
A Computational–Experimental Investigation of the Molecular Mechanism of Interleukin-6-Piperine Interaction


 and
and
Abstract
:1. Introduction
2. Results and Discussion
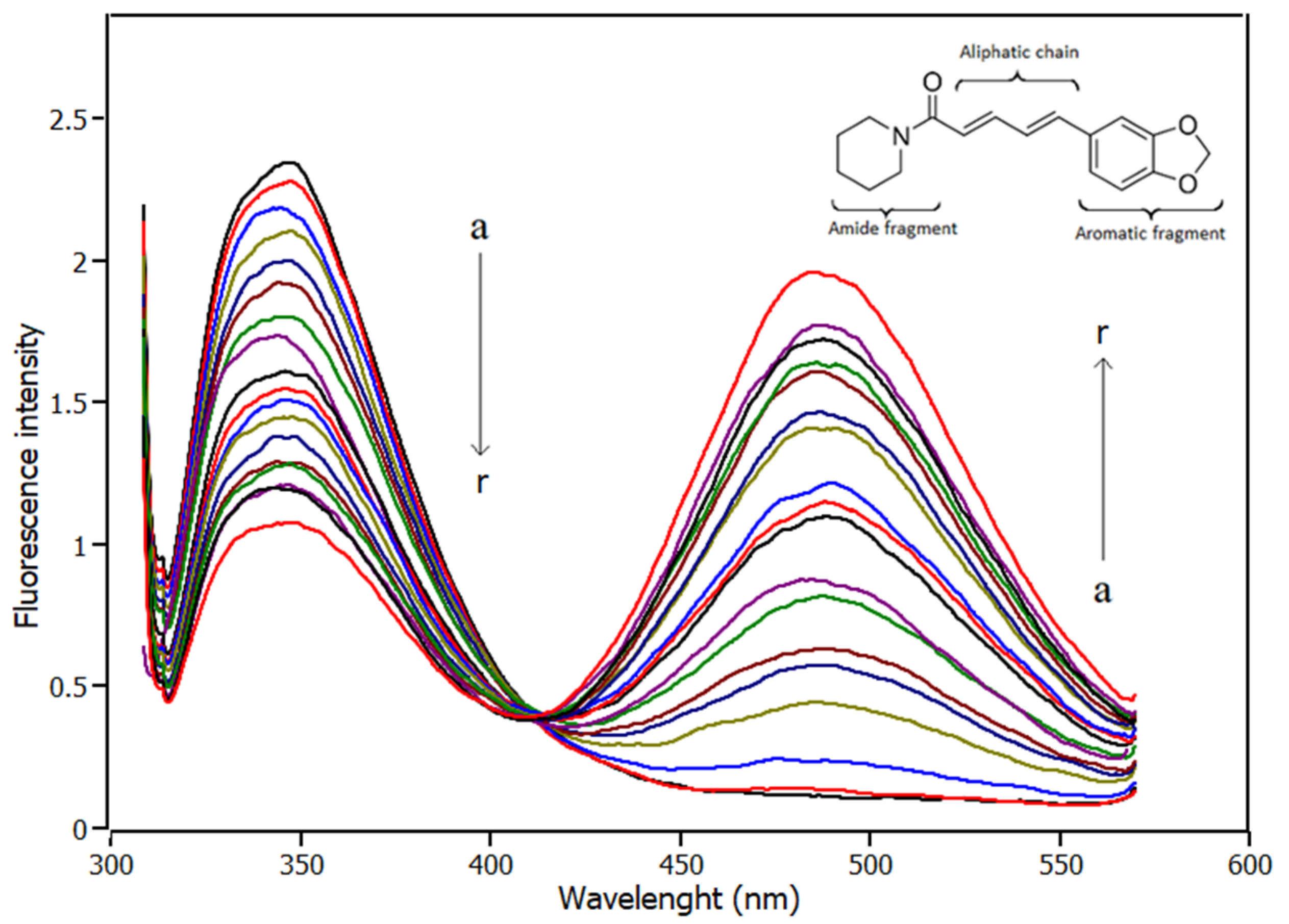
2.1. Fluorescence Spectroscopy
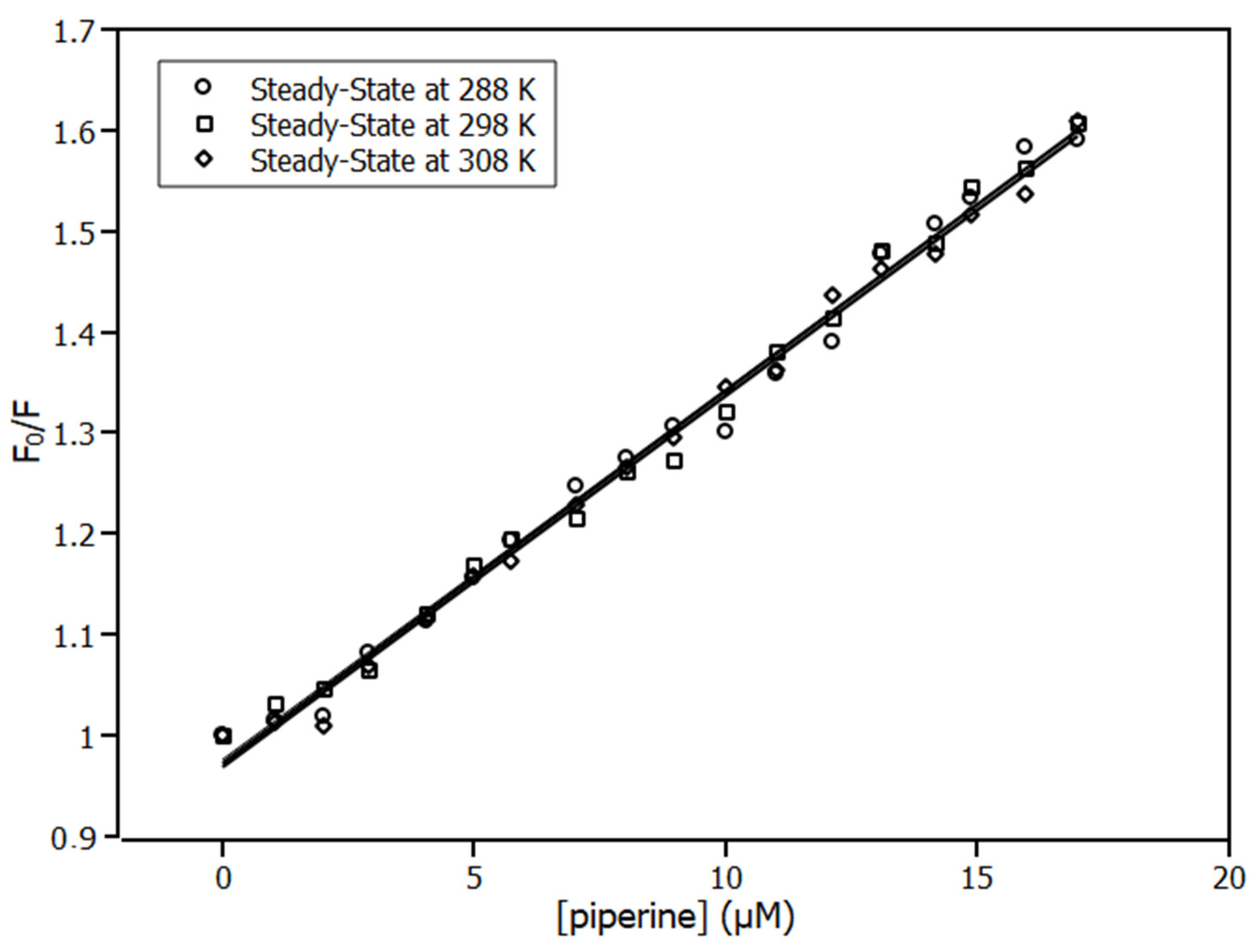
2.2. Thermodynamic Parameters
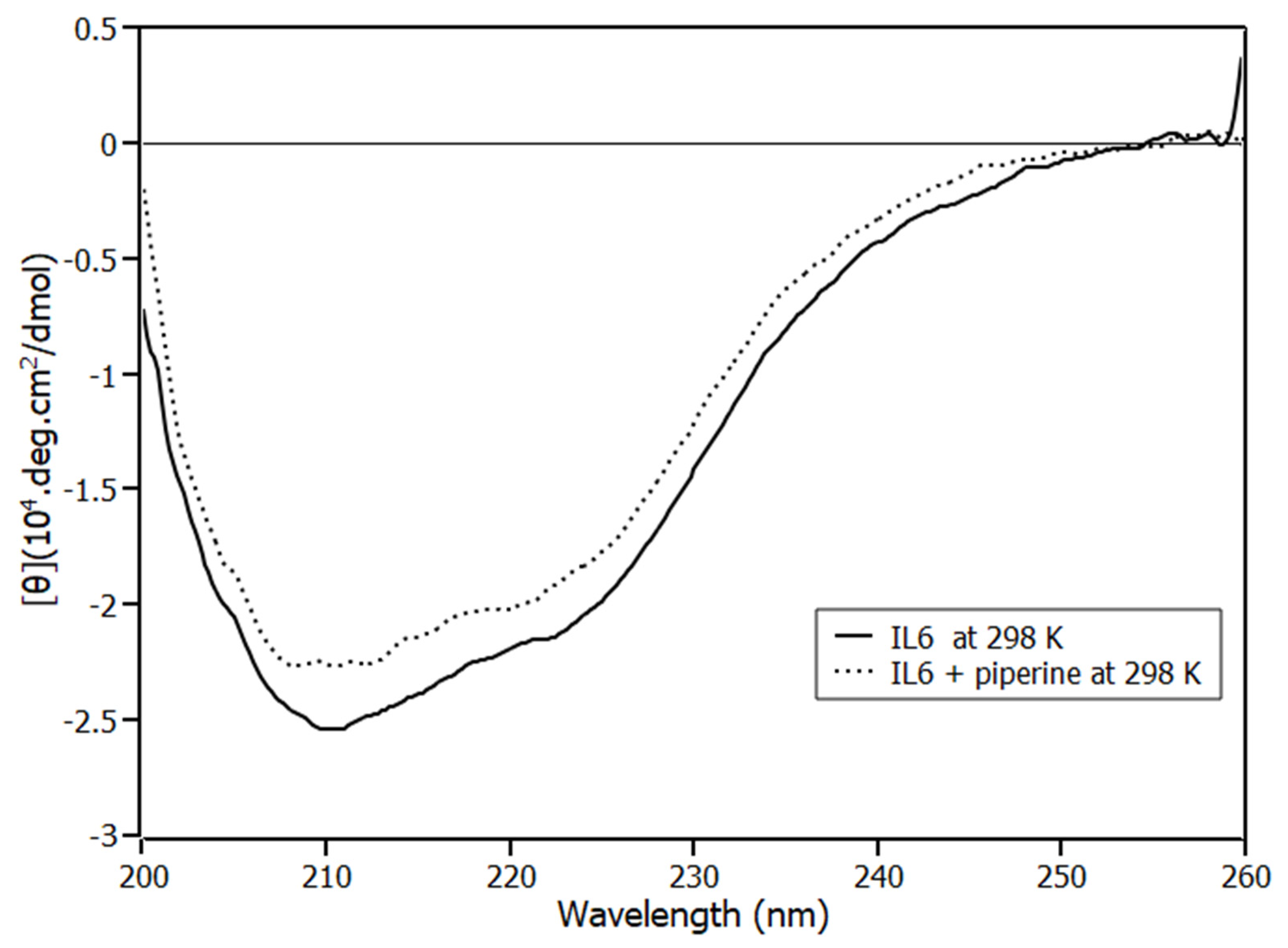
2.3. Circular Dichroism Spectroscopy
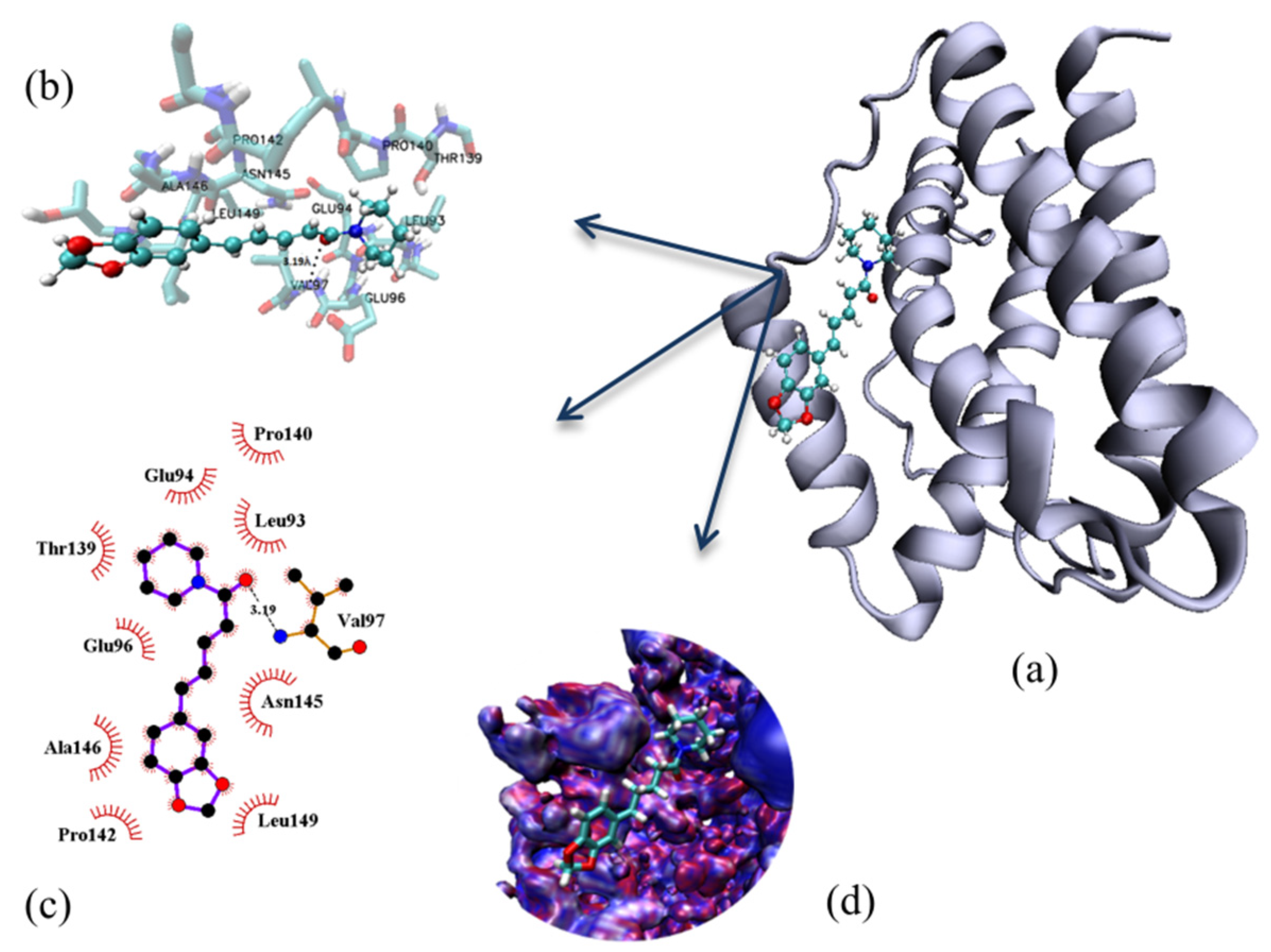
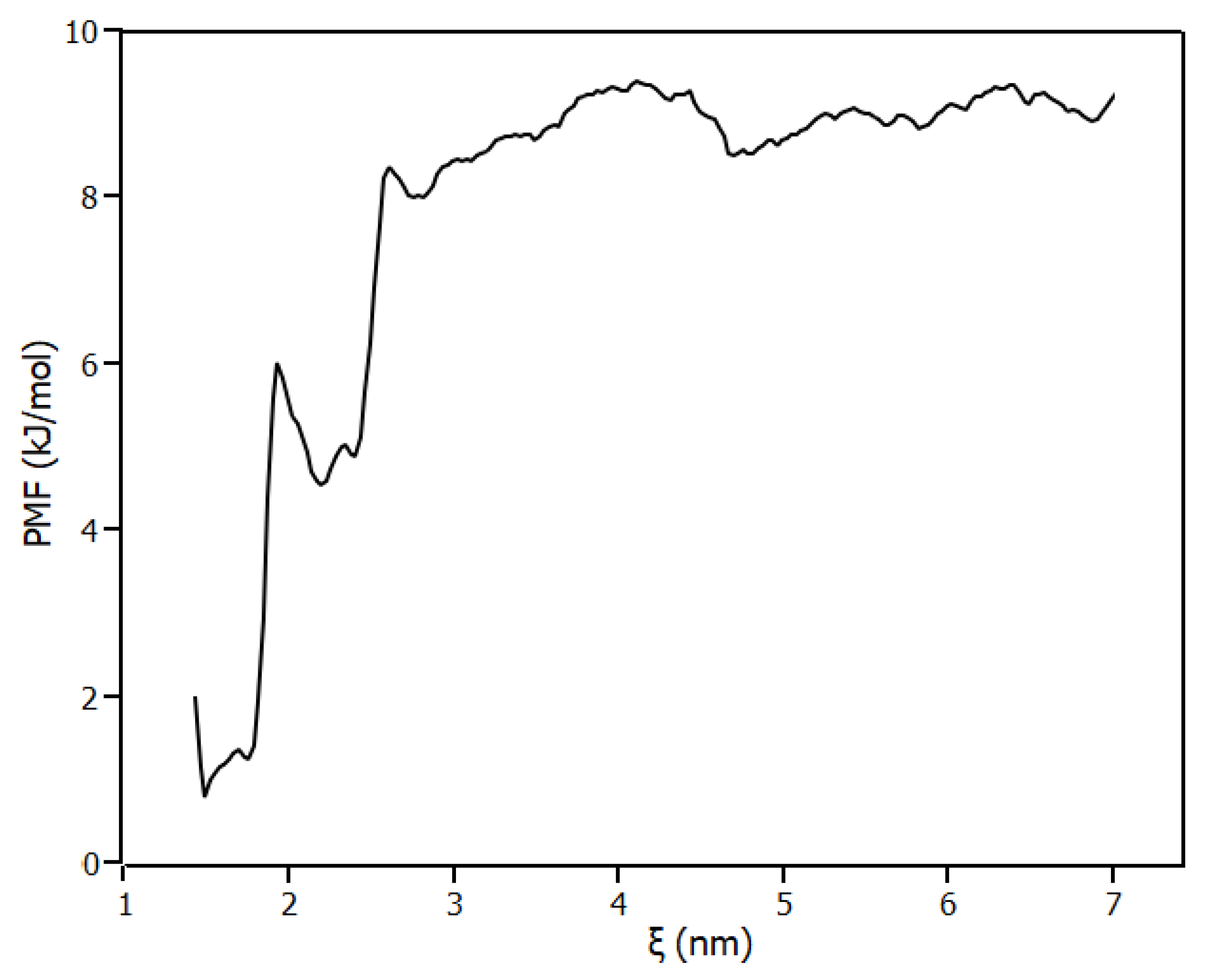
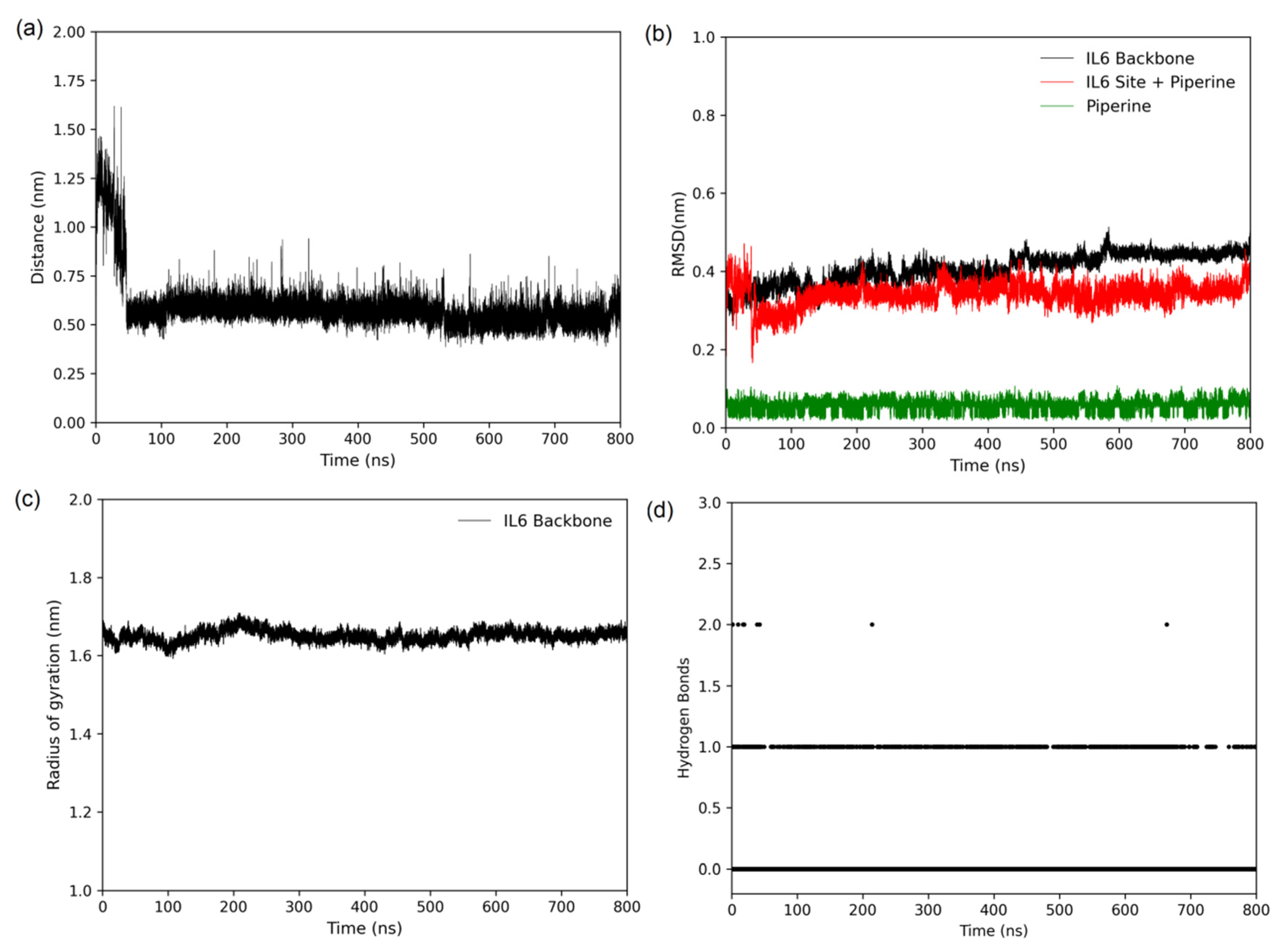
2.4. Molecular Modelling
3. Materials and Methods
3.1. Reagents
3.2. Steady-State Fluorescence Spectroscopy
3.3. Circular Dichroism Spectroscopy
3.4. Molecular Docking
3.5. Molecular Dynamics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kishimoto, T. Interleukin-6: Discovery of a pleiotropic cytokine. Arthritis Res. Ther. 2006, 8, S2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, X.; Huang, J.; Zhong, H.; Shen, N.; Faggioni, R.; Fung, M.; Yao, Y. Targeting interleukin-6 in inflammatory autoimmune diseases and cancers. Pharmacol. Ther. 2014, 141, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Lotz, M. Interleukin-6: A comprehensive review. Cytokines Interleukins Recept. 1995, 80, 209–233. [Google Scholar]
- Angriman, F.; Ferreyro, B.L.; Burry, L.; Fan, E.; Ferguson, N.D.; Husain, S.; Keshavjee, S.H.; Lupia, E.; Munshi, L.; Renzi, S.; et al. Interleukin-6 receptor blockade in patients with COVID-19: Placing clinical trials into context. Lancet Respir. Med. 2021, 9, 655–664. [Google Scholar] [CrossRef]
- Libermann, T.A.; Baltimore, D. Activation of interleukin-6 gene expression through the NF-kappa B transcription factor. Mol. Cell. Biol. 1990, 10, 2327–2334. [Google Scholar] [CrossRef]
- Chen, L.Y.C.; Hoiland, R.L.; Stukas, S.; Wellington, C.L.; Sekhon, M.S. Assessing the importance of interleukin-6 in COVID-19. Lancet Respir. Med. 2021, 9, e13. [Google Scholar] [CrossRef]
- Nasonov, E.; Samsonov, M. The role of Interleukin 6 inhibitors in therapy of severe COVID-19. Biomed. Pharmacother. 2020, 131, 110698. [Google Scholar] [CrossRef]
- Tan, H.; Miao, R.; Liu, T.; Cao, X.; Wu, X.; Xie, L.; Huang, Z.; Peng, W.; Gan, B. Enhancing the thermal resistance of a novel acidobacteria-derived phytase by engineering of disulfide bridges. J. Microbiol. Biotechnol. 2016, 26, 1717–1722. [Google Scholar] [CrossRef] [Green Version]
- Ding, H.; Gao, F.; Liu, D.; Li, Z.; Xu, X.; Wu, M.; Zhao, Y. Significant improvement of thermal stability of glucose 1-dehydrogenase by introducing disulfide bonds at the tetramer interface. Enzyme Microb. Technol. 2013, 53, 365–372. [Google Scholar] [CrossRef]
- Somers, W.; Stahl, M.; Seehra, J.S. 1.9 Å crystal structure of interleukin 6: Implications for a novel mode of receptor dimerization and signaling. EMBO J. 1997, 16, 989–997. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.-Y.; Yu, H.-A.; Hong, J.; Stahl, M.; McDonagh, T.; Kay, L.E.; Cumming, D.A. Solution structure of recombinant human interleukin-6. J. Mol. Biol. 1997, 268, 468–481. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.H.; Hughes, S.T.; Figueras, A.C.; Jones, S.A. Unravelling the broader complexity of IL-6 involvement in health and disease. Cytokine 2021, 148, 155684. [Google Scholar] [CrossRef] [PubMed]
- Aher, A.; Udhwani, T.; Khandelwal, R.; Limaye, A.; Hussain, T.; Nayarisseri, A.; Singh, S.K. In silico insights on IL-6: A potential target for multicentric castleman disease. Curr. Comput. Aided. Drug Des. 2020, 16, 641–653. [Google Scholar] [CrossRef]
- Meghwal, M.; Goswami, T.K. Piper nigrum and piperine: An update. Phyther. Res. 2013, 27, 1121–1130. [Google Scholar] [CrossRef]
- Zazeri, G.; Povinelli, A.P.R.; Le Duff, C.S.; Tang, B.; Cornelio, M.L.; Jones, A.M. Synthesis and Spectroscopic Analysis of Piperine-and Piperlongumine-Inspired Natural Product Scaffolds and Their Molecular Docking with IL-1β and NF-κB Proteins. Molecules 2020, 25, 2841. [Google Scholar] [CrossRef]
- Ying, X.; Chen, X.; Cheng, S.; Shen, Y.; Peng, L.; Xu, H.Z. Piperine inhibits IL-β induced expression of inflammatory mediators in human osteoarthritis chondrocyte. Int. Immunopharmacol. 2013, 17, 293–299. [Google Scholar] [CrossRef]
- Ying, X.; Yu, K.; Chen, X.; Chen, H.; Hong, J.; Cheng, S.; Peng, L. Piperine inhibits LPS induced expression of inflammatory mediators in RAW 264.7 cells. Cell. Immunol. 2013, 285, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Bang, J.S.; Oh, D.H.; Choi, H.M.; Sur, B.J.; Lim, S.J.; Kim, J.Y.; Yang, H.I.; Yoo, M.C.; Hahm, D.H.; Kim, K.S. Anti-inflammatory and antiarthritic effects of piperine in human interleukin 1β-stimulated fibroblast-like synoviocytes and in rat arthritis models. Arthritis Res. Ther. 2009, 11, R49. [Google Scholar] [CrossRef] [Green Version]
- Zsila, F.; Hazai, E.; Sawyer, L. Binding of the pepper alkaloid piperine to bovine β-lactoglobulin: Circular dichroism spectroscopy and molecular modeling study. J. Agric. Food Chem. 2005, 53, 10179–10185. [Google Scholar] [CrossRef]
- Zazeri, G.; Povinelli, A.P.R.; Lima, M.D.F.; Cornélio, M.L. The Cytokine IL-1β and Piperine Complex Surveyed by Experimental and Computational Molecular Biophysics. Biomolecules 2020, 10, 1337. [Google Scholar] [CrossRef]
- Tripathi, A.K.; Ray, A.K.; Mishra, S.K. Molecular and pharmacological aspects of piperine as a potential molecule for disease prevention and management: Evidence from clinical trials. Beni-Suef Univ. J. Basic Appl. Sci. 2022, 11, 16. [Google Scholar] [CrossRef] [PubMed]
- Zazeri, G.; Povinelli, A.P.R.; Lima, M.D.F.; Cornélio, M.L. Detailed Characterization of the Cooperative Binding of Piperine with Heat Shock Protein 70 by Molecular Biophysical Approaches. Biomedicines 2020, 8, 629. [Google Scholar] [CrossRef] [PubMed]
- Povinelli, A.P.; Zazeri, G.; Lima, M.F.; Cornélio, M.L. Details of the cooperative binding of piperlongumine with rat serum albumin obtained by spectroscopic and computational analyses. Sci. Rep. 2019, 9, 15667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Povinelli, A.P.R.; Zazeri, G.; Jones, A.M.; Cornélio, M.L. Unravelling the Interaction of Piperlongumine with the Nucleotide-Binding Domain of HSP70: A Spectroscopic and In Silico Study. Pharmaceuticals 2021, 14, 1298. [Google Scholar] [CrossRef]
- Povinelli, A.P.R.; Zazeri, G.; Cornélio, M.L. Molecular Mechanism of Flavonoids Using Fluorescence Spectroscopy and Computational Tools. In Flavonoids-A Coloring Model For Cheering Up Life; IntechOpen: Rijeka, Croatia, 2019. [Google Scholar]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, (1999); Kluwer Academic/Plenum Publishers: New York, NY, USA, 2004. [Google Scholar]
- Soares, S.; Mateus, N.; de Freitas, V. Interaction of different polyphenols with bovine serum albumin (BSA) and human salivary α-amylase (HSA) by fluorescence quenching. J. Agric. Food Chem. 2007, 55, 6726–6735. [Google Scholar] [CrossRef]
- Ross, P.D.; Subramanian, S. Thermodynamics of protein association reactions: Forces contributing to stability. Biochemistry 1981, 20, 3096–3102. [Google Scholar] [CrossRef]
- Zazeri, G.; Povinelli, A.P.R.; Lima, M.D.F.; Cornélio, M.L. Experimental approaches and computational modeling of rat serum albumin and its interaction with piperine. Int. J. Mol. Sci. 2019, 20, 2856. [Google Scholar] [CrossRef] [Green Version]
- Zazeri, G.; Povinelli, A.P.R.; Pavan, N.M.; de Carvalho, D.R.; Cardoso, C.L.; Ximenes, V.F. Experimental studies and computational modeling on cytochrome c reduction by quercetin: The role of oxidability and binding affinity. J. Mol. Struct. 2021, 1244, 130995. [Google Scholar] [CrossRef]
- Krüttgen, A.; Rose-John, S.; Möller, C.; Wroblowski, B.; Wollmer, A.; Müllberg, J.; Hirano, T.; Kishimoto, T.; Heinrich, P.C. Structure-function analysis of human interleukin-6. Evidence for the involvement of the carboxy-terminus in function. FEBS Lett. 1990, 262, 323–326. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.-G.; Moritz, R.L.; Reid, G.E.; Ward, L.D.; Simpson, R.J. Purification and characterization of a recombinant murine interleukin-6: Isolation of N- and C-terminally truncated forms. Eur. J. Biochem. 1992, 207, 903–913. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, J.T.; Martinez, H.M. Determination of the secondary structures of proteins by circular dichroism and optical rotatory dispersion. Biochemistry 1972, 11, 4120–4131. [Google Scholar] [CrossRef] [PubMed]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K.; et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2018, 27, 112–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Goodsell, D.S.; Pique, M.E.; Lindstrom, W.L.; Huey, R.; Forli, S.; Hart, W.E.; Halliday, S.; Belew, R.; Olson, A.J. User Guide AutoDock version 4.2. Automated Docking of Flexible Ligands to Flexible Receptors. 2010.
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. Des. Sel. 1995, 8, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Oostenbrink, C.; Villa, A.; Mark, A.E.; Van Gunsteren, W.F. A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 2004, 25, 1656–1676. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Malde, A.K.; Zuo, L.; Breeze, M.; Stroet, M.; Poger, D.; Nair, P.C.; Oostenbrink, C.; Mark, A.E. An automated force field topology builder (ATB) and repository: Version 1.0. J. Chem. Theory Comput. 2011, 7, 4026–4037. [Google Scholar] [CrossRef]
- Wu, Y.; Tepper, H.L.; Voth, G.A. Flexible simple point-charge water model with improved liquid-state properties. J. Chem. Phys. 2006, 124, 24503. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 14101. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Batcho, P.F.; Case, D.A.; Schlick, T. Optimized particle-mesh Ewald/multiple-time step integration for molecular dynamics simulations. J. Chem. Phys. 2001, 115, 4003–4018. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Roux, B. The calculation of the potential of mean force using computer simulations. Comput. Phys. Commun. 1995, 91, 275–282. [Google Scholar] [CrossRef]
- Kumar, S.; Rosenberg, J.M.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 1992, 13, 1011–1021. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Temperature (K) | Stern–Volmer (KSV) × 104 M−1 | Binding (Ka) × 104 M−1 |
|---|---|---|
| 288 | 3.69 ± 0.09 | 4.2 ± 0.3 |
| 298 | 3.67 ± 0.08 | 4.3 ± 0.5 |
| 308 | 3.68 ± 0.07 | 4.3 ± 0.5 |
| T (K) | ΔG (kJ/mol) | ΔH (kJ/mol) | T.ΔS (kJ/mol) |
|---|---|---|---|
| 288 | −14 ± 3 | 2.5 ± 0.2 | 16 ± 3 |
| 298 | −14 ± 3 | 2.5 ± 0.2 | 17 ± 3 |
| 308 | −15 ± 3 | 2.5 ± 0.2 | 17 ± 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Povinelli, A.P.R.; Zazeri, G.; Jones, A.M.; Cornélio, M.L. A Computational–Experimental Investigation of the Molecular Mechanism of Interleukin-6-Piperine Interaction. Int. J. Mol. Sci. 2022, 23, 7994. https://doi.org/10.3390/ijms23147994
Povinelli APR, Zazeri G, Jones AM, Cornélio ML. A Computational–Experimental Investigation of the Molecular Mechanism of Interleukin-6-Piperine Interaction. International Journal of Molecular Sciences. 2022; 23(14):7994. https://doi.org/10.3390/ijms23147994
Chicago/Turabian StylePovinelli, Ana Paula Ribeiro, Gabriel Zazeri, Alan M. Jones, and Marinônio Lopes Cornélio. 2022. "A Computational–Experimental Investigation of the Molecular Mechanism of Interleukin-6-Piperine Interaction" International Journal of Molecular Sciences 23, no. 14: 7994. https://doi.org/10.3390/ijms23147994
APA StylePovinelli, A. P. R., Zazeri, G., Jones, A. M., & Cornélio, M. L. (2022). A Computational–Experimental Investigation of the Molecular Mechanism of Interleukin-6-Piperine Interaction. International Journal of Molecular Sciences, 23(14), 7994. https://doi.org/10.3390/ijms23147994