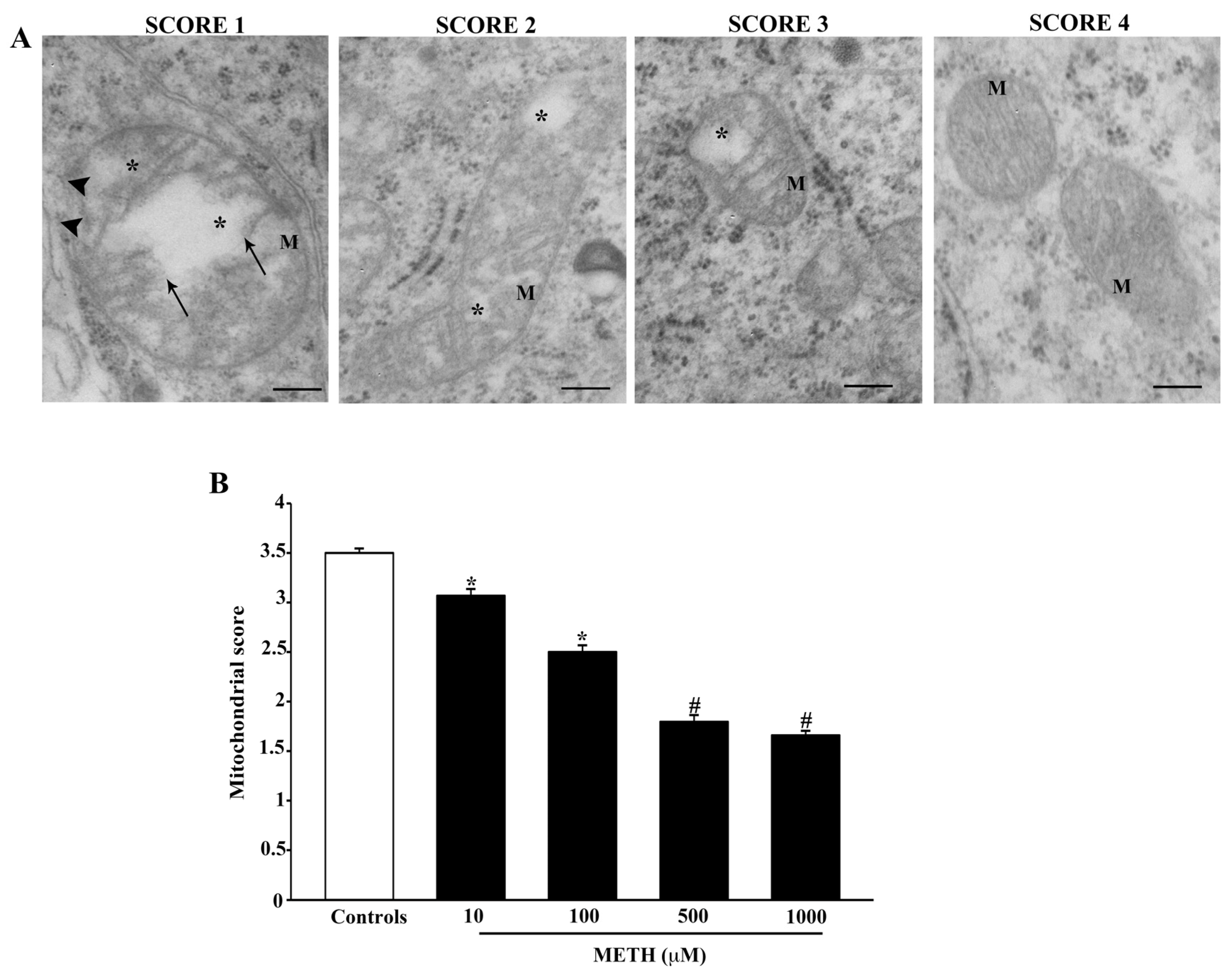
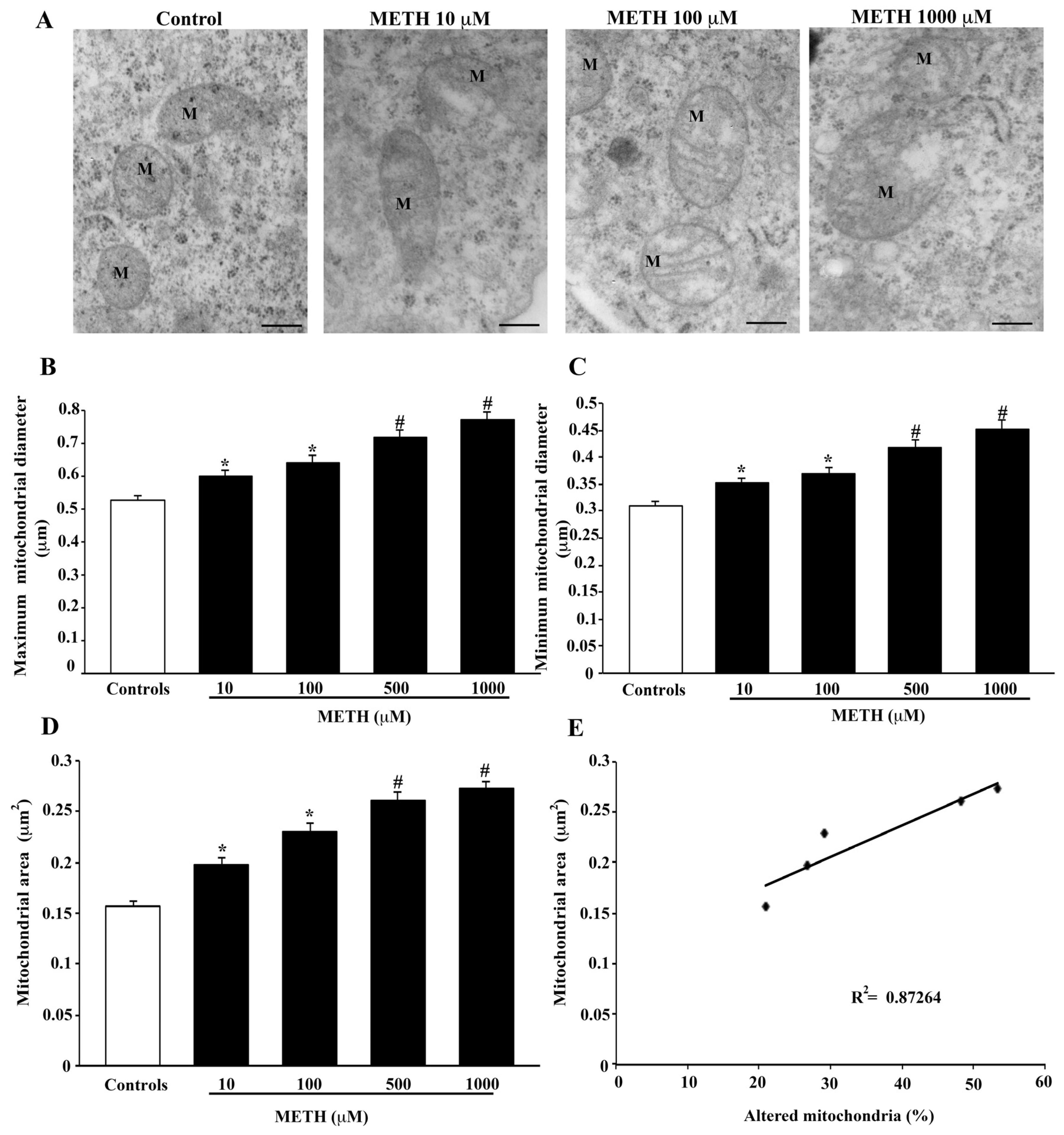
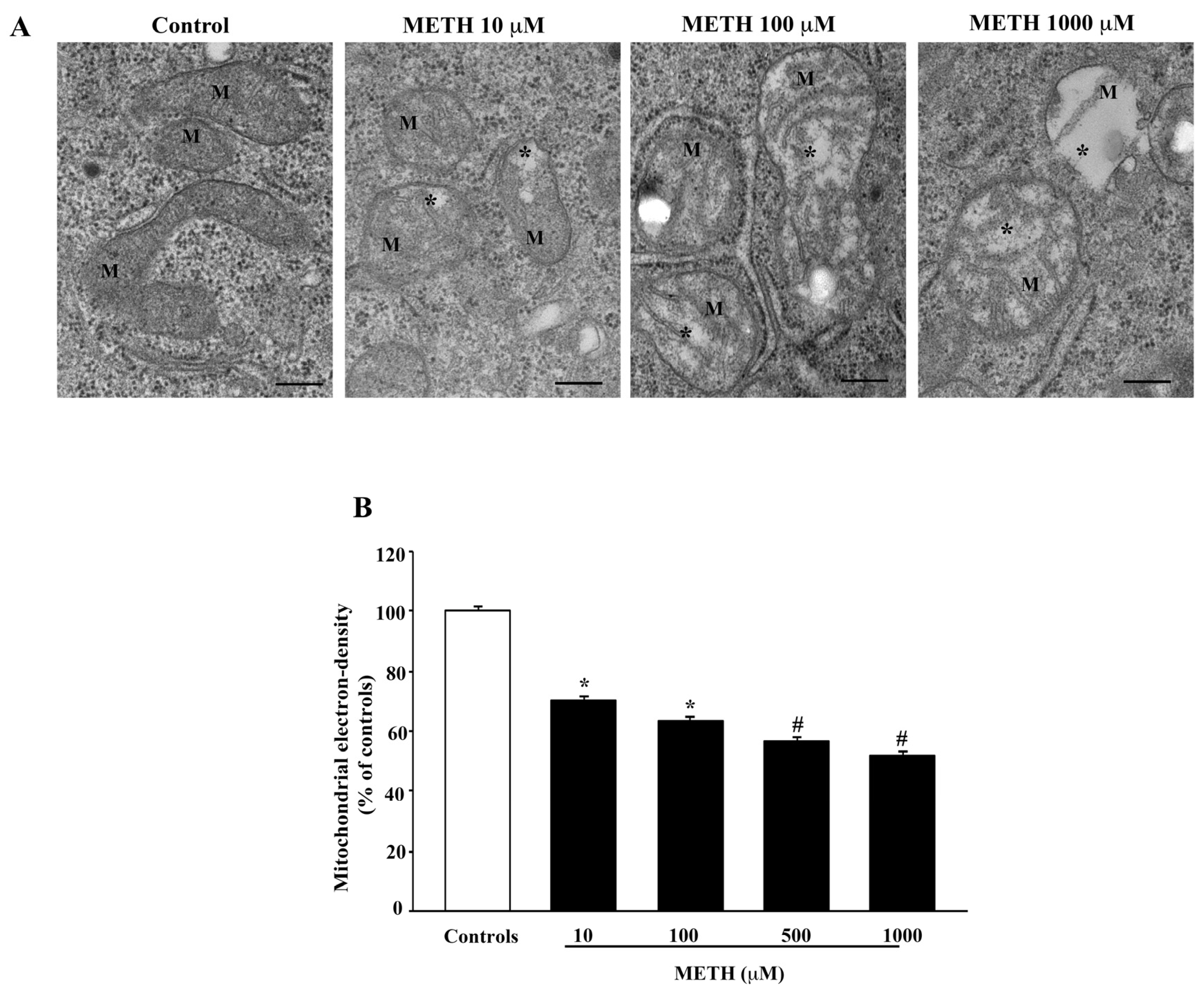
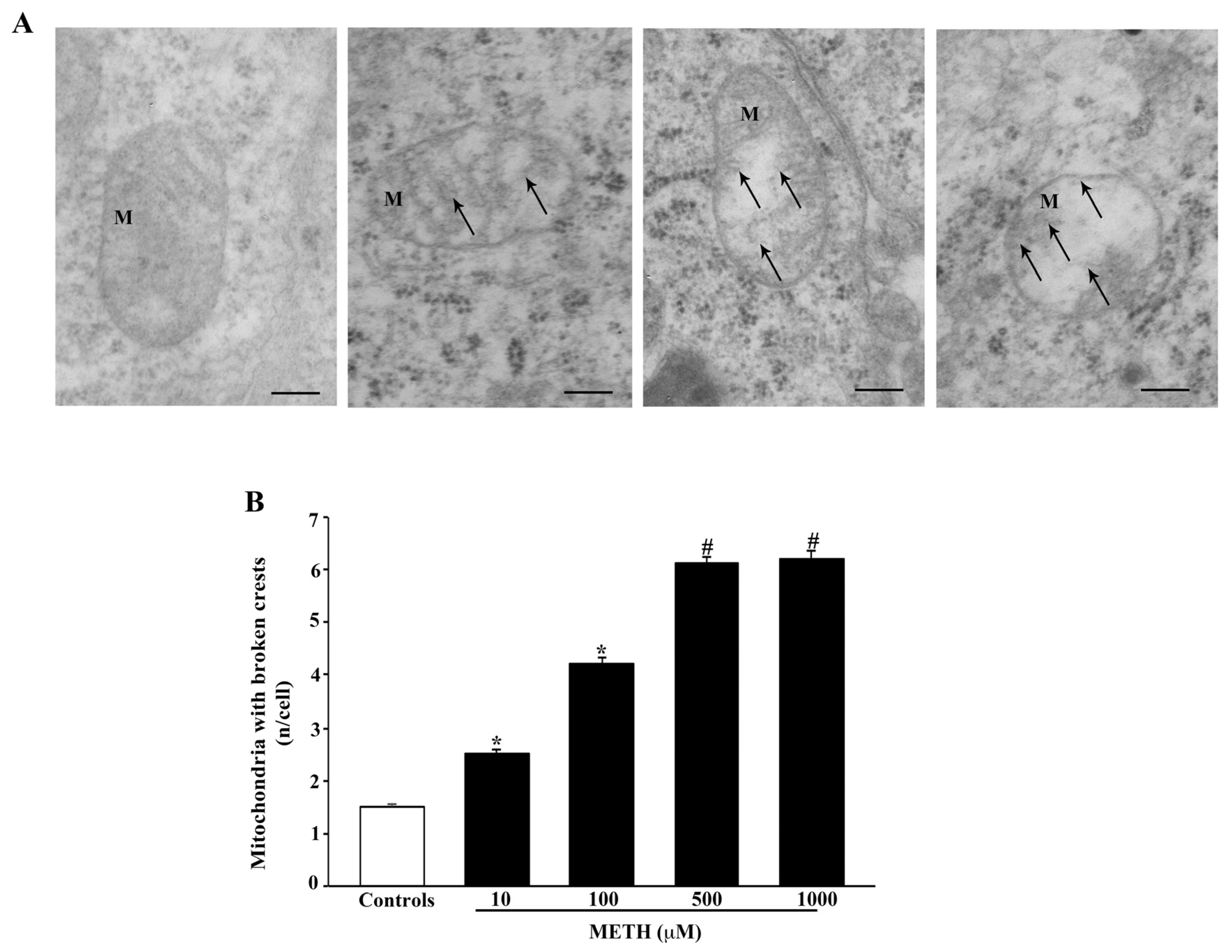
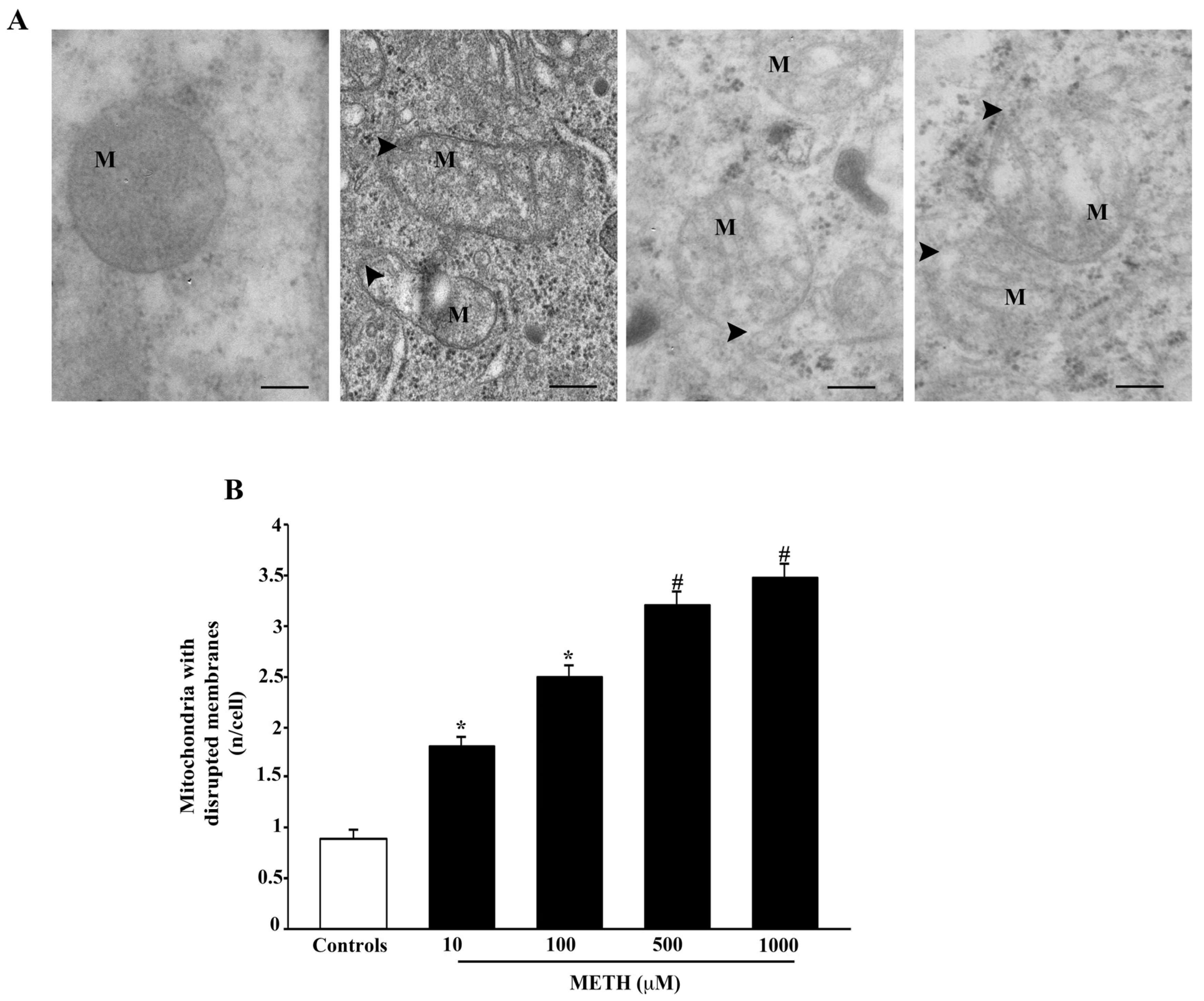
2.4. Dose-Dependent Effects of METH on Mitochondral Alterations Detected at TEM
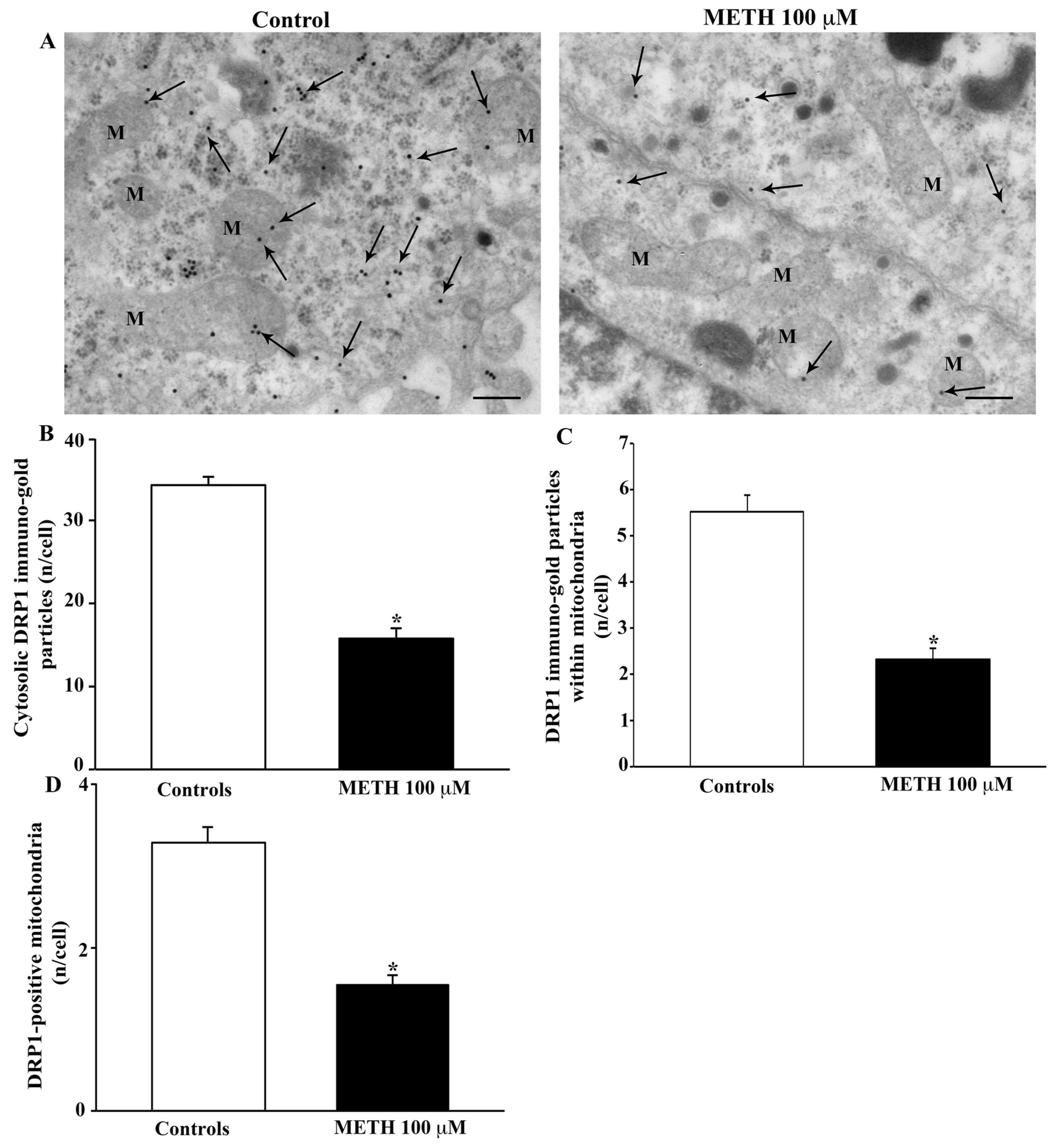
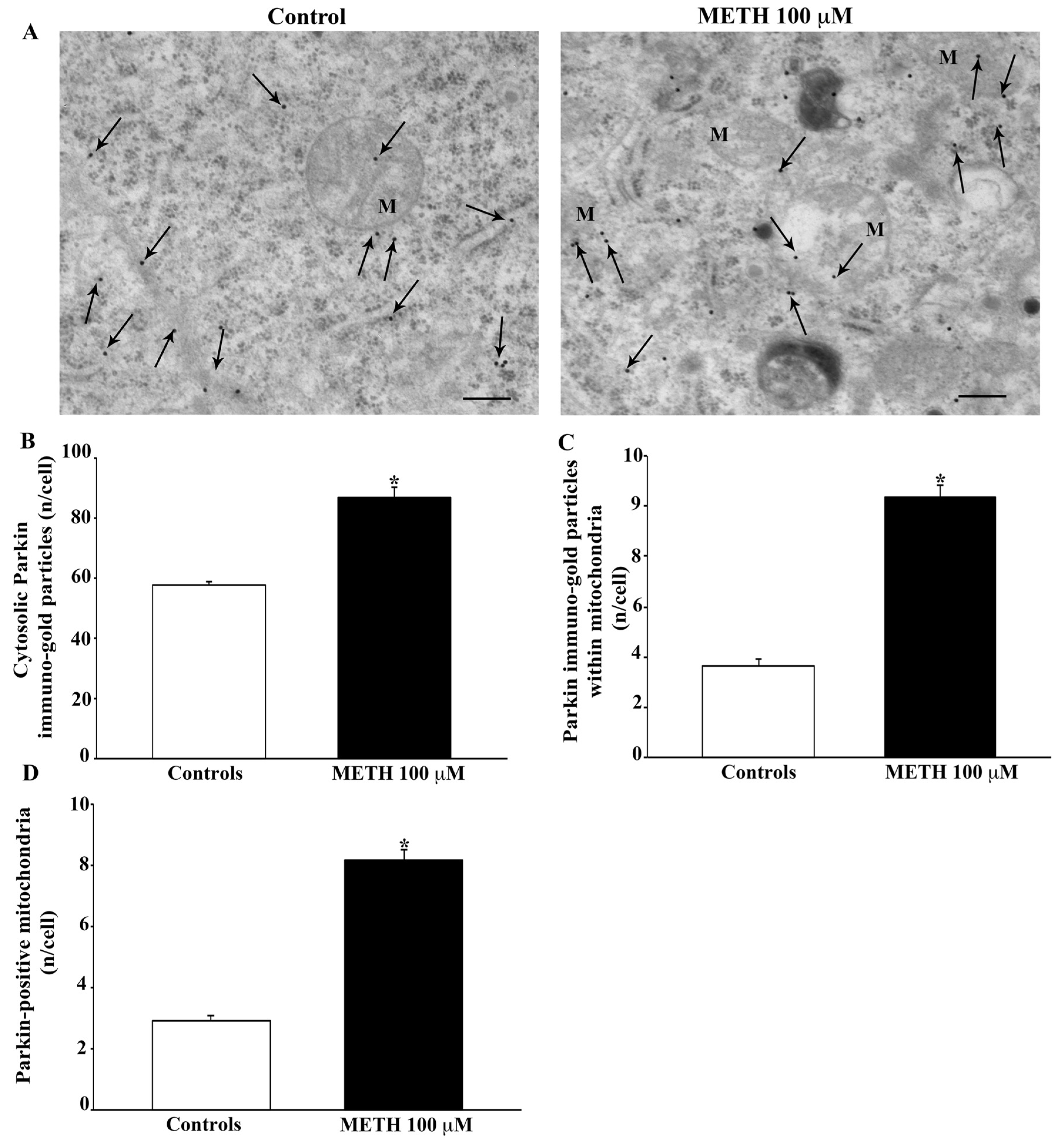
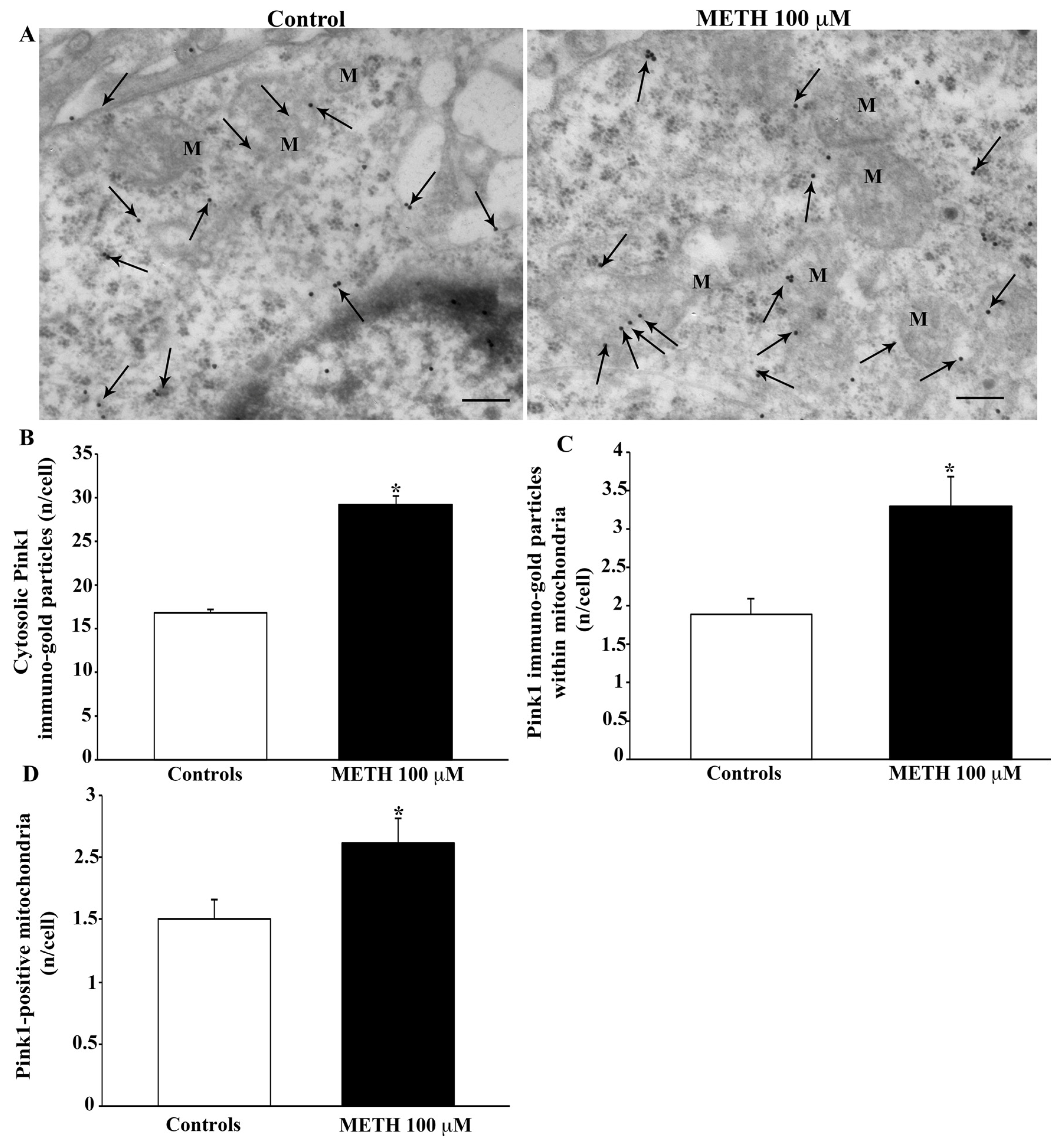
2.5. Effects of METH In Situ on Ultrastructural Stoichiometry of Proteins Affecting Mitochondrial Dynamics
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. Hematoxylin and Eosin (H&E) Histochemistry
4.3. Fluoro-Jade B (FJB) Histofluorescence
4.4. Trypan Blue (TB) Staining
4.5. Mitochondrial Labeling
4.6. Immunocytochemistry by Light Microscopy
4.7. Transmission Electron Microscopy (TEM)
4.8. Ultrastructural Morphometry of Mitochondria
4.9. Immunoelectron Microscopy
4.10. Western Blotting
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ricaurte, G.A.; Guillery, R.W.; Seiden, L.S.; Schuster, C.R.; Moore, R.Y. Dopamine nerve terminal degeneration produced by high doses of methylamphetamine in the rat brain. Brain Res. 1982, 235, 93–103. [Google Scholar] [CrossRef]
- Wilson, J.M.; Kalasinsky, K.S.; Levey, A.I.; Bergeron, C.; Reiber, G.; Anthony, R.M.; Schmunk, G.A.; Shannak, K.; Haycock, J.W.; Kish, S.J. Striatal dopamine nerve terminal markers in human, chronic methamphetamine users. Nat. Med. 1996, 2, 699–703. [Google Scholar] [CrossRef]
- Moratalla, R.; Khairnar, A.; Simola, N.; Granado, N.; García-Montes, J.R.; Porceddu, P.F.; Tizabi, Y.; Costa, G.; Morelli, M. Amphetamine-related drugs neurotoxicity in humans and in experimental animals: Main mechanisms. Prog. Neurobiol. 2017, 155, 149–170. [Google Scholar] [CrossRef] [Green Version]
- Jayanthi, S.; Daiwile, A.P.; Cadet, J.L. Neurotoxicity of methamphetamine: Main effects and mechanisms. Exp. Neurol. 2021, 344, 113795. [Google Scholar] [CrossRef]
- Limanaqi, F.; Busceti, C.L.; Celli, R.; Biagioni, F.; Fornai, F. Autophagy as a gateway for the effects of methamphetamine: From neurotransmitter release and synaptic plasticity to psychiatric and neurodegenerative disorders. Prog. Neurobiol. 2021, 204, 102112. [Google Scholar] [CrossRef]
- Fleckenstein, A.E.; Metzger, R.R.; Gibb, J.W.; Hanson, G.R. A rapid and reversible change in dopamine transporters induced by methamphetamine. Eur. J. Pharmacol. 1997, 323, R9–R10. [Google Scholar] [CrossRef]
- Rothman, R.B.; Partilla, J.S.; Baumann, M.H.; Dersch, C.M.; Carroll, F.I.; Rice, K.C. Neurochemical neutralization of methamphetamine with high-affinity nonselective inhibitors of biogenic amine transporters: A pharmacological strategy for treating stimulant abuse. Synapse 2000, 35, 222–227. [Google Scholar] [CrossRef] [Green Version]
- Volz, T.J.; Hanson, G.R.; Fleckenstein, A.E. The role of the plasmalemmal dopamine and vesicular monoamine transporters in methamphetamine-induced dopaminergic deficits. J. Neurochem. 2007, 101, 883–888. [Google Scholar] [CrossRef]
- Kokoshka, J.M.; Vaughan, R.A.; Hanson, G.R.; Fleckenstein, A.E. Nature of methamphetamine-induced rapid and reversible changes in dopamine transporters. Eur. J. Pharmacol. 1998, 361, 269–275. [Google Scholar] [CrossRef]
- Sandoval, V.; Riddle, E.L.; Hanson, G.R.; Fleckenstein, A.E. Methylphenidate Redistributes Vesicular Monoamine Transporter-2: Role of Dopamine Receptors. J. Neurosci. 2002, 22, 8705–8710. [Google Scholar] [CrossRef] [Green Version]
- Sandoval, V.; Riddle, E.L.; Hanson, G.R.; Fleckenstein, A.E. Methylphenidate alters vesicular monoamine transport and prevents methamphetamine-induced dopaminergic deficits. J. Pharmacol. Exp. Ther. 2003, 304, 1181–1187. [Google Scholar] [CrossRef] [Green Version]
- Sulzer, D.; Rayport, S. Amphetamine and other psychostimulants reduce pH gradients in midbrain dopaminergic neurons and chromaffin granules: A mechanism of action. Neuron 1990, 5, 797–808. [Google Scholar] [CrossRef]
- Sulzer, D.; Pothos, E.; Sung, H.M.; Maidment, N.T.; Hoebel, B.G.; Rayport, S. Weak base model of amphetamine action. Ann. N. Y. Acad. Sci. 1992, 654, 525–528. [Google Scholar] [CrossRef] [PubMed]
- Cubells, J.F.; Rayport, S.; Rajendran, G.; Sulzer, D. Methamphetamine neurotoxicity involves vacuolation of endocytic organelles and dopamine-dependent intracellular oxidative stress. J. Neurosci. 1994, 14, 2260–2271. [Google Scholar] [CrossRef] [PubMed]
- Kogan, F.J.; Nichols, W.K.; Gibb, J.W. Influence of methamphetamine on nigral and striatal tyrosine hydroxylase activity and on striatal dopamine levels. Eur. J. Pharmacol. 1976, 36, 363–371. [Google Scholar] [CrossRef]
- O’Dell, S.J.; Weihmuller, F.B.; Marshall, J.F. Multiple methamphetamine injections induce marked increases in extracellular striatal dopamine which correlate with subsequent neurotoxicity. Brain Res. 1991, 564, 256–260. [Google Scholar] [CrossRef]
- Green, A.L.; El Hait, M.A.S. p-Methoxyamphetamine, a potent reversible inhibitor of type-A monoamine oxidase in vitro and in vivo. J. Pharm. Pharmacol. 1980, 32, 262–266. [Google Scholar] [CrossRef]
- Suzuki, O.; Hattori, H.; Asano, M.; Oya, M.; Katsumata, Y. Inhibition of monoamine oxidase by d-methamphetamine. Biochem. Pharmacol. 1980, 29, 2071–2073. [Google Scholar] [CrossRef]
- Hastings, T.G.; Lewis, D.A.; Zigmond, M.J. Role of oxidation in the neurotoxic effects of intrastriatal dopamine injections. Proc. Natl. Acad. Sci. USA 1996, 93, 1956–1961. [Google Scholar] [CrossRef] [Green Version]
- Kita, T.; Miyazaki, I.; Asanuma, M.; Takeshima, M.; Wagner, G.C. Dopamine-induced behavioral changes and oxidative stress in methamphetamine-induced neurotoxicity. Int. Rev. Neurobiol. 2009, 88, 43–64. [Google Scholar] [CrossRef]
- Graham, D.G.; Tiffany, S.M.; Vogel, F.S. The Toxicity of Melanin Precursors. J. Investig. Dermatol. 1978, 70, 113–116. [Google Scholar] [CrossRef] [Green Version]
- Cohen, G. Oxy-radical toxicity in catecholamine neurons. Neurotoxicology 1984, 5, 77–82. [Google Scholar]
- LaVoie, M.J.; Hastings, T.G. Dopamine quinone formation and protein modification associated with the striatal neurotoxicity of methamphetamine: Evidence against a role for extracellular dopamine. J. Neurosci. 1999, 19, 1484–1491. [Google Scholar] [CrossRef] [PubMed]
- Fornai, F.; Lazzeri, G.; Lenzi, P.; Gesi, M.; Ferrucci, M.; Soldani, P.; Pellegrini, A.; Capobianco, L.; De Blasi, A.; Ruggieri, S.; et al. Amphetamines induce ubiquitin-positive inclusions within striatal cells. J. Neurosci. 2003, 24, 182–183. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, G.; Fornai, F.; Busceti, C.L.; Aloisi, G.; Cerrito, F.; De Blasi, A.; Melchiorri, D.; Nicoletti, F. Selective blockade of mGlu5 metabotropic glutamate receptors is protective against methamphetamine neurotoxicity. J. Neurosci. 2002, 22, 2135–2141. [Google Scholar] [CrossRef]
- Riddle, E.L.; Fleckenstein, A.E.; Hanson, G.R. Mechanisms of methamphetamine-induced dopaminergic neurotoxicity. AAPS J. 2006, 8, E413–E418. [Google Scholar] [CrossRef]
- Lazzeri, G.; Lenzi, P.; Busceti, C.L.; Ferrucci, M.; Falleni, A.; Bruno, V.; Paparelli, A.; Fornai, F. Mechanisms involved in the formation of dopamine-induced intracellular bodies within striatal neurons. J. Neurochem. 2007, 101, 1414–1427. [Google Scholar] [CrossRef]
- Yang, X.; Wang, Y.; Li, Q.; Zhong, Y.; Chen, L.; Du, Y.; He, J.; Liao, L.; Xiong, K.; Yi, C.X.; et al. The main molecular mechanisms underlying methamphetamine- induced neurotoxicity and implications for pharmacological treatment. Front. Mol. Neurosci. 2018, 11, 186. [Google Scholar] [CrossRef] [PubMed]
- Graves, S.M.; Xie, Z.; Stout, K.A.; Zampese, E.; Burbulla, L.F.; Shih, J.C.; Kondapalli, J.; Patriarchi, T.; Tian, L.; Brichta, L.; et al. Dopamine metabolism by a monoamine oxidase mitochondrial shuttle activates the electron transport chain. Nat. Neurosci. 2020, 23, 15–20, Erratum in: Nat. Neurosci. 2020, 23, 293. [Google Scholar] [CrossRef]
- Limanaqi, F.; Gambardella, S.; Biagioni, F.; Busceti, C.L.; Fornai, F. Epigenetic effects induced by methamphetamine and methamphetamine-dependent oxidative stress. Oxid. Med. Cell. Longev. 2018, 2018, 28. [Google Scholar] [CrossRef] [Green Version]
- Bazylianska, V.; Sharma, A.; Chauhan, H.; Schneider, B.; Moszczynska, A. Dopamine and Methamphetamine Differentially Affect Electron Transport Chain Complexes and Parkin in Rat Striatum: New Insight into Methamphetamine Neurotoxicity. Int. J. Mol. Sci. 2021, 23, 363. [Google Scholar] [CrossRef] [PubMed]
- Graves, S.M.; Schwarzschild, S.E.; Tai, R.A.; Chen, Y.; Surmeier, D.J. Mitochondrial oxidant stress mediates methamphetamine neurotoxicity in substantia nigra dopaminergic neurons. Neurobiol. Dis. 2021, 156, 105409. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Bazylianska, V.; Moszczynska, A. Parkin-deficient rats are resistant to neurotoxicity of chronic high-dose methamphetamine. Exp. Neurol. 2021, 345, 113811. [Google Scholar] [CrossRef]
- Calma, I.D.; Persons, A.L.; Napier, T.C. Mitochondrial function influences expression of methamphetamine-induced behavioral sensitization. Sci. Rep. 2021, 11, 24529. [Google Scholar] [CrossRef] [PubMed]
- Fornai, F.; Lenzi, P.; Lazzeri, G.; Ferrucci, M.; Fulceri, F.; Giorgi, F.S.; Falleni, A.; Ruggieri, S.; Paparelli, A. Fine ultrastructure and biochemistry of PC12 cells: A comparative approach to understand neurotoxicity. Brain Res. 2007, 1129, 174–190. [Google Scholar] [CrossRef]
- Strober, W. Trypan blue exclusion test of cell viability. Curr. Protoc. Immunol. 2001, 3, Appendix 3B. [Google Scholar] [CrossRef] [PubMed]
- Schmued, L.C.; Hopkins, K.J. Fluoro-Jade B: A high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000, 874, 123–130. [Google Scholar] [CrossRef] [Green Version]
- Flameng, W.; Borgers, M.; Daenen, W.; Stalpaert, G. Ultrastructural and cytochemical correlates of myocardial protection by cardiac hypothermia in man. J. Thorac. Cardiovasc. Surg. 1980, 79, 413–424. [Google Scholar] [CrossRef]
- Feng, X.; Yin, W.; Wang, J.; Feng, L.; Kang, Y.J. Mitophagy promotes the stemness of bone marrow-derived mesenchymal stem cells. Exp. Biol. Med. 2021, 246, 97–105. [Google Scholar] [CrossRef]
- Lazzeri, G.; Biagioni, F.; Fulceri, F.; Busceti, C.L.; Scavuzzo, M.C.; Ippolito, C.; Salvetti, A.; Lenzi, P.; Fornai, F. mTOR Modulates Methamphetamine-Induced Toxicity through Cell Clearing Systems. Oxid. Med. Cell. Longev. 2018, 2018, 6124745. [Google Scholar] [CrossRef] [Green Version]
- Cruz, C.; Cairrao, E.; Silvestre, S.; Breitenfeld, L.; Almeida, P.; Queiroz, J.A. Targeting of mitochondria-endoplasmic reticulum by fluorescent macrocyclic compounds. PLoS ONE 2011, 6, e27078. [Google Scholar] [CrossRef] [PubMed]
- Melega, A.; Cho, K.; Harvey, D.; Laćan, G. Methamphetamine blood concentrations in human abusers: Application to pharmacokinetic modelling. Synapse 2007, 61, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Larsen, K.E.; Fon, E.A.; Hastings, T.G.; Edwards, R.H.; Sulzer, D. Methamphetamine-induced degeneration of dopaminergic neurons involves autophagy and upregulation of dopamine synthesis. J. Neurosci. 2002, 22, 8951–8960. [Google Scholar] [CrossRef] [PubMed]
- Fornai, F.; Lenzi, P.; Gesi, M.; Soldani, P.; Ferrucci, M.; Lazzeri, G.; Capobianco, L.; Battaglia, G.; De Blasi, A.; Nicoletti, F.; et al. Methamphetamine produces neuronal inclusions in the nigrostriatal system and in PC12 cells. J. Neurochem. 2004, 88, 114–123. [Google Scholar] [CrossRef] [Green Version]
- Castino, R.; Lazzeri, G.; Lenzi, P.; Bellio, N.; Follo, C.; Ferrucci, M.; Fornai, F.; Isidoro, C. Suppression of autophagy precipitates neuronal cell death following low doses of methamphetamine. J. Neurochem. 2008, 106, 1426–1439. [Google Scholar] [CrossRef]
- Shin, E.J.; Tran, H.Q.; Nguyen, P.T.; Jeong, J.H.; Nah, S.Y.; Jang, C.G.; Nabeshima, T.; Kim, H.C. Role of Mitochondria in Methamphetamine-Induced Dopaminergic Neurotoxicity: Involvement in Oxidative Stress, Neuroinflammation, and Pro-apoptosis-A Review. Neurochem. Res. 2018, 43, 66–78. [Google Scholar] [CrossRef]
- Zhu, L.N.; Qiao, H.H.; Chen, L.; Sun, L.P.; Hui, J.L.; Lian, Y.L.; Xie, W.B.; Ding, J.Y.; Meng, Y.L.; Zhu, B.F.; et al. SUMOylation of Alpha-Synuclein Influences on Alpha-Synuclein Aggregation Induced by Methamphetamine. Front. Cell. Neurosci. 2018, 12, 262. [Google Scholar] [CrossRef]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [Green Version]
- Lenzi, P.; Marongiu, R.; Falleni, A.; Gelmetti, V.; Busceti, C.L.; Michiorri, S.; Valente, E.M.; Fornai, F. A subcellular analysis of genetic modulation of PINK1 on mitochondrial alterations, autophagy and cell death. Arch. Ital. Biol. 2012, 150, 194–217. [Google Scholar] [CrossRef]
- Yoo, S.M.; Jung, Y.K. A Molecular Approach to Mitophagy and Mitochondrial Dynamics. Mol. Cells 2018, 41, 18–26. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarffe, L.A.; Stevens, D.A.; Dawson, V.L.; Dawson, T.M. Parkin and PINK1: Much more than mitophagy. Trends Neurosci. 2014, 37, 315–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barazzuol, L.; Giamogante, F.; Brini, M.; Calì, T. PINK1/Parkin Mediated Mitophagy, Ca2+ Signalling, and ER-Mitochondria Contacts in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dagda, R.K.; Cherra, S.J., 3rd; Kulich, S.M.; Tandon, A.; Park, D.; Chu, C.T. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J. Biol. Chem. 2009, 284, 13843–13855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutz, A.K.; Exner, N.; Fett, M.E.; Schlehe, J.S.; Kloos, K.; Lämmermann, K.; Brunner, B.; Kurz-Drexler, A.; Vogel, F.; Reichert, A.S.; et al. Loss of parkin or PINK1 function increases Drp1-dependent mitochondrial fragmentation. J. Biol. Chem. 2009, 284, 22938–22951. [Google Scholar] [CrossRef] [Green Version]
- Burman, J.L.; Pickles, S.; Wang, C.; Sekine, S.; Vargas, J.N.S.; Zhang, Z.; Youle, A.M.; Nezich, C.L.; Wu, X.; Hammer, J.A.; et al. Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J. Cell Biol. 2017, 216, 3231–3247. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.M.; Quinton, M.S.; Yamamoto, B.K. Methamphetamine-induced inhibition of mitochondrial complex II: Roles of glutamate and peroxynitrite. J. Neurochem. 2005, 95, 429–436. [Google Scholar] [CrossRef]
- Ryskalin, L.; Biagioni, F.; Busceti, C.L.; Polzella, M.; Lenzi, P.; Frati, A.; Ferrucci, M.; Fornai, F. Lactoferrin Protects against Methamphetamine Toxicity by Modulating Autophagy and Mitochondrial Status. Nutrients 2021, 13, 3356. [Google Scholar] [CrossRef]
- Song, X.; Violin, J.D.; Seidler, F.J.; Slotkin, T.A. Modeling the Developmental Neurotoxicity of Chlorpyrifosin Vitro: Macromolecule Synthesis in PC12 Cells. Toxicol. Appl. Pharmacol. 1998, 151, 182–191. [Google Scholar] [CrossRef]
- Qiao, D.; Seidler, F.J.; Slotkin, T.A. Developmental neurotoxicity of chlorpyrifos modeled in vitro: Comparative effects of metabolites and other cholinesterase inhibitors on DNA synthesis in PC12 and C6 cells. Environ. Health Perspect. 2001, 109, 909–913. [Google Scholar] [CrossRef]
- Lazzeri, G.; Busceti, C.L.; Biagioni, F.; Fabrizi, C.; Morucci, G.; Giorgi, F.S.; Ferrucci, M.; Lenzi, P.; Puglisi-Allegra, S.; Fornai, F. Norepinephrine Protects against Methamphetamine Toxicity through β2-Adrenergic Receptors Promoting LC3 Compartmentalization. Int. J. Mol. Sci. 2021, 22, 7232. [Google Scholar] [CrossRef] [PubMed]
- Pendergrass, W.; Wolf, N.; Poot, M. Efficacy of MitoTracker Green and CMXrosamine to measure changes in mitochondrial membrane potentials in living cells and tissues. Cytom. A 2004, 61, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Gautam, N.; Sankaran, S.; Yason, J.A.; Tan, K.S.W.; Gascoigne, N.R.J. A high content imaging flow cytometry approach to study mitochondria in T cells: MitoTracker Green FM dye concentration optimization. Methods 2018, 134–135, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Ferese, R.; Lenzi, P.; Fulceri, F.; Biagioni, F.; Fabrizi, C.; Gambardella, S.; Familiari, P.; Frati, A.; Limanaqi, F.; Fornai, F. Quantitative Ultrastructural Morphometry and Gene Expression of mTOR-Related Mitochondriogenesis within Glioblastoma Cells. Int. J. Mol. Sci. 2020, 21, 4570. [Google Scholar] [CrossRef]
- Ghadially, F.N. Ultrastructural Pathology of the Cell and Matrix, 3rd ed.; Wellington Butterworths: London, UK, 1988; pp. 191–328. [Google Scholar]
- Natale, G.; Lenzi, P.; Lazzeri, G.; Falleni, A.; Biagioni, F.; Ryskalin, L.; Fornai, F. Compartment-dependent mitochondrial alterations in experimental ALS, the effects of mitophagy and mitochondriogenesis. Front. Cell Neurosci. 2015, 9, 434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenzi, P.; Lazzeri, G.; Biagioni, F.; Busceti, C.L.; Gambardella, S.; Salvetti, A.; Fornai, F. The Autophagoproteasome a Novel Cell Clearing Organelle in Baseline and Stimulated Conditions. Front. Neuroanat. 2016, 10, 78. [Google Scholar] [CrossRef] [Green Version]
- Bendayan, M.; Zollinger, M. Ultrastructural localization of antigenic sites on osmium-fixed tissues applying the protein A-gold technique. J. Histochem. Cytochem. 1983, 31, 101–109. [Google Scholar] [CrossRef] [Green Version]
- Lucocq, J.M.; Habermann, A.; Watt, S.; Backer, J.M.; Mayhew, T.M.; Griffiths, G. A rapid method for assessing the distribution of gold labeling on thin sections. J. Histochem. Cytochem. 2004, 52, 991–1000. [Google Scholar] [CrossRef]
- Stirling, J.W.; Graff, P.S. Antigen unmasking for immunoelectron microscopy: Labeling is improved by treating with sodium ethoxide or sodium metaperiodate, then heating on retrieval medium. J. Histochem. Cytochem. 1995, 43, 115–123. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lenzi, P.; Biagioni, F.; Busceti, C.L.; Lazzeri, G.; Polzella, M.; Frati, A.; Ferrucci, M.; Fornai, F. Alterations of Mitochondrial Structure in Methamphetamine Toxicity. Int. J. Mol. Sci. 2022, 23, 8926. https://doi.org/10.3390/ijms23168926
Lenzi P, Biagioni F, Busceti CL, Lazzeri G, Polzella M, Frati A, Ferrucci M, Fornai F. Alterations of Mitochondrial Structure in Methamphetamine Toxicity. International Journal of Molecular Sciences. 2022; 23(16):8926. https://doi.org/10.3390/ijms23168926
Chicago/Turabian StyleLenzi, Paola, Francesca Biagioni, Carla L. Busceti, Gloria Lazzeri, Maico Polzella, Alessandro Frati, Michela Ferrucci, and Francesco Fornai. 2022. "Alterations of Mitochondrial Structure in Methamphetamine Toxicity" International Journal of Molecular Sciences 23, no. 16: 8926. https://doi.org/10.3390/ijms23168926
APA StyleLenzi, P., Biagioni, F., Busceti, C. L., Lazzeri, G., Polzella, M., Frati, A., Ferrucci, M., & Fornai, F. (2022). Alterations of Mitochondrial Structure in Methamphetamine Toxicity. International Journal of Molecular Sciences, 23(16), 8926. https://doi.org/10.3390/ijms23168926