
Complexing the Oncolytic Adenoviruses Ad∆∆ and Ad-3∆-A20T with Cationic Nanoparticles Enhances Viral Infection and Spread in Prostate and Pancreatic Cancer Models

 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
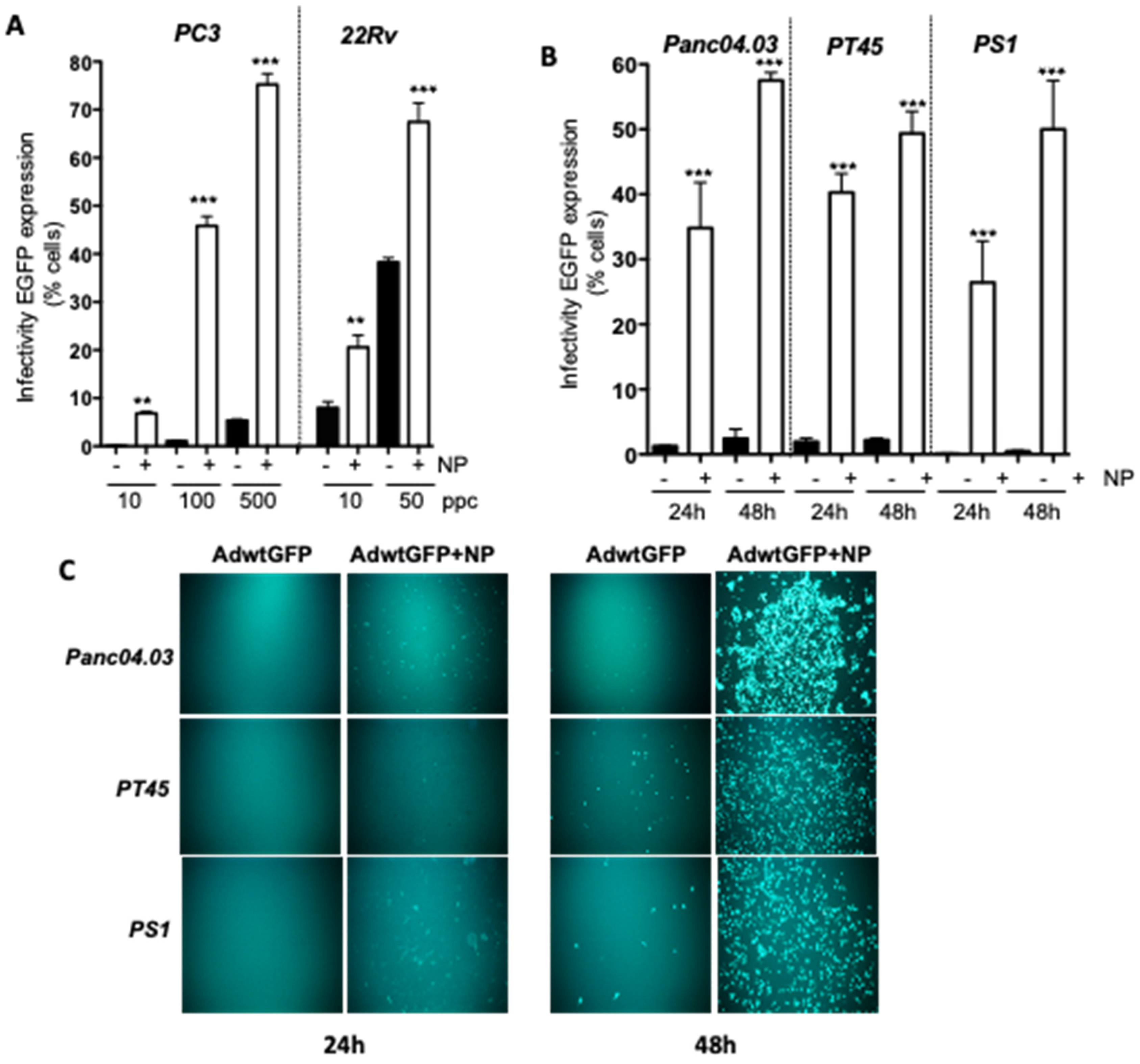
2.1. Gold Nanoparticles (AuNP) Enhance Adenovirus Uptake in PCa and PDAC Adenocarcinoma Cells
2.2. The AuNP-Dependent Increased Viral Uptake in PCa and PDAC Cells Results in Enhanced Ad∆∆-Induced Cell Killing
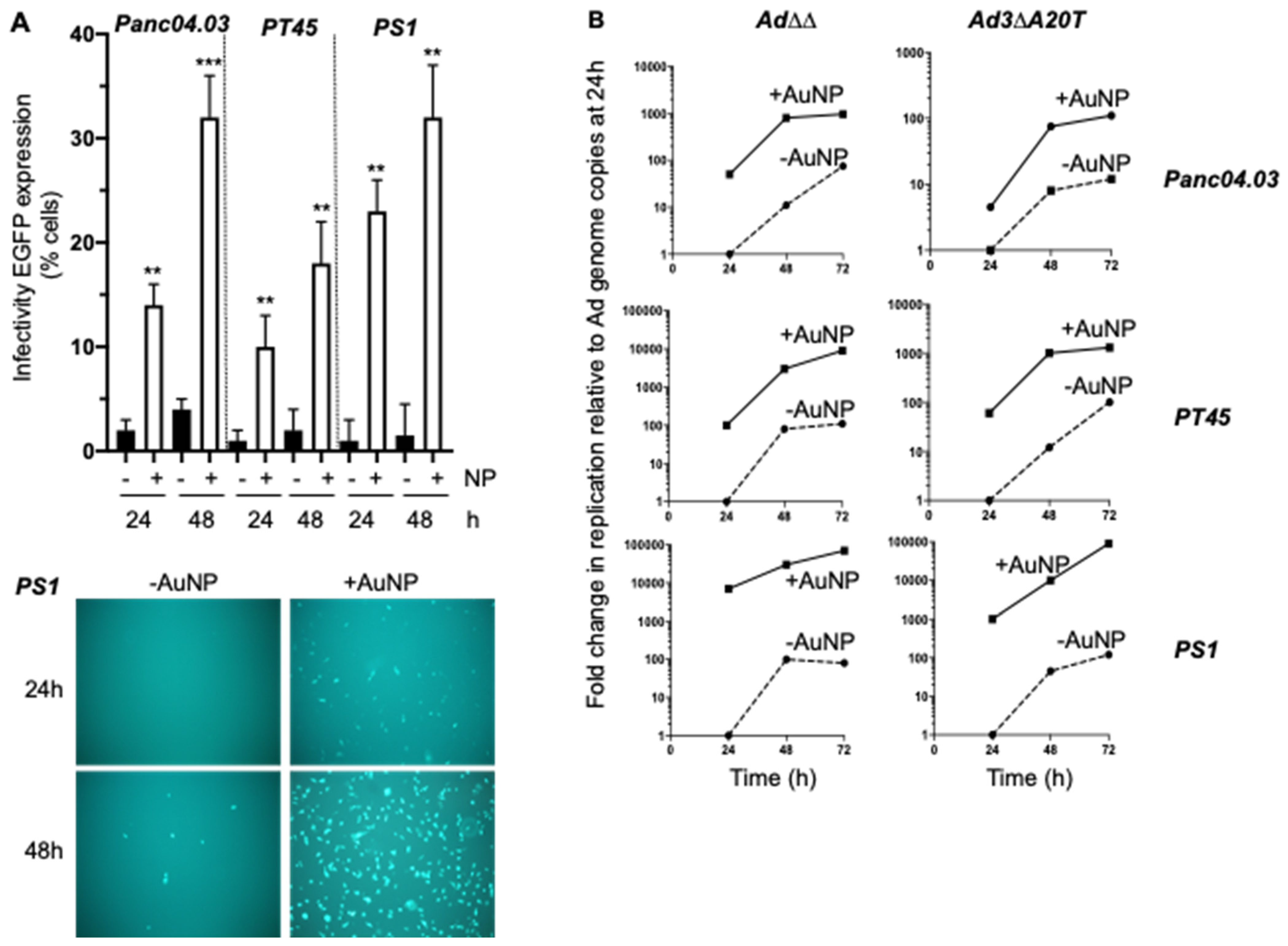
2.3. Complexing of Ad∆∆ with AuNPs Increases Ad∆∆ Replication in All Tested Cell Lines
2.4. Efficient Elimination of Panc04.03 Cells in Organotypic Co-Cultures with PS1 Cells after Infection with Ad5wt and Ad∆∆ Precoated with AuNPs
2.5. Infectivity and Replication of the αvß6-Integrin Targeted Viral Mutant Ad-3∆-A20T Is Improved in Combination with AuNPs
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture Conditions
4.2. Viruses
4.3. Gold Nanoparticles (AuNP) and Ad-AuNPs Complexes
4.4. Flow Cytometry
4.5. Cell Viability Assays
4.6. Viral Replication Assay by TCID50
4.7. Viral Genome Amplification by qPCR
4.8. Organotypic 3-Dimensional (3D) Co-Culture Models
4.9. Immunohistochemistry
4.10. In Vivo Tumour Growth
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rawla, P. Epidemiology of Prostate Cancer. World J. Oncol. 2019, 10, 63–89. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Rozengurt, E.; Eibl, G. Crosstalk between KRAS, SRC and YAP Signaling in Pancreatic Cancer: Interactions Leading to Aggressive Disease and Drug Resistance. Cancers 2021, 13, 5126. [Google Scholar] [CrossRef] [PubMed]
- Bienkowski, M.; Tomasik, B.; Braun, M.; Jassem, J. PARP inhibitors for metastatic castration-resistant prostate cancer: Biological rationale and current evidence. Cancer Treat. Rev. 2022, 104, 102359. [Google Scholar] [CrossRef] [PubMed]
- Biegert, G.W.G.; Rosewell Shaw, A.; Suzuki, M. Current development in adenoviral vectors for cancer immunotherapy. Mol. Oncolytics 2021, 23, 571–581. [Google Scholar] [CrossRef]
- Labani-Motlagh, A.; Naseri, S.; Wenthe, J.; Eriksson, E.; Loskog, A. Systemic immunity upon local oncolytic virotherapy armed with immunostimulatory genes may be supported by tumor-derived exosomes. Mol. Oncolytics 2021, 20, 508–518. [Google Scholar] [CrossRef]
- Tripodi, L.; Vitale, M.; Cerullo, V.; Pastore, L. Oncolytic Adenoviruses for Cancer Therapy. Int. J. Mol. Sci. 2021, 22, 2517. [Google Scholar] [CrossRef]
- Kanaya, N.; Kuroda, S.; Kakiuchi, Y.; Kumon, K.; Tsumura, T.; Hashimoto, M.; Morihiro, T.; Kubota, T.; Aoyama, K.; Kikuchi, S.; et al. Immune Modulation by Telomerase-Specific Oncolytic Adenovirus Synergistically Enhances Antitumor Efficacy with Anti-PD1 Antibody. Mol. Ther. 2020, 28, 794–804. [Google Scholar] [CrossRef]
- Havunen, R.; Santos, J.M.; Sorsa, S.; Rantapero, T.; Lumen, D.; Siurala, M.; Airaksinen, A.J.; Cervera-Carrascon, V.; Tähtinen, S.; Kanerva, A.; et al. Abscopal Effect in Non-injected Tumors Achieved with Cytokine-Armed Oncolytic Adenovirus. Mol. Oncolytics 2018, 11, 109–121. [Google Scholar] [CrossRef]
- Kakiuchi, Y.; Kuroda, S.; Kanaya, N.; Kumon, K.; Tsumura, T.; Hashimoto, M.; Yagi, C.; Sugimoto, R.; Hamada, Y.; Kikuchi, S.; et al. Local oncolytic adenovirotherapy produces an abscopal effect via tumor-derived extracellular vesicles. Mol. Ther. 2021, 29, 2920–2930. [Google Scholar] [CrossRef] [PubMed]
- Freytag, S.O.; Stricker, H.; Lu, M.; Elshaikh, M.; Aref, I.; Pradhan, D.; Levin, K.; Kim, J.H.; Peabody, J.; Siddiqui, F.; et al. Prospective randomized phase 2 trial of intensity modulated radiation therapy with or without oncolytic adenovirus-mediated cytotoxic gene therapy in intermediate-risk prostate cancer. Int. J. Radiat. Oncol. Biol. Phys. 2014, 89, 268–276. [Google Scholar] [CrossRef]
- Freytag, S.O.; Stricker, H.; Movsas, B.; Kim, J.H. Prostate cancer gene therapy clinical trials. Mol. Ther. 2007, 15, 1042–1052. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, Y.; Liu, M.M.; Zhou, H.; Chowdhury, W.; Lupold, S.E.; Deweese, T.L.; Rodriguez, R. Evaluation of continuous low dose rate versus acute single high dose rate radiation combined with oncolytic viral therapy for prostate cancer. Int. J. Radiat. Biol. 2010, 86, 220–229. [Google Scholar] [CrossRef][Green Version]
- Azab, B.M.; Dash, R.; Das, S.K.; Bhutia, S.K.; Sarkar, S.; Shen, X.N.; Quinn, B.A.; Dent, P.; Dmitriev, I.P.; Wang, X.Y.; et al. Enhanced prostate cancer gene transfer and therapy using a novel serotype chimera cancer terminator virus (Ad.5/3-CTV). J. Cell Physiol. 2014, 229, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Bazan-Peregrino, M.; Garcia-Carbonero, R.; Laquente, B.; Alvarez, R.; Mato-Berciano, A.; Gimenez-Alejandre, M.; Morgado, S.; Rodriguez-Garcia, A.; Maliandi, M.V.; Riesco, M.C.; et al. VCN-01 disrupts pancreatic cancer stroma and exerts antitumor effects. J. Immunother. Cancer 2021, 9, e003254. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Shin, D.W.; Park, H.; Kim, J.; Youn, Y.; Kim, J.H.; Kim, J.; Hwang, J.H. Tolerability and safety of EUS-injected adenovirus-mediated double-suicide gene therapy with chemotherapy in locally advanced pancreatic cancer: A phase 1 trial. Gastrointest. Endosc. 2020, 92, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Mulvihill, S.; Warren, R.; Venook, A.; Adler, A.; Randlev, B.; Heise, C.; Kirn, D. Safety and feasibility of injection with an E1B-55 kDa gene-deleted, replication-selective adenovirus (ONYX-015) into primary carcinomas of the pancreas: A phase I trial. Gene Ther. 2001, 8, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Shayakhmetov, D.M.; Gaggar, A.; Ni, S.; Li, Z.Y.; Lieber, A. Adenovirus binding to blood factors results in liver cell infection and hepatotoxicity. J. Virol. 2005, 79, 7478–7491. [Google Scholar] [CrossRef]
- Parker, A.L.; Waddington, S.N.; Nicol, C.G.; Shayakhmetov, D.M.; Buckley, S.M.; Denby, L.; Kemball-Cook, G.; Ni, S.; Lieber, A.; McVey, J.H.; et al. Multiple vitamin K-dependent coagulation zymogens promote adenovirus-mediated gene delivery to hepatocytes. Blood 2006, 108, 2554–2561. [Google Scholar] [CrossRef] [PubMed]
- Öberg, D.; Yanover, E.; Sweeney, K.; Adam, V.; Costas, C.; Lemoine, N.R.; Halldén, G. Improved potency and selectivity of an oncolytic E1ACR2 and E1B19K deleted adenoviral mutant (Ad∆∆) in prostate and pancreatic cancers. Clin. Cancer Res. 2010, 16, 541–553. [Google Scholar] [CrossRef]
- Leitner, S.; Sweeney, K.; Oberg, D.; Davies, D.; Miranda, E.; Lemoine, N.R.; Hallden, G. Oncolytic adenoviral mutants with E1B19K gene deletions enhance gemcitabine-induced apoptosis in pancreatic carcinoma cells and anti-tumor efficacy in vivo. Clin. Cancer Res. 2009, 15, 1730–1740. [Google Scholar] [CrossRef]
- Cherubini, G.; Kallin, C.; Mozetic, A.; Hammaren-Busch, K.; Muller, H.; Lemoine, N.R.; Hallden, G. The oncolytic adenovirus AdDeltaDelta enhances selective cancer cell killing in combination with DNA-damaging drugs in pancreatic cancer models. Gene Ther. 2011, 18, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Adam, V.; Ekblad, M.; Sweeney, K.; Muller, H.; Busch, K.H.; Johnsen, C.T.; Kang, N.R.; Lemoine, N.R.; Hallden, G. Synergistic and Selective Cancer Cell Killing Mediated by the Oncolytic Adenoviral Mutant AdDeltaDelta and Dietary Phytochemicals in Prostate Cancer Models. Hum. Gene Ther. 2012, 23, 1003–1015. [Google Scholar] [CrossRef] [PubMed]
- Aguirre-Hernandez, C.; Maya-Pineda, H.; Millan, J.S.; Man, Y.K.S.; Lu, Y.J.; Hallden, G. Sensitisation to mitoxantrone-induced apoptosis by the oncolytic adenovirus Ad through Bcl-2-dependent attenuation of autophagy. Oncogenesis 2018, 7, 6. [Google Scholar] [CrossRef]
- Liu, T.C.; Hallden, G.; Wang, Y.; Brooks, G.; Francis, J.; Lemoine, N.; Kirn, D. An E1B-19 kDa gene deletion mutant adenovirus demonstrates tumor necrosis factor-enhanced cancer selectivity and enhanced oncolytic potency. Mol. Ther. 2004, 9, 786–803. [Google Scholar] [CrossRef]
- Radhakrishnan, S.; Miranda, E.; Ekblad, M.; Holford, A.; Pizarro, M.T.; Lemoine, N.R.; Hallden, G. Efficacy of oncolytic mutants targeting pRb and p53 pathways is synergistically enhanced when combined with cytotoxic drugs in prostate cancer cells and tumor xenografts. Hum. Gene Ther. 2010, 21, 1311–1325. [Google Scholar] [CrossRef] [PubMed]
- Pantelidou, C.; Cherubini, G.; Lemoine, N.R.; Hallden, G. The E1B19K-deleted oncolytic adenovirus mutant AdDelta19K sensitizes pancreatic cancer cells to drug-induced DNA-damage by down-regulating Claspin and Mre11. Oncotarget 2016, 7, 15703–15724. [Google Scholar] [CrossRef] [PubMed]
- Carlisle, R.C.; Di, Y.; Cerny, A.M.; Sonnen, A.F.; Sim, R.B.; Green, N.K.; Subr, V.; Ulbrich, K.; Gilbert, R.J.; Fisher, K.D.; et al. Human erythrocytes bind and inactivate type 5 adenovirus by presenting Coxsackie virus-adenovirus receptor and complement receptor 1. Blood 2009, 113, 1909–1918. [Google Scholar] [CrossRef] [PubMed]
- Man, Y.K.S.; Davies, J.A.; Coughlan, L.; Pantelidou, C.; Blazquez-Moreno, A.; Marshall, J.F.; Parker, A.L.; Hallden, G. The Novel Oncolytic Adenoviral Mutant Ad5-3Delta-A20T Retargeted to alphavbeta6 Integrins Efficiently Eliminates Pancreatic Cancer Cells. Mol. Cancer 2018, 17, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, L.; Vallath, S.; Gros, A.; Gimenez-Alejandre, M.; Van Rooijen, N.; Thomas, G.J.; Baker, A.H.; Cascallo, M.; Alemany, R.; Hart, I.R. Combined fiber modifications both to target alpha(v)beta(6) and detarget the coxsackievirus-adenovirus receptor improve virus toxicity profiles in vivo but fail to improve antitumoral efficacy relative to adenovirus serotype 5. Hum. Gene Ther. 2012, 23, 960–979. [Google Scholar] [CrossRef] [PubMed]
- Quigley, N.G.; Steiger, K.; Hoberuck, S.; Czech, N.; Zierke, M.A.; Kossatz, S.; Pretze, M.; Richter, F.; Weichert, W.; Pox, C.; et al. PET/CT imaging of head-and-neck and pancreatic cancer in humans by targeting the “Cancer Integrin” alphavbeta6 with Ga-68-Trivehexin. Eur. J. Nucl. Med. Mol. Imaging 2021, 49, 1136–1147. [Google Scholar] [CrossRef]
- Wang, Y.; Hallden, G.; Hill, R.; Anand, A.; Liu, T.C.; Francis, J.; Brooks, G.; Lemoine, N.; Kirn, D. E3 gene manipulations affect oncolytic adenovirus activity in immunocompetent tumor models. Nat. Biotechnol. 2003, 21, 1328–1335. [Google Scholar] [CrossRef]
- Stella Man, Y.K.; Foster, J.; Carapuca, E.; Davies, J.A.; Parker, A.L.; Sosabowski, J.; Hallden, G. Systemic delivery and SPECT/CT in vivo imaging of (125)I-labelled oncolytic adenoviral mutants in models of pancreatic cancer. Sci. Rep. 2019, 9, 12840. [Google Scholar] [CrossRef]
- Wang, C.H.; Chan, L.W.; Johnson, R.N.; Chu, D.S.; Shi, J.; Schellinger, J.G.; Lieber, A.; Pun, S.H. The transduction of Coxsackie and Adenovirus Receptor-negative cells and protection against neutralizing antibodies by HPMA-co-oligolysine copolymer-coated adenovirus. Biomaterials 2011, 32, 9536–9545. [Google Scholar] [CrossRef]
- Hernandez, Y.; Gonzalez-Pastor, R.; Belmar-Lopez, C.; Mendoza, G.; de la Fuente, J.; Martin-Duque, P. Gold nanoparticle coatings as efficient adenovirus carriers to non-infectable stem cells. RSC Adv. 2019, 9, 1327–1334. [Google Scholar] [CrossRef]
- Gonzalez-Pastor, R.; Hernandez, Y.; Gimeno, M.; de Martino, A.; Man, Y.K.S.; Hallden, G.; Quintanilla, M.; de la Fuente, J.M.; Martin-Duque, P. Coating an adenovirus with functionalized gold nanoparticles favors uptake, intracellular trafficking and anti-cancer therapeutic efficacy. Acta Biomater. 2021, 134, 593–604. [Google Scholar] [CrossRef]
- Neuzillet, C.; Tijeras-Raballand, A.; Ragulan, C.; Cros, J.; Patil, Y.; Martinet, M.; Erkan, M.; Kleeff, J.; Wilson, J.; Apte, M.; et al. Inter- and intra-tumoural heterogeneity in cancer-associated fibroblasts of human pancreatic ductal adenocarcinoma. J. Pathol. 2019, 248, 51–65. [Google Scholar] [CrossRef]
- Waddington, S.N.; McVey, J.H.; Bhella, D.; Parker, A.L.; Barker, K.; Atoda, H.; Pink, R.; Buckley, S.M.; Greig, J.A.; Denby, L.; et al. Adenovirus serotype 5 hexon mediates liver gene transfer. Cell 2008, 132, 397–409. [Google Scholar] [CrossRef]
- Alba, R.; Bradshaw, A.C.; Coughlan, L.; Denby, L.; McDonald, R.A.; Waddington, S.N.; Buckley, S.M.; Greig, J.A.; Parker, A.L.; Miller, A.M.; et al. Biodistribution and retargeting of FX-binding ablated adenovirus serotype 5 vectors. Blood 2010, 116, 2656–2664. [Google Scholar] [CrossRef]
- Parker, A.L.; Waddington, S.N.; Buckley, S.M.; Custers, J.; Havenga, M.J.; van Rooijen, N.; Goudsmit, J.; McVey, J.H.; Nicklin, S.A.; Baker, A.H. Effect of neutralizing sera on factor x-mediated adenovirus serotype 5 gene transfer. J. Virol. 2009, 83, 479–483. [Google Scholar] [CrossRef]
- Zheng, S.; Ulasov, I.V.; Han, Y.; Tyler, M.A.; Zhu, Z.B.; Lesniak, M.S. Fiber-knob modifications enhance adenoviral tropism and gene transfer in malignant glioma. J. Gene Med. 2007, 9, 151–160. [Google Scholar] [CrossRef]
- Ahmed, A.U.; Thaci, B.; Tobias, A.L.; Auffinger, B.; Zhang, L.; Cheng, Y.; Kim, C.K.; Yunis, C.; Han, Y.; Alexiades, N.G.; et al. A preclinical evaluation of neural stem cell-based cell carrier for targeted antiglioma oncolytic virotherapy. J. Natl. Cancer Inst. 2013, 105, 968–977. [Google Scholar] [CrossRef]
- Mooney, R.; Majid, A.A.; Batalla-Covello, J.; Machado, D.; Liu, X.; Gonzaga, J.; Tirughana, R.; Hammad, M.; Lesniak, M.S.; Curiel, D.T.; et al. Enhanced Delivery of Oncolytic Adenovirus by Neural Stem Cells for Treatment of Metastatic Ovarian Cancer. Mol. Oncolytics 2019, 12, 79–92. [Google Scholar] [CrossRef]
- Mader, E.K.; Butler, G.; Dowdy, S.C.; Mariani, A.; Knutson, K.L.; Federspiel, M.J.; Russell, S.J.; Galanis, E.; Dietz, A.B.; Peng, K.W. Optimizing patient derived mesenchymal stem cells as virus carriers for a phase I clinical trial in ovarian cancer. J. Transl. Med. 2013, 11, 20. [Google Scholar] [CrossRef]
- Muhammad, T.; Sakhawat, A.; Khan, A.A.; Ma, L.; Gjerset, R.A.; Huang, Y. Mesenchymal stem cell-mediated delivery of therapeutic adenoviral vectors to prostate cancer. Stem Cell Res. 2019, 10, 190. [Google Scholar] [CrossRef]
- Zhang, L.; Wei, Q.; Liu, X.; Zhang, T.; Wang, S.; Zhou, L.; Zou, L.; Fan, F.; Chi, H.; Sun, J.; et al. Exosomal microRNA-98-5p from hypoxic bone marrow mesenchymal stem cells inhibits myocardial ischemia-reperfusion injury by reducing TLR4 and activating the PI3K/Akt signaling pathway. Int. Immunopharmacol. 2021, 101, 107592. [Google Scholar] [CrossRef]
- Ramirez, M.; Garcia-Castro, J.; Melen, G.J.; Gonzalez-Murillo, A.; Franco-Luzon, L. Patient-derived mesenchymal stem cells as delivery vehicles for oncolytic virotherapy: Novel state-of-the-art technology. Oncolytic Virother 2015, 4, 149–155. [Google Scholar] [CrossRef]
- Mugaka, B.P.; Hu, Y.; Ma, Y.; Ding, Y. Surface Modification of Gold Nanoparticles for Targeted Drug Delivery. In Surface Modification of Nanoparticles for Targeted Drug Delivery; Pathak, Y.V., Ed.; Springer International Publishing: Cham, Switzerland, 2019; pp. 391–403. [Google Scholar]
- Weklak, D.; Pembaur, D.; Koukou, G.; Jönsson, F.; Hagedorn, C.; Kreppel, F. Genetic and Chemical Capsid Modifications of Adenovirus Vectors to Modulate Vector-Host Interactions. Viruses 2021, 13, 1300. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Man, Y.K.S.; Aguirre-Hernandez, C.; Fernandez, A.; Martin-Duque, P.; González-Pastor, R.; Halldén, G. Complexing the Oncolytic Adenoviruses Ad∆∆ and Ad-3∆-A20T with Cationic Nanoparticles Enhances Viral Infection and Spread in Prostate and Pancreatic Cancer Models. Int. J. Mol. Sci. 2022, 23, 8884. https://doi.org/10.3390/ijms23168884
Man YKS, Aguirre-Hernandez C, Fernandez A, Martin-Duque P, González-Pastor R, Halldén G. Complexing the Oncolytic Adenoviruses Ad∆∆ and Ad-3∆-A20T with Cationic Nanoparticles Enhances Viral Infection and Spread in Prostate and Pancreatic Cancer Models. International Journal of Molecular Sciences. 2022; 23(16):8884. https://doi.org/10.3390/ijms23168884
Chicago/Turabian StyleMan, Yang Kee Stella, Carmen Aguirre-Hernandez, Adrian Fernandez, Pilar Martin-Duque, Rebeca González-Pastor, and Gunnel Halldén. 2022. "Complexing the Oncolytic Adenoviruses Ad∆∆ and Ad-3∆-A20T with Cationic Nanoparticles Enhances Viral Infection and Spread in Prostate and Pancreatic Cancer Models" International Journal of Molecular Sciences 23, no. 16: 8884. https://doi.org/10.3390/ijms23168884
APA StyleMan, Y. K. S., Aguirre-Hernandez, C., Fernandez, A., Martin-Duque, P., González-Pastor, R., & Halldén, G. (2022). Complexing the Oncolytic Adenoviruses Ad∆∆ and Ad-3∆-A20T with Cationic Nanoparticles Enhances Viral Infection and Spread in Prostate and Pancreatic Cancer Models. International Journal of Molecular Sciences, 23(16), 8884. https://doi.org/10.3390/ijms23168884