
miR-143-3p Inhibits Aberrant Tau Phosphorylation and Amyloidogenic Processing of APP by Directly Targeting DAPK1 in Alzheimer’s Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
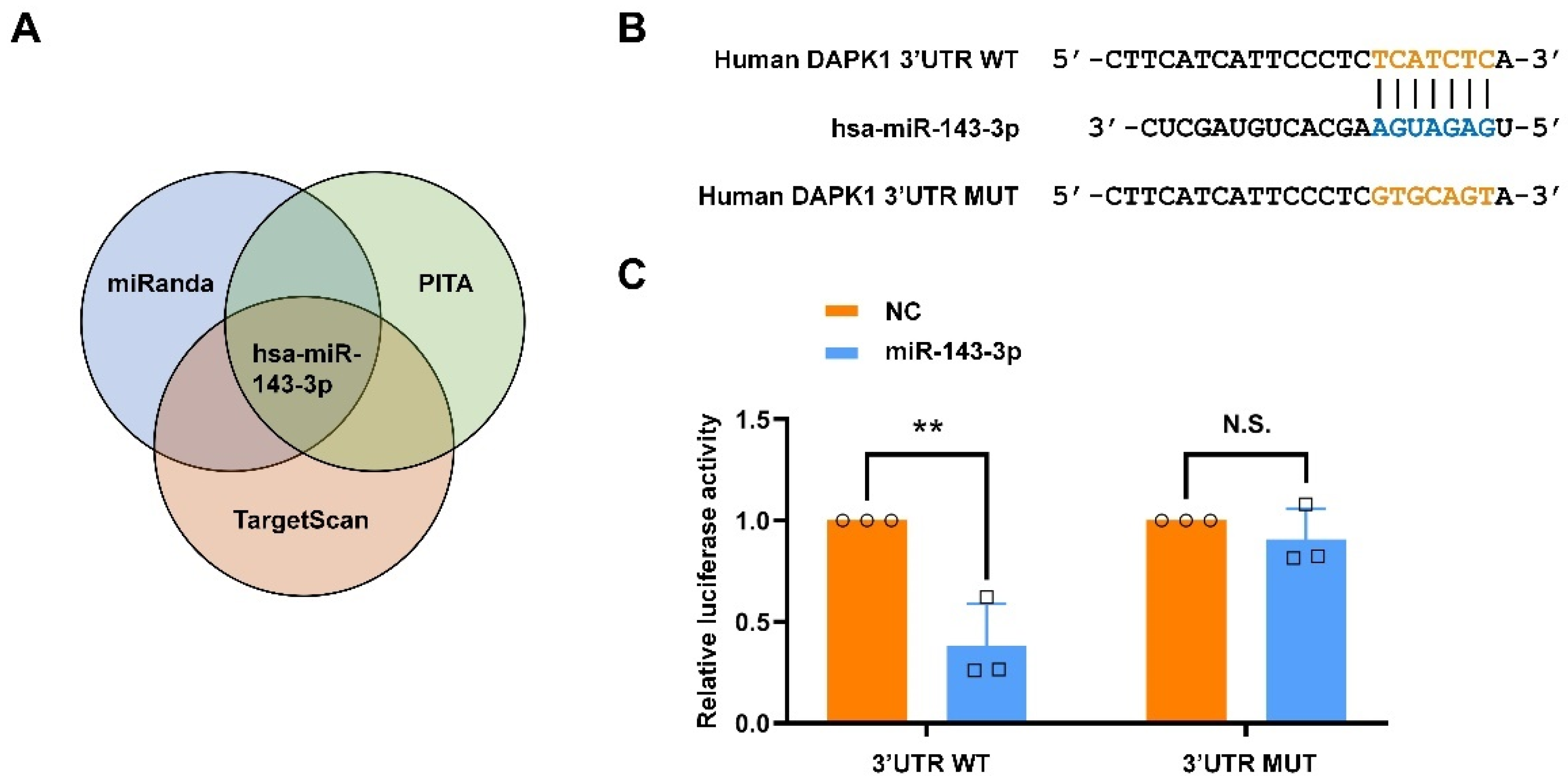
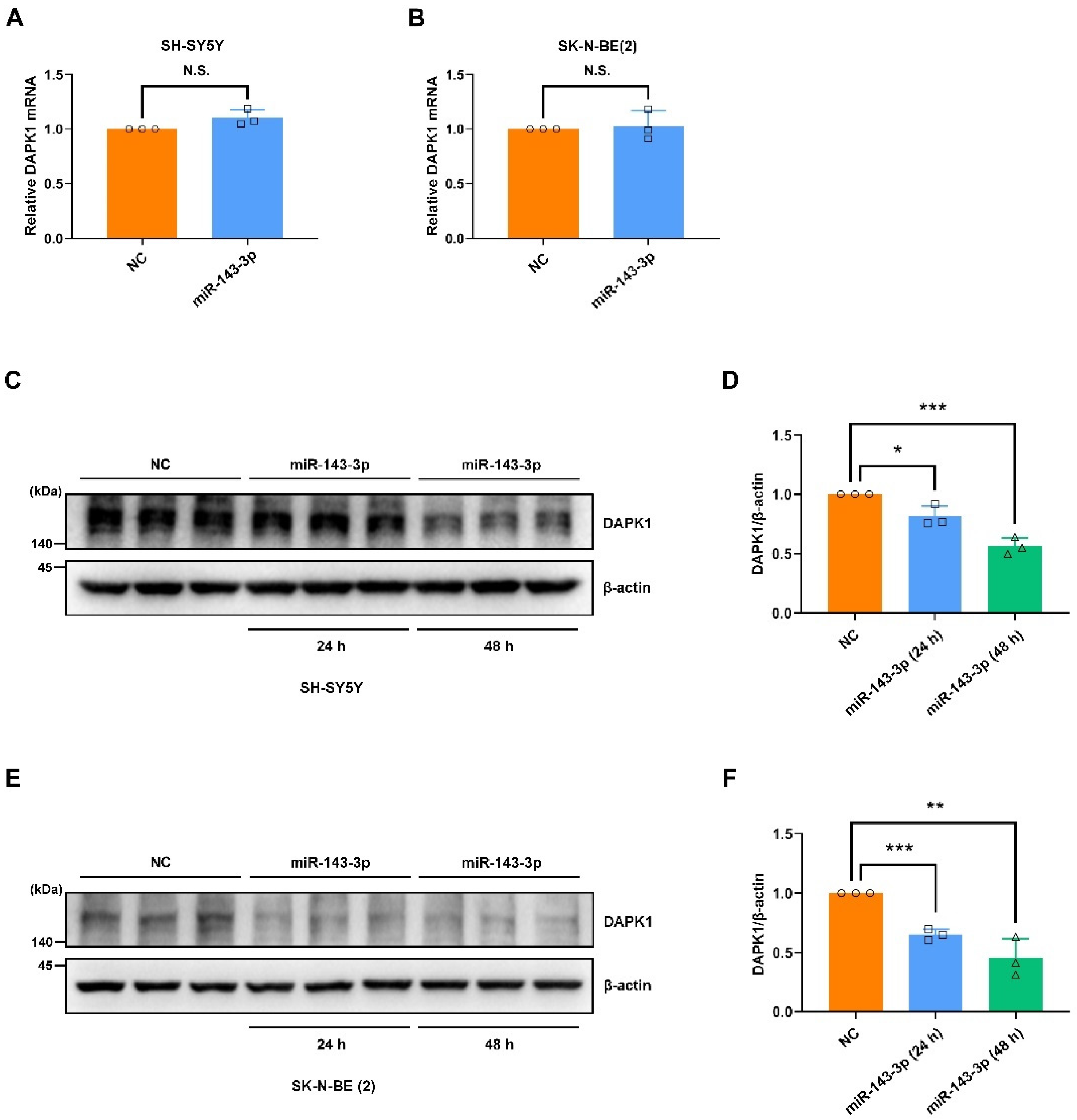
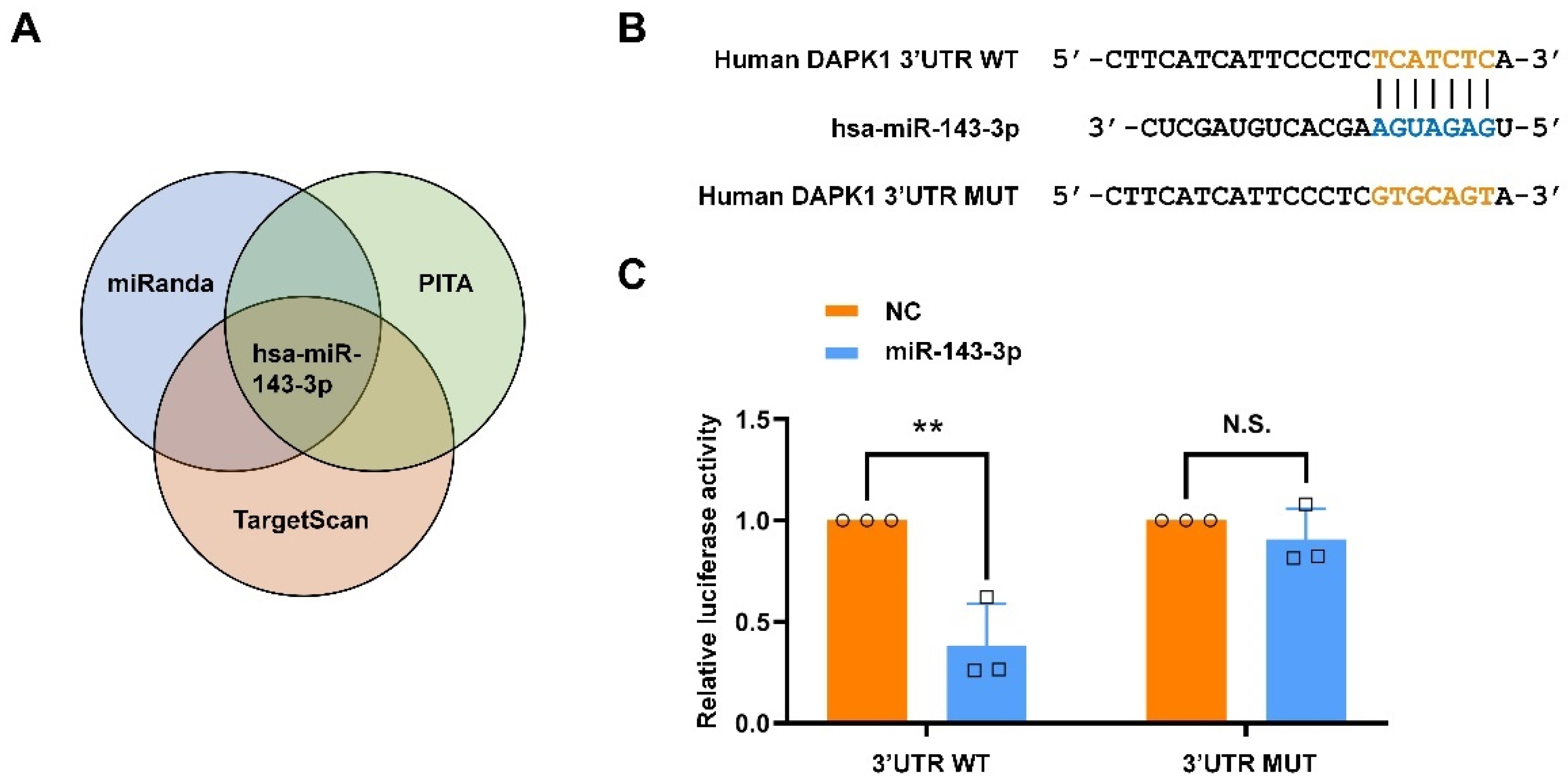
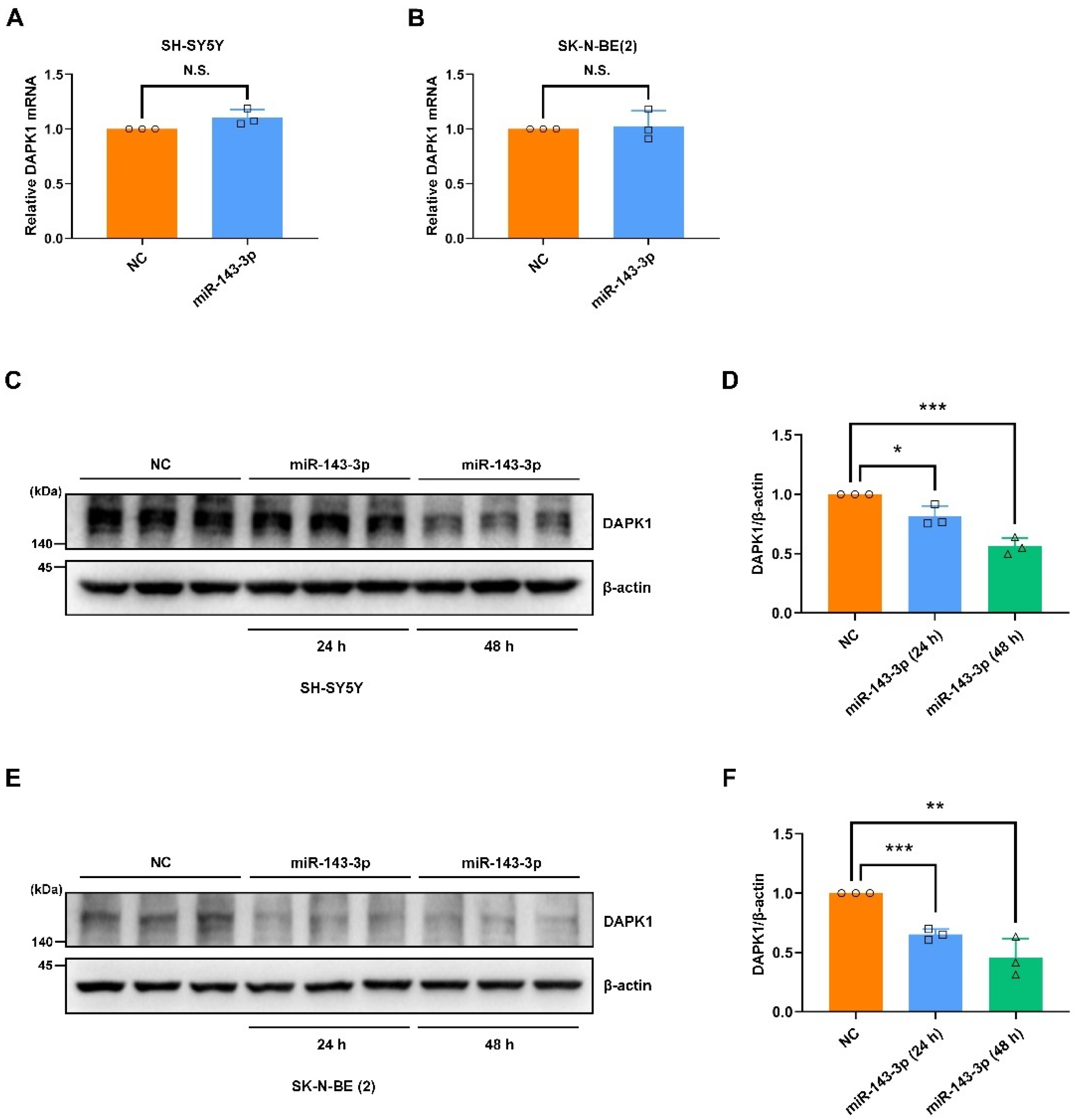
2.1. Hsa-miR-143-3p Directly Targets Human DAPK1 to Suppress Its Protein Expression
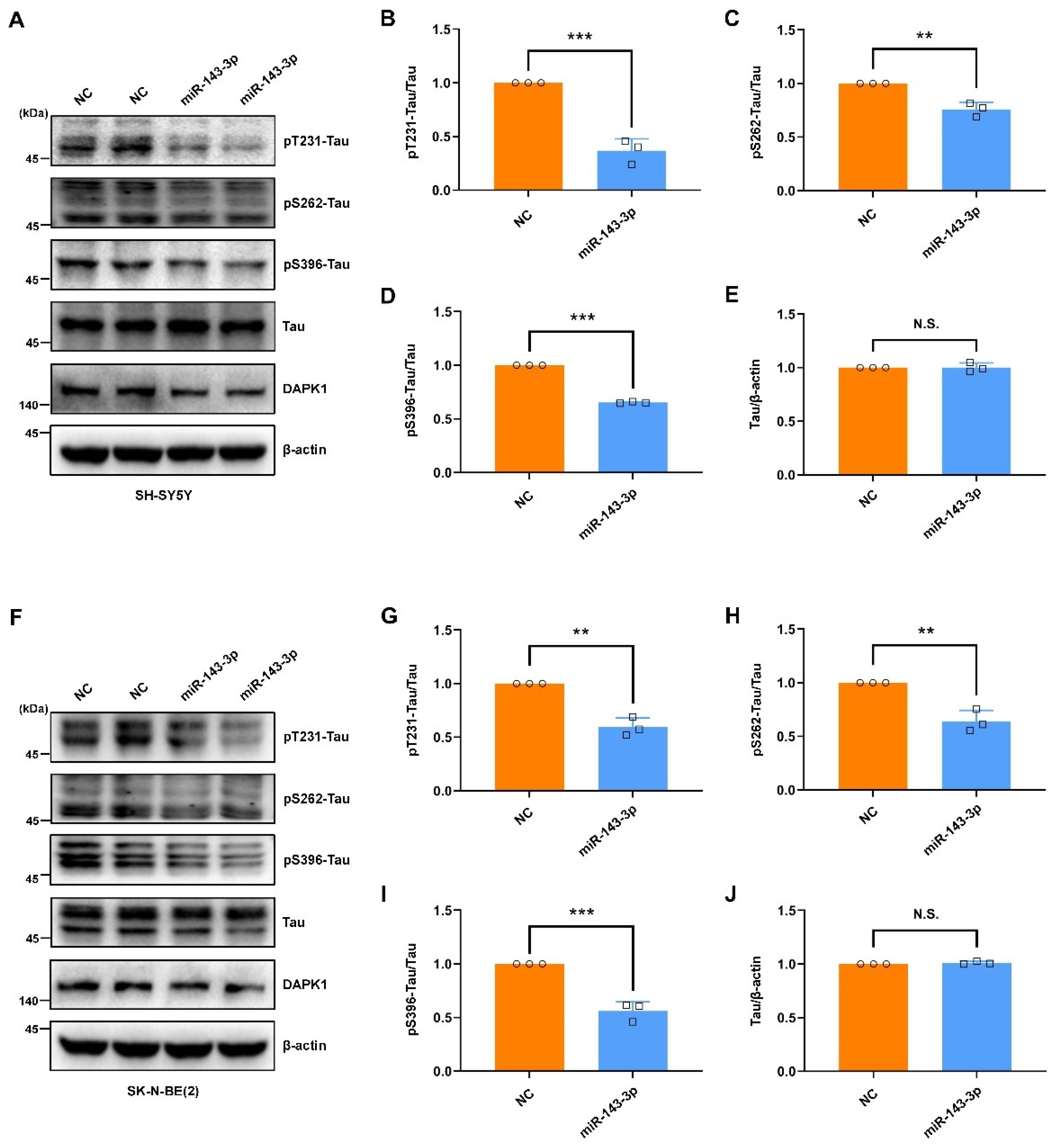
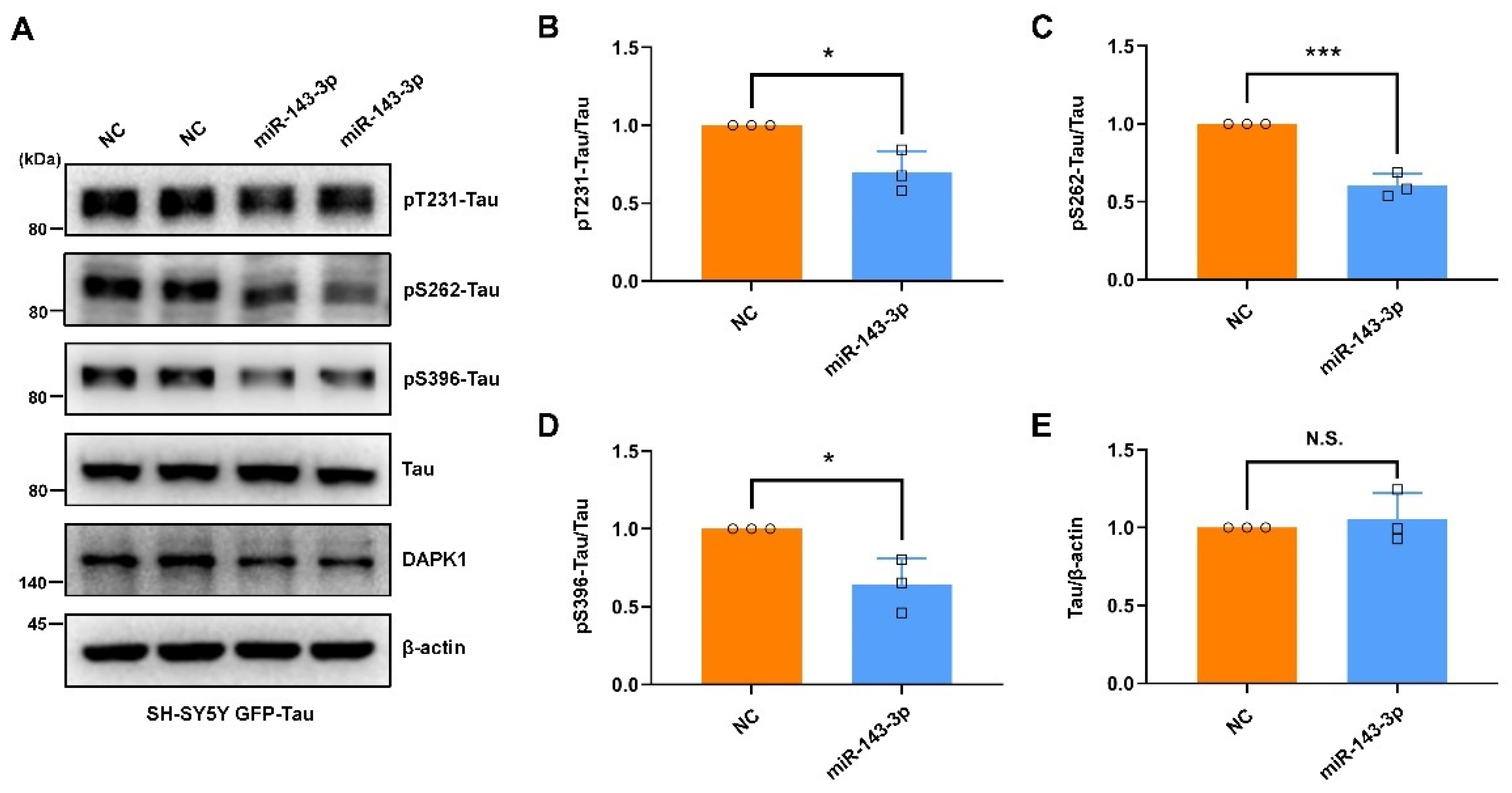
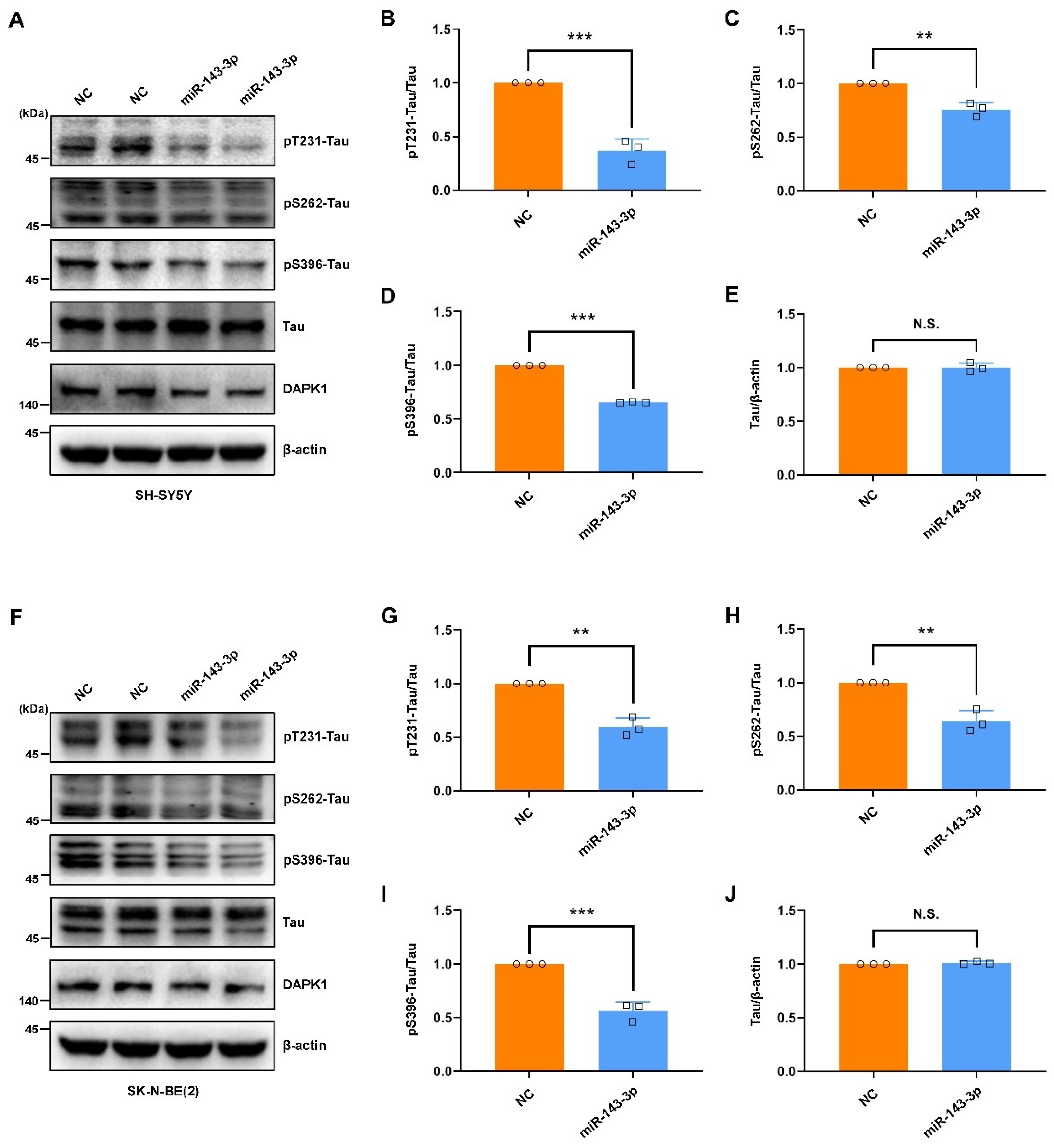
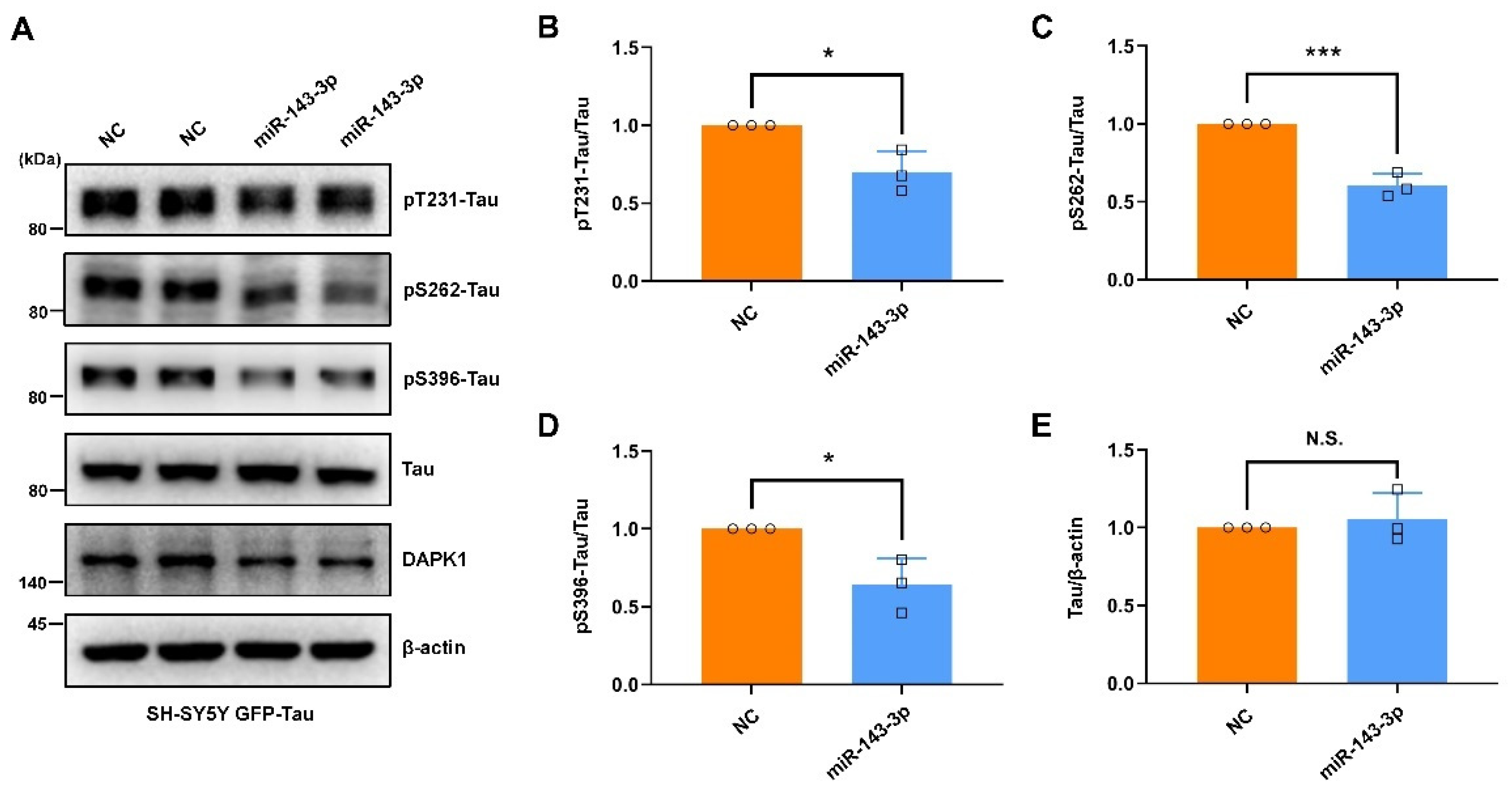
2.2. Hsa-miR-143-3p Inhibits Tau Phosphorylation
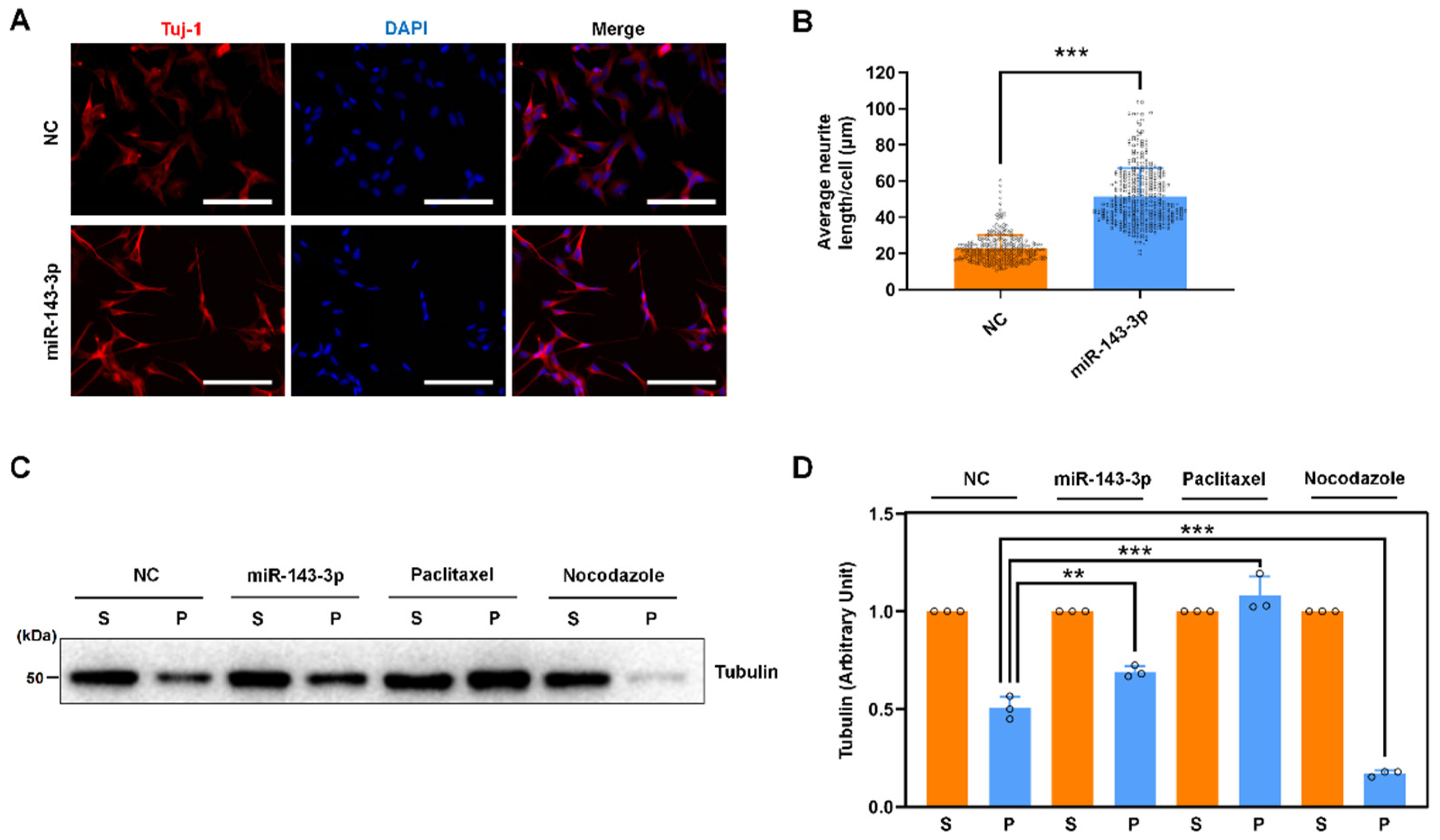
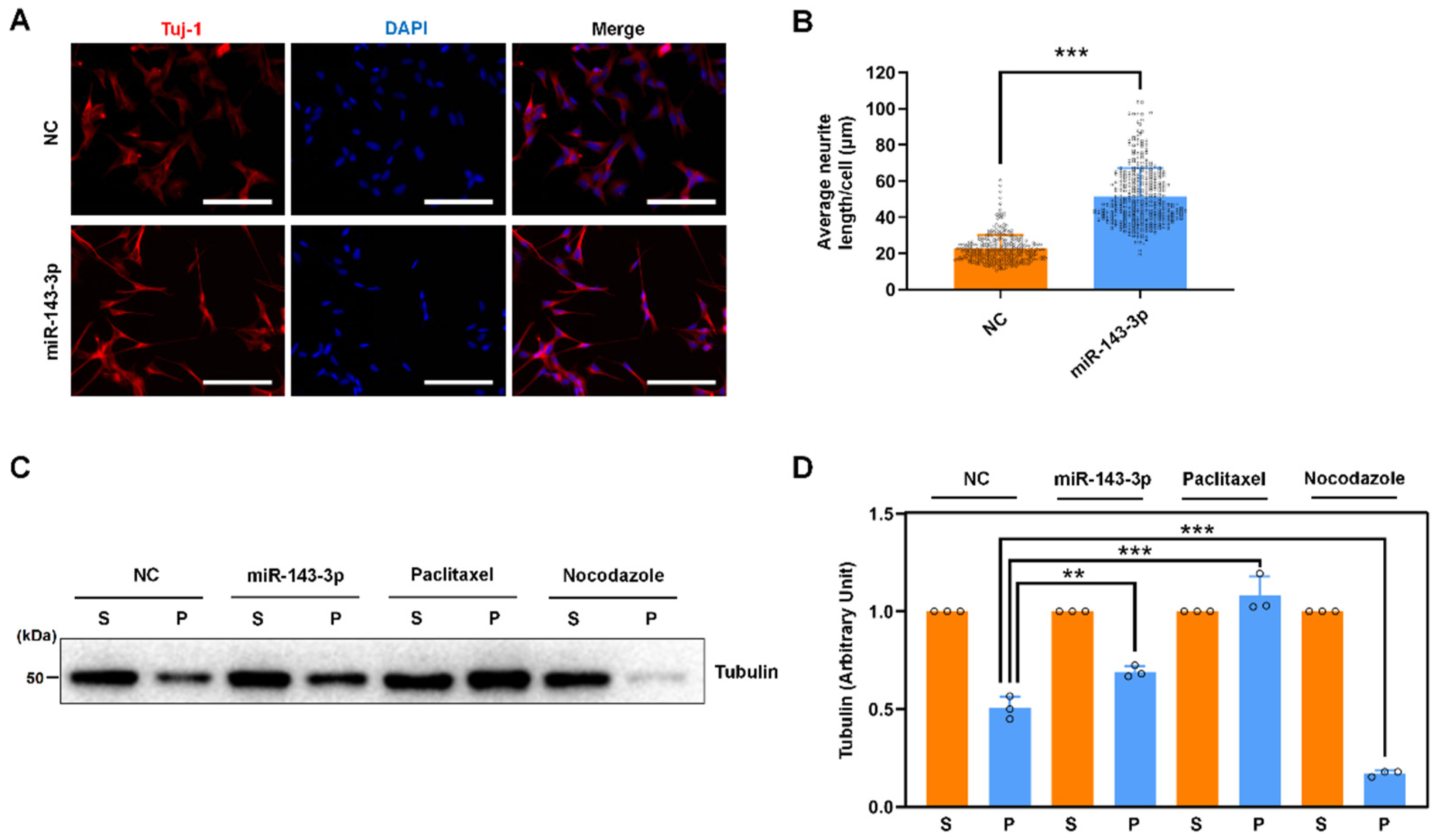
2.3. Hsa-miR-143-3p Promotes Neurite Outgrowth and Microtubule Assembly
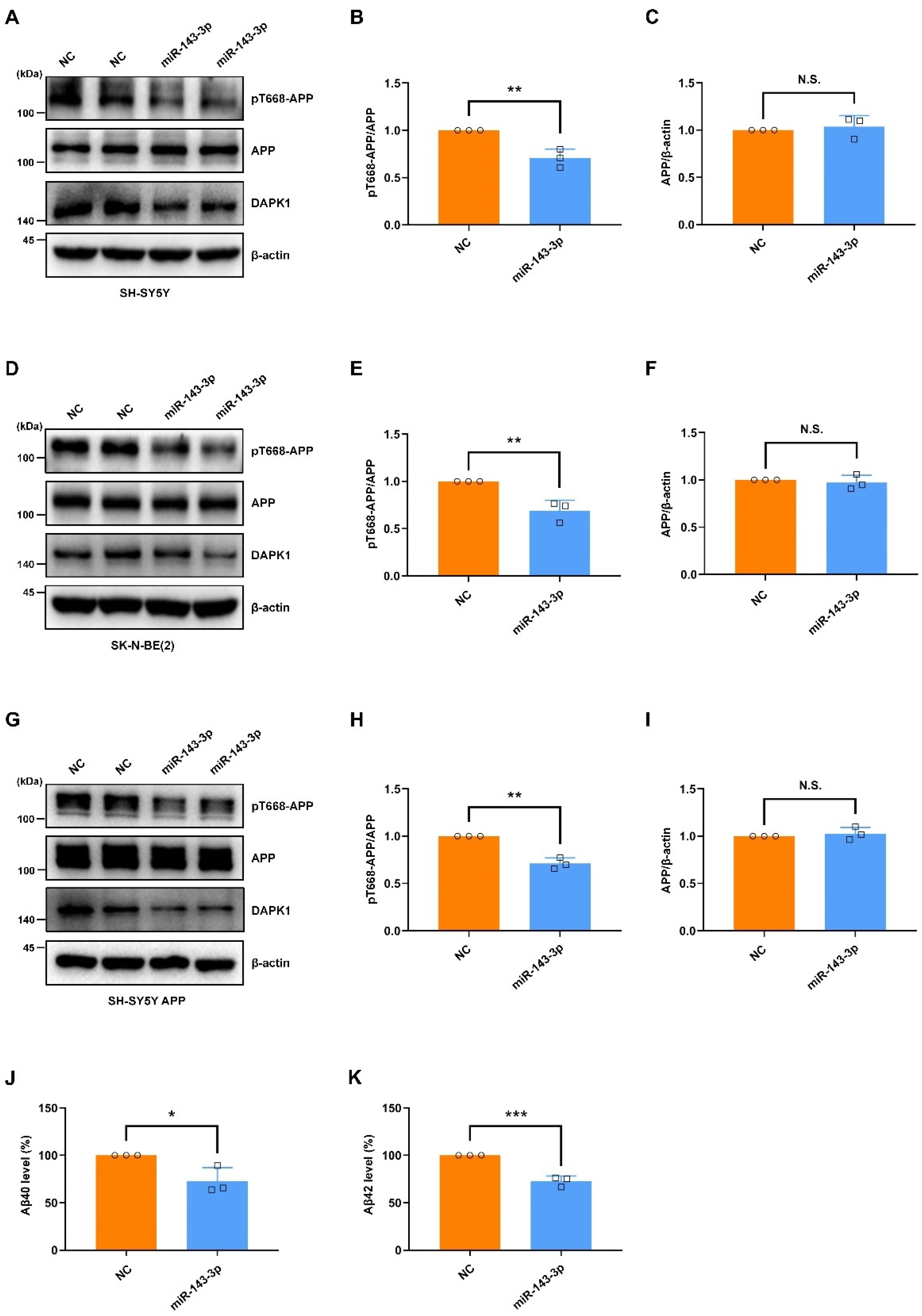
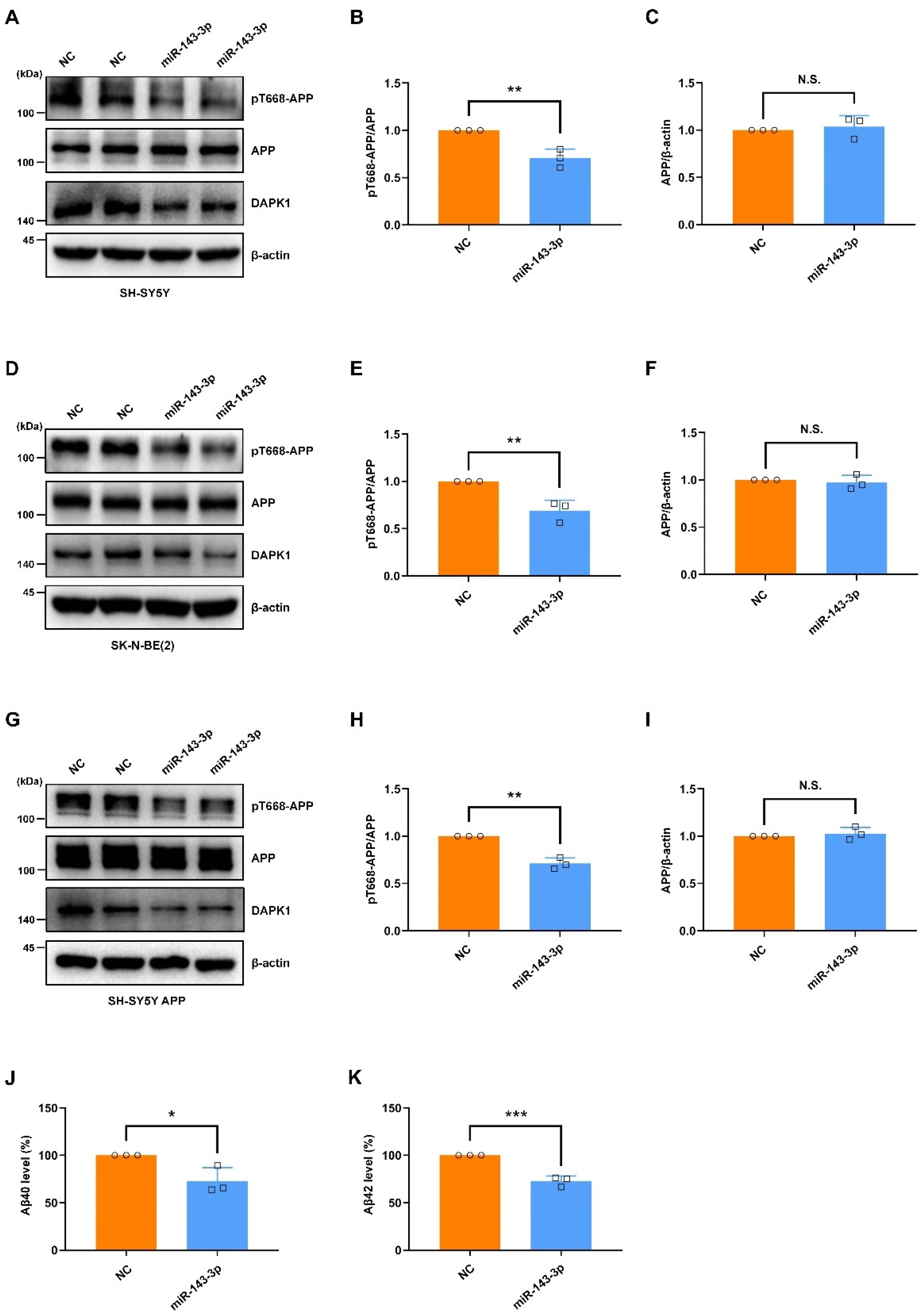
2.4. Hsa-miR-143-3p Attenuates APP Phosphorylation and Inhibits the Secretion of Aβ40 and Aβ42
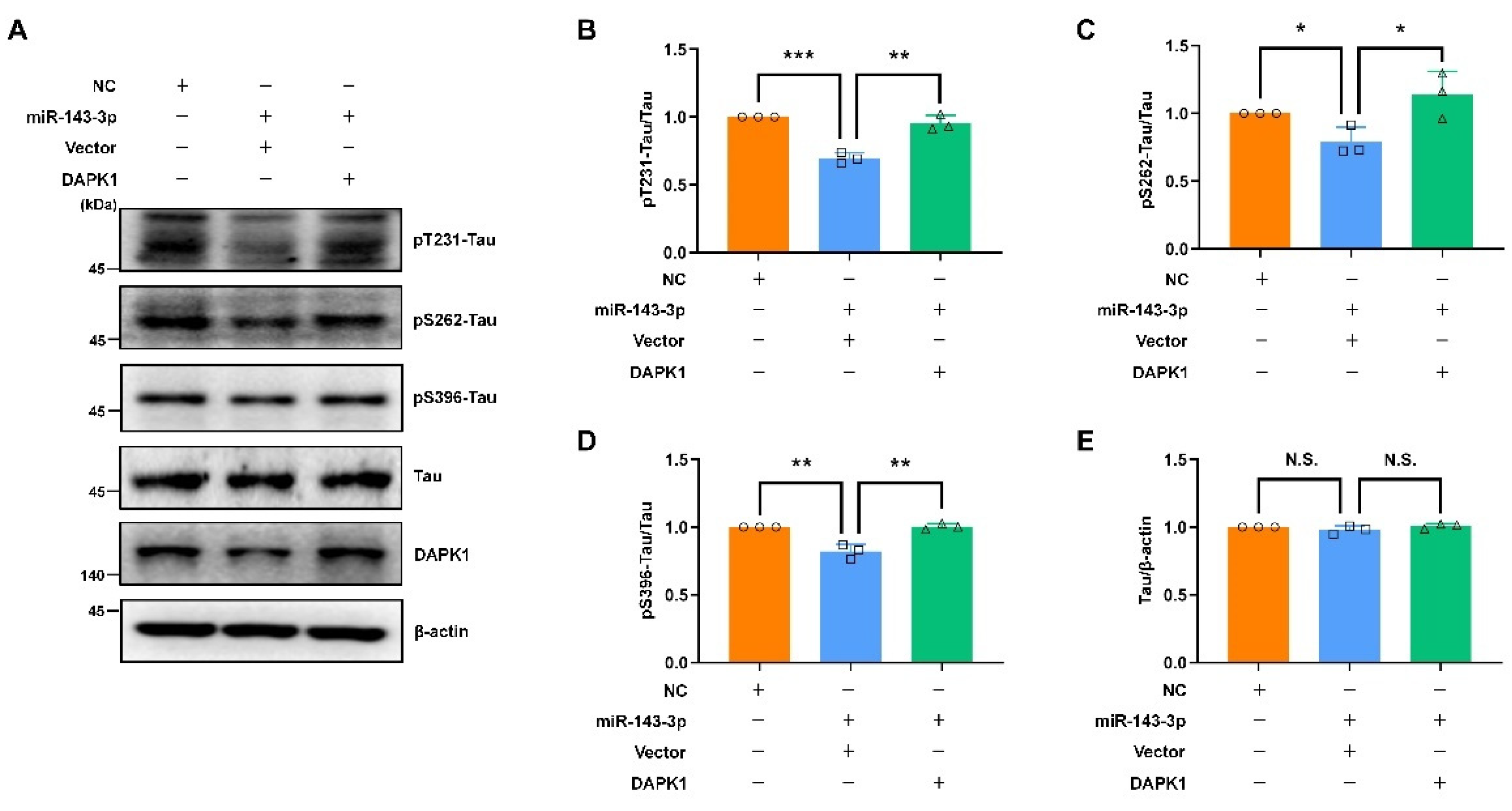
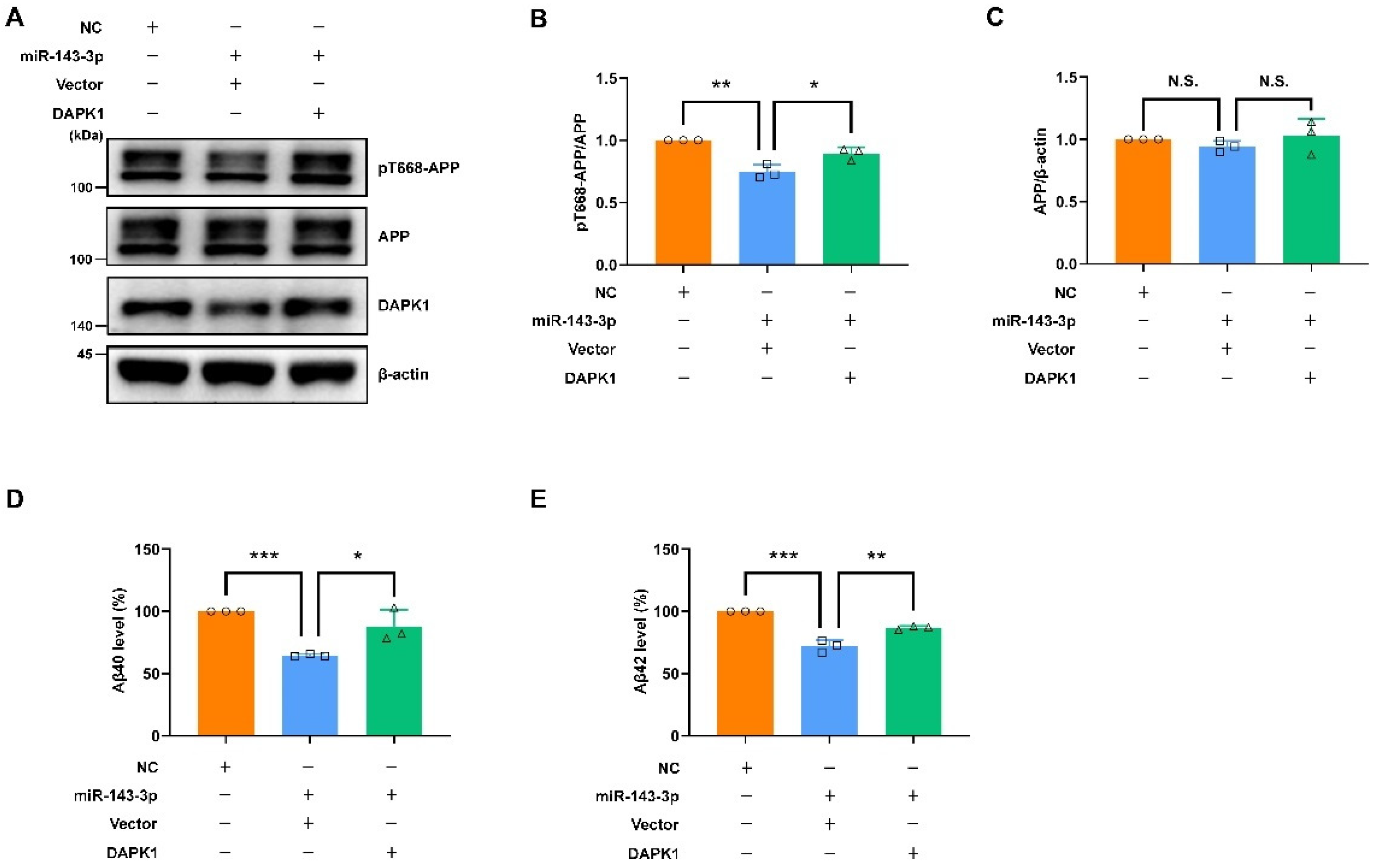
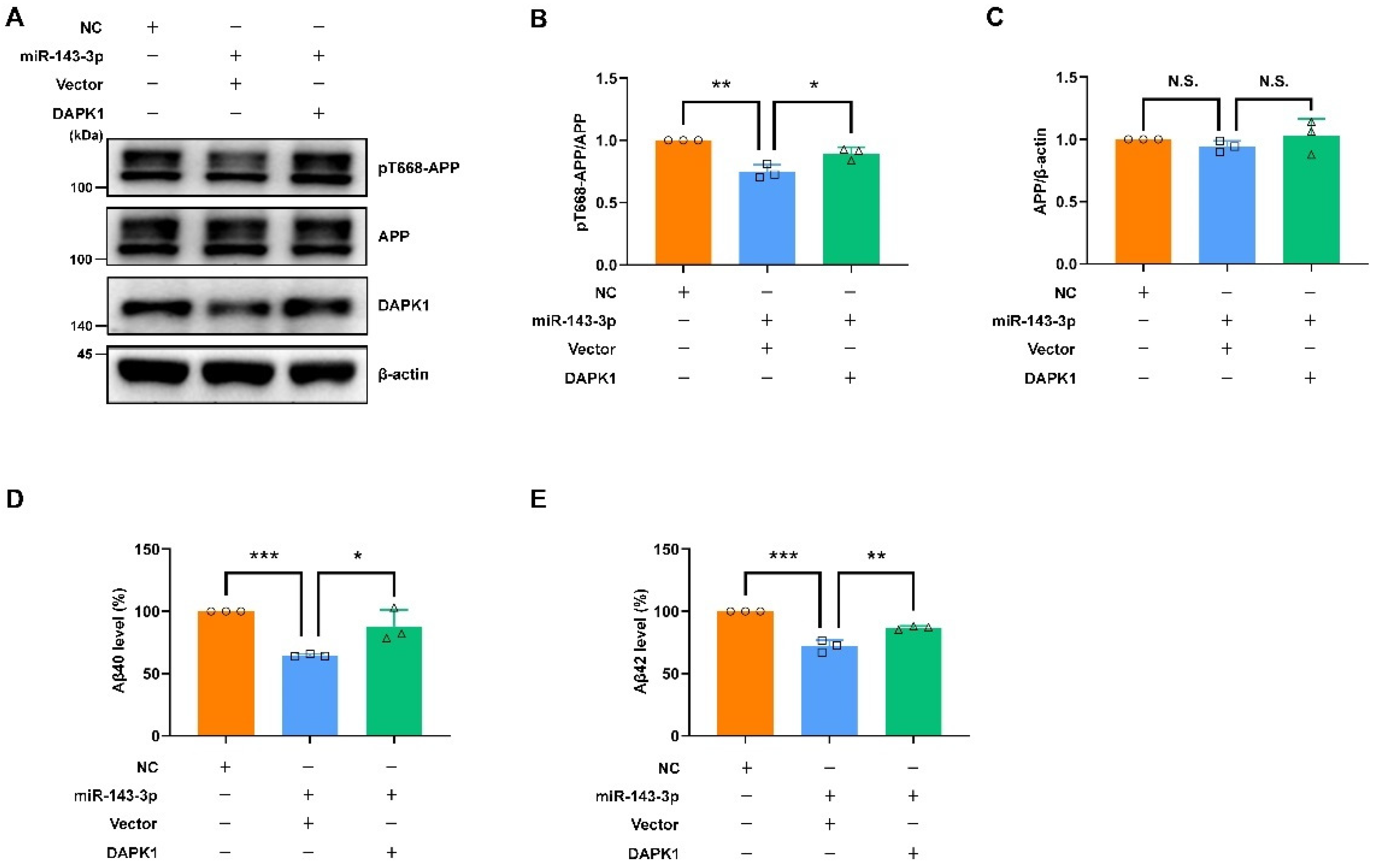
2.5. Restoring DAPK1 Antagonizes the Effects of hsa-miR-143-3p in Attenuating Tau Phosphorylation and Amyloidogenic Processing of APP
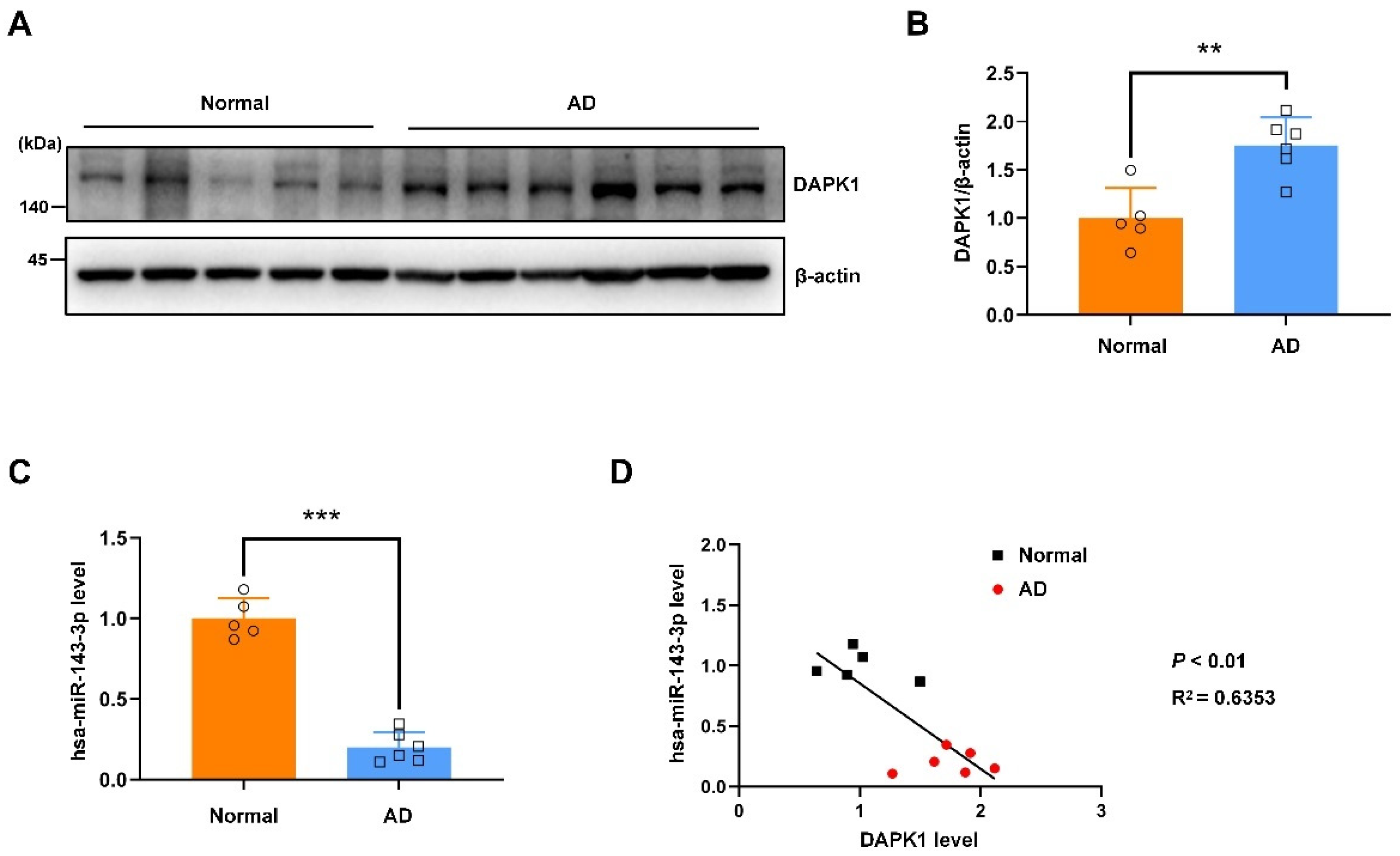
2.6. The hsa-miR-143-3p Levels Are Decreased and Inversely Correlated with the DAPK1 Protein Levels in the Hippocampal Tissues of Patients with AD
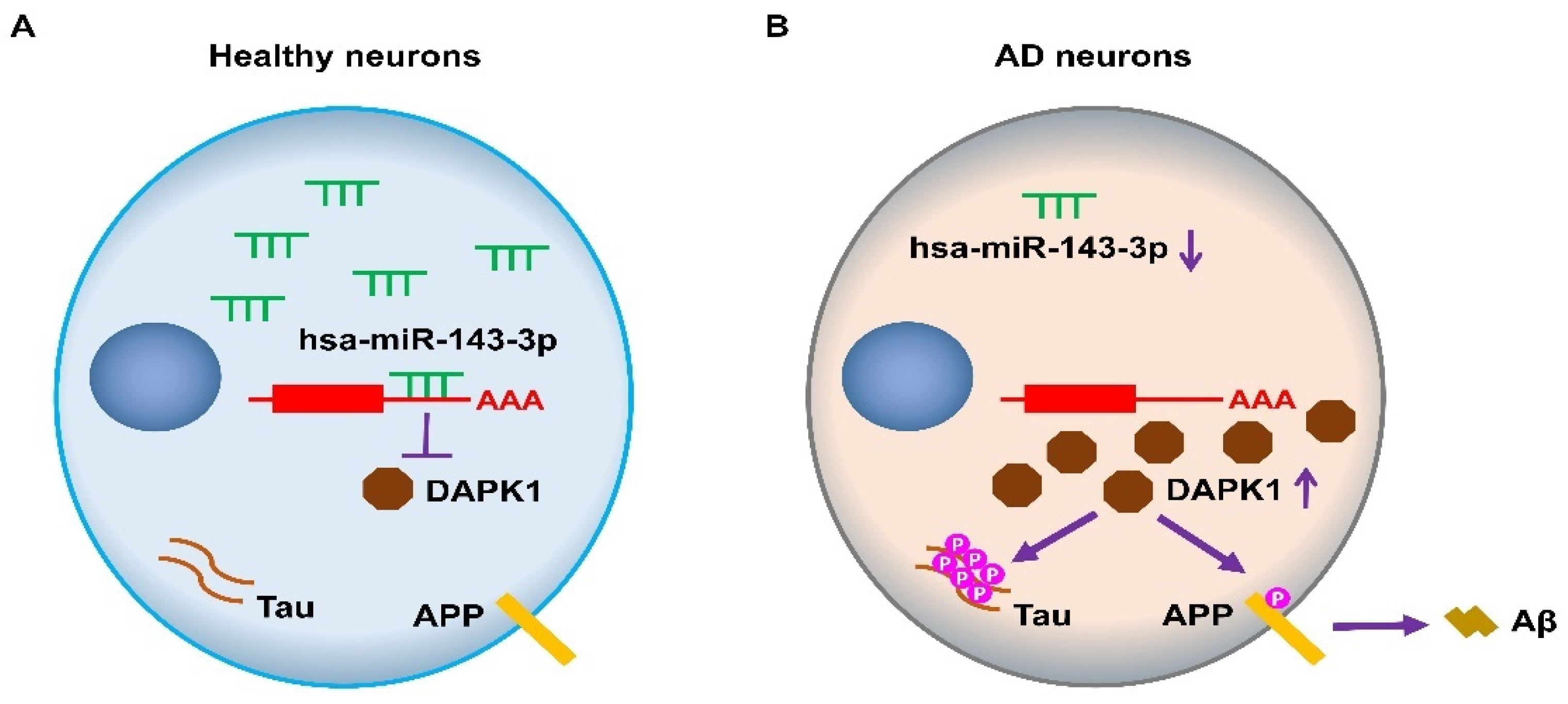
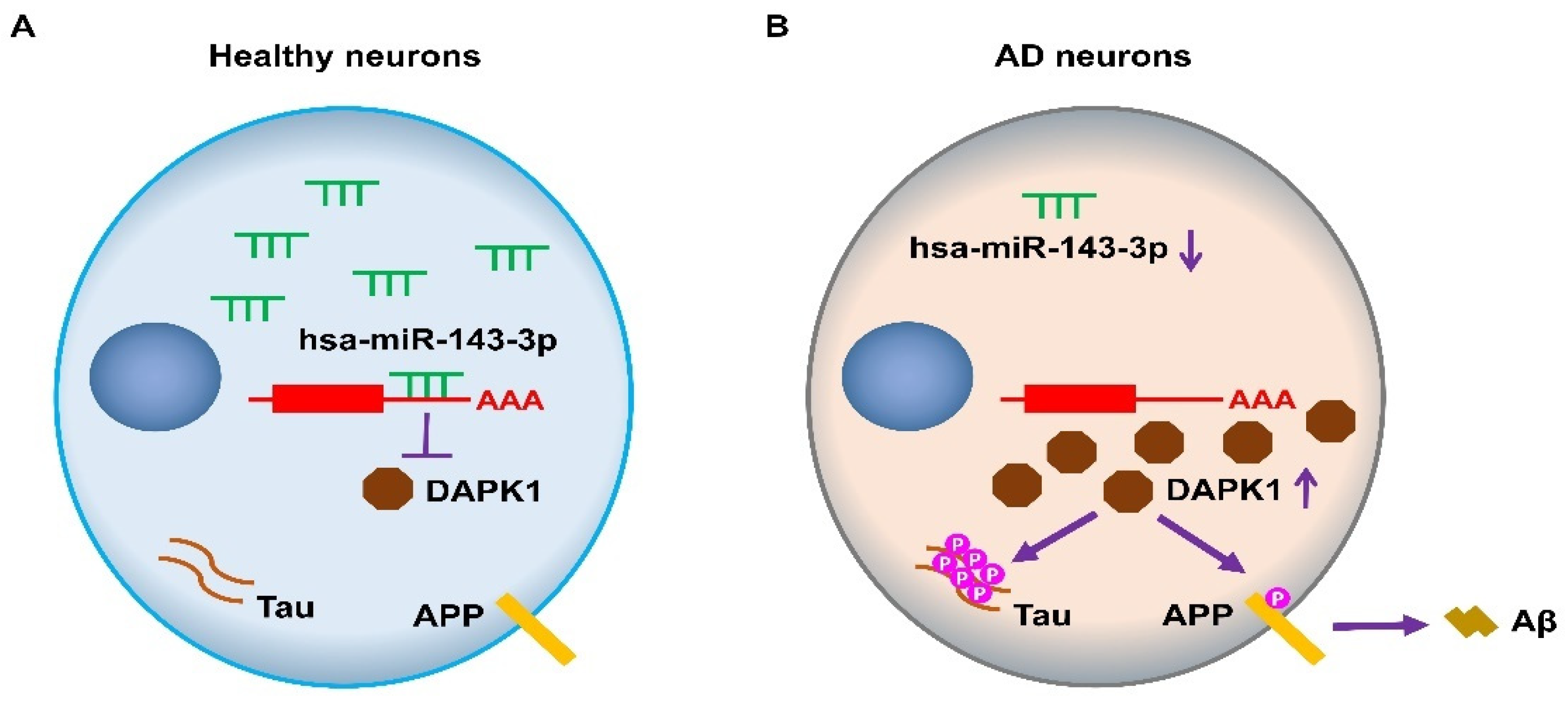
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Plasmids
4.3. Transfection
4.4. RNA Extraction and Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR) Assay
4.5. Immunofluorescence and Immunoblotting Analyses
4.6. Luciferase Reporter Assay
4.7. Neurite Outgrowth Assay
4.8. Microtubule Assembly Assay
4.9. Solid-Phase Sandwich Enzyme-Linked Immunosorbent Assay (ELISA) Analysis of Secreted Aβ40 and Aβ42
4.10. Brain Samples
4.11. Construction of the Protein–Protein Interaction Network
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Alzheimer’s Association. 2022 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2022, 18, 700–789. [Google Scholar] [CrossRef]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.G. A Century of Alzheimer’s Disease. Science 2006, 314, 777–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ittner, L.M.; Götz, J. Amyloid-β and tau—A toxic pas de deux in Alzheimer’s disease. Nat. Rev. Neurosci. 2011, 12, 67–72. [Google Scholar] [CrossRef]
- Deiss, L.P.; Feinstein, E.; Berissi, H.; Cohen, O.; Kimchi, A. Identification of a novel serine/threonine kinase and a novel 15-kD protein as potential mediators of the gamma interferon-induced cell death. Genes Dev. 1995, 9, 15–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bialik, S.; Kimchi, A. The Death-Associated Protein Kinases: Structure, Function, and Beyond. Annu. Rev. Biochem. 2006, 75, 189–210. [Google Scholar] [CrossRef] [Green Version]
- Michie, A.M.; McCaig, A.M.; Nakagawa, R.; Vukovic, M. Death-associated protein kinase (DAPK) and signal transduction: Regulation in cancer. FEBS J. 2009, 277, 74–80. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.-Y.; Lee, Y.-R.; Chen, R.-H. The functions and regulations of DAPK in cancer metastasis. Apoptosis 2013, 19, 364–370. [Google Scholar] [CrossRef]
- Lai, M.-Z.; Chen, R.-H. Regulation of inflammation by DAPK. Apoptosis 2013, 19, 357–363. [Google Scholar] [CrossRef]
- Kim, B.M.; You, M.-H.; Chen, C.-H.; Lee, S.; Hong, Y.; Kimchi, A.; Zhou, X.Z.; Lee, T.H. Death-associated protein kinase 1 has a critical role in aberrant tau protein regulation and function. Cell Death Dis. 2014, 5, e1237. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.M.; You, M.-H.; Chen, C.-H.; Suh, J.; Tanzi, R.E.; Lee, T.H. Inhibition of death-associated protein kinase 1 attenuates the phosphorylation and amyloidogenic processing of amyloid precursor protein. Hum. Mol. Genet. 2016, 25, 2498–2513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Grupe, A.; Rowland, C.; Nowotny, P.; Kauwe, J.S.; Smemo, S.; Hinrichs, A.; Tacey, K.; Toombs, T.A.; Kwok, S.; et al. DAPK1 variants are associated with Alzheimer’s disease and allele-specific expression. Hum. Mol. Genet. 2006, 15, 2560–2568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Wetten, S.; Li, L.; Jean, P.L.S.; Upmanyu, R.; Surh, L.; Hosford, D.; Barnes, M.R.; Briley, J.D.; Borrie, M.; et al. Candidate Single-Nucleotide Polymorphisms From a Genomewide Association Study of Alzheimer Disease. Arch. Neurol. 2008, 65, 45–53. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Zhou, X.Z.; Lee, T.H. Death-Associated Protein Kinase 1 as a Promising Drug Target in Cancer and Alzheimer’s Disease. Recent Pat. Anti-Cancer Drug Discov. 2019, 14, 144–157. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Chen, D.; Zhou, X.Z.; Lee, T.H. Death-Associated Protein Kinase 1 Phosphorylation in Neuronal Cell Death and Neurodegenerative Disease. Int. J. Mol. Sci. 2019, 20, 3131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, M.-H.; Kim, B.M.; Chen, C.-H.; Begley, M.J.; Cantley, L.C.; Lee, T.H. Death-associated protein kinase 1 phosphorylates NDRG2 and induces neuronal cell death. Cell Death Differ. 2016, 24, 238–250. [Google Scholar] [CrossRef] [Green Version]
- Duan, D.-X.; Chai, G.-S.; Ni, Z.-F.; Hu, Y.; Luo, Y.; Cheng, X.-S.; Chen, N.-N.; Wang, J.-Z.; Liu, G.-P. Phosphorylation of Tau by Death-Associated Protein Kinase 1 Antagonizes the Kinase-Induced Cell Apoptosis. J. Alzheimer’s Dis. 2013, 37, 795–808. [Google Scholar] [CrossRef]
- Pei, P.; Wang, S.; Jin, H.; Bi, L.; Wei, N.; Yan, H.; Yang, X.; Yao, C.; Xu, M.; Shu, S.; et al. A Novel Mechanism of Spine Damages in Stroke via DAPK1 and Tau. Cereb. Cortex 2015, 25, 4559–4571. [Google Scholar] [CrossRef] [Green Version]
- Qiu, C.; Albayram, O.; Kondo, A.; Wang, B.; Kim, N.; Arai, K.; Tsai, C.-Y.; Bassal, M.A.; Herbert, M.K.; Washida, K.; et al. Cis P-tau underlies vascular contribution to cognitive impairment and dementia and can be effectively targeted by immunotherapy in mice. Sci. Transl. Med. 2021, 13, eaaz7615. [Google Scholar] [CrossRef]
- Kim, N.; Wang, B.; Koikawa, K.; Nezu, Y.; Qiu, C.; Lee, T.H.; Zhou, X.Z. Inhibition of death-associated protein kinase 1 attenuates cis P-tau and neurodegeneration in traumatic brain injury. Prog. Neurobiol. 2021, 203, 102072. [Google Scholar] [CrossRef]
- Chen, D.; Mei, Y.; Kim, N.; Lan, G.; Gan, C.; Fan, F.; Zhang, T.; Xia, Y.; Wang, L.; Lin, C.; et al. Melatonin directly binds and inhibits death-associated protein kinase 1 function in Alzheimer’s disease. J. Pineal Res. 2020, 69, e12665. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Zhang, T.; Lee, T.H. Cellular Mechanisms of Melatonin: Insight from Neurodegenerative Diseases. Biomolecules 2020, 10, 1158. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Xia, Y.; Hu, L.; Chen, D.; Gan, C.-L.; Wang, L.; Mei, Y.; Lan, G.; Shui, X.; Tian, Y.; et al. Death-associated protein kinase 1 mediates Aβ42 aggregation-induced neuronal apoptosis and tau dysregulation in Alzheimer’s disease. Int. J. Biol. Sci. 2022, 18, 693–706. [Google Scholar] [CrossRef]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef]
- Bazrgar, M.; Khodabakhsh, P.; Mohagheghi, F.; Prudencio, M.; Ahmadiani, A. Brain microRNAs dysregulation: Implication for missplicing and abnormal post-translational modifications of tau protein in Alzheimer’s disease and related tauopathies. Pharmacol. Res. 2020, 155, 104729. [Google Scholar] [CrossRef]
- Abuelezz, N.Z.; Nasr, F.E.; AbdulKader, M.A.; Bassiouny, A.R.; Zaky, A. MicroRNAs as Potential Orchestrators of Alzheimer’s Disease-Related Pathologies: Insights on Current Status and Future Possibilities. Front. Aging Neurosci. 2021, 13, 743573. [Google Scholar] [CrossRef]
- Samadian, M.; Gholipour, M.; Hajiesmaeili, M.; Taheri, M.; Ghafouri-Fard, S. The Eminent Role of microRNAs in the Pathogenesis of Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 641080. [Google Scholar] [CrossRef]
- Walgrave, H.; Balusu, S.; Snoeck, S.; Eynden, E.V.; Craessaerts, K.; Thrupp, N.; Wolfs, L.; Horré, K.; Fourne, Y.; Ronisz, A.; et al. Restoring miR-132 expression rescues adult hippocampal neurogenesis and memory deficits in Alzheimer’s disease. Cell Stem Cell 2021, 28, 1805–1821.e8. [Google Scholar] [CrossRef]
- Madadi, S.; Schwarzenbach, H.; Saidijam, M.; Mahjub, R.; Soleimani, M. Potential microRNA-related targets in clearance pathways of amyloid-β: Novel therapeutic approach for the treatment of Alzheimer’s disease. Cell Biosci. 2019, 9, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iio, A.; Nakagawa, Y.; Hirata, I.; Naoe, T.; Akao, Y. Identification of non-coding RNAs embracing microRNA-143/145 cluster. Mol. Cancer 2010, 9, 136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kent, O.A.; McCall, M.N.; Cornish, T.C.; Halushka, M.K. Lessons from miR-143/145: The importance of cell-type localization of miRNAs. Nucleic Acids Res. 2014, 42, 7528–7538. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.; Doecke, J.; A Sharples, R.; Villemagne, V.L.; Fowler, C.J.; Rembach, A.; Martins, R.; Rowe, C.C.; Macaulay, S.L.; Masters, C.; et al. Prognostic serum miRNA biomarkers associated with Alzheimer’s disease shows concordance with neuropsychological and neuroimaging assessment. Mol. Psychiatry 2014, 20, 1188–1196. [Google Scholar] [CrossRef] [Green Version]
- Dong, H.; Li, J.; Huang, L.; Chen, X.; Li, D.; Wang, T.; Hu, C.; Xu, J.; Zhang, C.; Zen, K.; et al. Serum MicroRNA Profiles Serve as Novel Biomarkers for the Diagnosis of Alzheimer’s Disease. Dis. Markers 2015, 2015, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, A.; Ravan, H.; Mehrabani, M. Multiplex monitoring of Alzheimer associated miRNAs based on the modular logic circuit operation and doping of catalytic hairpin assembly. Biosens. Bioelectron. 2020, 170, 112710. [Google Scholar] [CrossRef]
- Jia, L.; Zhu, M.; Yang, J.; Pang, Y.; Wang, Q.; Li, Y.; Li, T.; Li, F.; Wang, Q.; Li, Y.; et al. Prediction of P-tau/Aβ42 in the cerebrospinal fluid with blood microRNAs in Alzheimer’s disease. BMC Med. 2021, 19, 264. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2020, 49, D605–D612. [Google Scholar] [CrossRef]
- Ballatore, C.; Lee, V.M.-Y.; Trojanowski, J.Q. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 2007, 8, 663–672. [Google Scholar] [CrossRef]
- Nelson, P.T.; Alafuzoff, I.; Bigio, E.H.; Bouras, C.; Braak, H.; Cairns, N.J.; Castellani, R.J.; Crain, B.J.; Davies, P.; Del Tredici, K.; et al. Correlation of Alzheimer Disease Neuropathologic Changes With Cognitive Status: A Review of the Literature. J. Neuropathol. Exp. Neurol. 2012, 71, 362–381. [Google Scholar] [CrossRef]
- Wu, P.-R.; Tsai, P.-I.; Chen, G.-C.; Chou, H.-J.; Huang, Y.-P.; Chen, Y.-H.; Lin, M.-Y.; Kimchi, A.; Chien, C.-T.; Chen, R.-H. DAPK activates MARK1/2 to regulate microtubule assembly, neuronal differentiation, and tau toxicity. Cell Death Differ. 2011, 18, 1507–1520. [Google Scholar] [CrossRef] [PubMed]
- Drubin, D.G.; Feinstein, S.C.; Shooter, E.M.; Kirschner, M.W. Nerve growth factor-induced neurite outgrowth in PC12 cells involves the coordinate induction of microtubule assembly and assembly-promoting factors. J. Cell Biol. 1985, 101, 1799–1807. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Cowan, N.; Kirschner, M. The Primary Structure and Heterogeneity of Tau Protein from Mouse Brain. Science 1988, 239, 285–288. [Google Scholar] [CrossRef]
- Lee, G.; Neve, R.L.; Kosik, K.S. The microtubule binding domain of tau protein. Neuron 1989, 2, 1615–1624. [Google Scholar] [CrossRef]
- Bramblett, G.T.; Goedert, M.; Jakes, R.; Merrick, S.E.; Trojanowski, J.Q.; Lee, V.M. Abnormal tau phosphorylation at Ser396 in alzheimer’s disease recapitulates development and contributes to reduced microtubule binding. Neuron 1993, 10, 1089–1099. [Google Scholar] [CrossRef]
- Esmaeli-Azad, B.; McCarty, J.; Feinstein, S. Sense and antisense transfection analysis of tau function: Tau influences net microtubule assembly, neurite outgrowth and neuritic stability. J. Cell Sci. 1994, 107, 869–879. [Google Scholar] [CrossRef]
- Mandell, J.W.; A Banker, G. Microtubule-associated proteins, phosphorylation gradients, and the establishment of neuronal polarity. Perspect. Dev. Neurobiol. 1996, 4, 125–135. [Google Scholar]
- Johnson, G.V.; Stoothoff, W.H. Tau phosphorylation in neuronal cell function and dysfunction. J. Cell Sci. 2004, 117, 5721–5729. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, K.; Zaidi, T.; Bancher, C.; Grundke-Iqbal, I. Alzheimer paired helical filaments Restoration of the biological activity by dephosphorylation. FEBS Lett. 1994, 349, 104–108. [Google Scholar] [CrossRef] [Green Version]
- Stoothoff, W.H.; Johnson, G.V. Tau phosphorylation: Physiological and pathological consequences. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2005, 1739, 280–297. [Google Scholar] [CrossRef] [Green Version]
- Hardy, J.; Selkoe, D.J. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, T.; Nakaya, T. Regulation of Amyloid β-Protein Precursor by Phosphorylation and Protein Interactions. J. Biol. Chem. 2008, 283, 29633–29637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.-S.; Kao, S.-C.; Lemere, C.A.; Xia, W.; Tseng, H.-C.; Zhou, Y.; Neve, R.; Ahlijanian, M.K.; Tsai, L.-H. APP processing is regulated by cytoplasmic phosphorylation. J. Cell Biol. 2003, 163, 83–95. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Frequency of Stages of Alzheimer-Related Lesions in Different Age Categories. Neurobiol. Aging 1997, 18, 351–357. [Google Scholar] [CrossRef]
- Price, J.L.; Morris, J.C. Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann. Neurol. 1999, 45, 358–368. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Shaw, L.M.; Aisen, P.S.; Weiner, M.W.; Petersen, R.C.; Trojanowski, J.Q. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010, 9, 119–128. [Google Scholar] [CrossRef] [Green Version]
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.S.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Clinical and Biomarker Changes in Dominantly Inherited Alzheimer’s Disease. N. Engl. J. Med. 2012, 367, 795–804. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, M.; Takahashi, H.; Nakamura, T.; Hioki, T.; Nagayama, S.; Ooashi, N.; Sun, X.; Ishii, T.; Kudo, Y.; Nakajima-Iijima, S.; et al. Developmental changes in distribution of death-associated protein kinase mRNAs. J. Neurosci. Res. 1999, 58, 674–683. [Google Scholar] [CrossRef]
- Lee, V.M.-Y.; Balin, B.J.; Otvos, L.; Trojanowski, J.Q. A68: A Major Subunit of Paired Helical Filaments and Derivatized Forms of Normal Tau. Science 1991, 251, 675–678. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.; Cairns, N.; Crowther, R. Tau proteins of alzheimer paired helical filaments: Abnormal phosphorylation of all six brain isoforms. Neuron 1992, 8, 159–168. [Google Scholar] [CrossRef]
- Matsuo, E.S.; Shin, R.-W.; Billingsley, M.L.; Van Devoorde, A.; O’Connor, M.; Trojanowski, J.Q.; Lee, V.M. Biopsy-derived adult human brain tau is phosphorylated at many of the same sites as Alzheimer’s disease paired helical filament tau. Neuron 1994, 13, 989–1002. [Google Scholar] [CrossRef]
- Iqbal, K.; Liu, F.; Gong, C.-X.; Alonso, A.D.C.; Grundke-Iqbal, I. Mechanisms of tau-induced neurodegeneration. Acta Neuropathol. 2009, 118, 53–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, K.; Liu, F.; Gong, C.-X. Tau and neurodegenerative disease: The story so far. Nat. Rev. Neurol. 2015, 12, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Luna-Muñoz, J.; Chávez-Macías, L.; García-Sierra, F.; Mena, R. Earliest Stages of Tau Conformational Changes are Related to the Appearance of a Sequence of Specific Phospho-Dependent Tau Epitopes in Alzheimer’s Disease1. J. Alzheimer’s Dis. 2007, 12, 365–375. [Google Scholar] [CrossRef]
- Lee, T.H.; Pastorino, L.; Lu, K.P. Peptidyl-prolyl cis–trans isomerase Pin1 in ageing, cancer and Alzheimer disease. Expert Rev. Mol. Med. 2011, 13, e21. [Google Scholar] [CrossRef]
- Wang, L.; Zhou, Y.; Chen, D.; Lee, T.H. Peptidyl-Prolyl Cis/Trans Isomerase Pin1 and Alzheimer’s Disease. Front. Cell Dev. Biol. 2020, 8, 355. [Google Scholar] [CrossRef]
- Hasegawa, M.; Morishima-Kawashima, M.; Takio, K.; Suzuki, M.; Titani, K.; Ihara, Y. Protein sequence and mass spectrometric analyses of tau in the Alzheimer’s disease brain. J. Biol. Chem. 1992, 267, 17047–17054. [Google Scholar] [CrossRef]
- Biernat, J.; Gustke, N.; Drewes, G.; Mandelkow, E. Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: Distinction between PHF-like immunoreactivity and microtubule binding. Neuron 1993, 11, 153–163. [Google Scholar] [CrossRef]
- Lauckner, J.; Frey, P.; Geula, C. Comparative distribution of tau phosphorylated at Ser262 in pre-tangles and tangles. Neurobiol. Aging 2003, 24, 767–776. [Google Scholar] [CrossRef]
- Mondragonrodriguez, S.; Perry, G.; Lunamunoz, J.; Acevedo-Aquino, M.C.; Williams, S.E. Phosphorylation of tau protein at sites Ser396-404is one of the earliest events in Alzheimer’s disease and Down syndrome. Neuropathol. Appl. Neurobiol. 2014, 40, 121–135. [Google Scholar] [CrossRef]
- Stathas, S.; Alvarez, V.E.; Xia, W.; Nicks, R.; Meng, G.; Daley, S.; Pothast, M.; Shah, A.; Kelley, H.; Esnault, C.; et al. Tau phosphorylation sites serine202 and serine396 are differently altered in chronic traumatic encephalopathy and Alzheimer’s disease. Alzheimer’s Dement. 2021. [Google Scholar] [CrossRef]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s Disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Chen, D.; Lee, T.H. Phosphorylation Signaling in APP Processing in Alzheimer’s Disease. Int. J. Mol. Sci. 2019, 21, 209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, K.-A.; Kim, H.-S.; Ha, T.-Y.; Ha, J.-W.; Shin, K.Y.; Jeong, Y.H.; Lee, J.-P.; Park, C.-H.; Kim, S.; Baik, T.-K.; et al. Phosphorylation of Amyloid Precursor Protein (APP) at Thr668 Regulates the Nuclear Translocation of the APP Intracellular Domain and Induces Neurodegeneration. Mol. Cell. Biol. 2006, 26, 4327–4338. [Google Scholar] [CrossRef] [Green Version]
- Iijima, K.-I.; Ando, K.; Takeda, S.; Satoh, Y.; Seki, T.; Itohara, S.; Greengard, P.; Kirino, Y.; Nairn, A.; Suzuki, T. Neuron-Specific Phosphorylation of Alzheimer’s β-Amyloid Precursor Protein by Cyclin-Dependent Kinase 5. J. Neurochem. 2002, 75, 1085–1091. [Google Scholar] [CrossRef]
- Drummond, E.; Wisniewski, T. Alzheimer’s disease: Experimental models and reality. Acta Neuropathol. 2016, 133, 155–175. [Google Scholar] [CrossRef]
- Veening-Griffioen, D.H.; Ferreira, G.S.; van Meer, P.J.K.; Boon, W.P.C.; Gispen-de Wied, C.C.; Moors, E.H.M.; Schellekens, H. Are some animal models more equal than others? A case study on the translational value of animal models of efficacy for Alzheimer’s disease. Eur. J. Pharmacol. 2019, 859, 172524. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Lan, G.; Li, R.; Mei, Y.; Shui, X.; Gu, X.; Wang, L.; Zhang, T.; Gan, C.-L.; Xia, Y.; et al. Melatonin ameliorates tau-related pathology via the miR-504-3p and CDK5 axis in Alzheimer’s disease. Transl. Neurodegener. 2022, 11, 1–19. [Google Scholar] [CrossRef]
- Gan, C.-L.; Zou, Y.; Xia, Y.; Zhang, T.; Chen, D.; Lan, G.; Mei, Y.; Wang, L.; Shui, X.; Hu, L.; et al. Inhibition of Death-associated Protein Kinase 1 protects against Epileptic Seizures in mice. Int. J. Biol. Sci. 2021, 17, 2356–2366. [Google Scholar] [CrossRef]
- Wisessaowapak, C.; Visitnonthachai, D.; Watcharasit, P.; Satayavivad, J. Prolonged arsenic exposure increases tau phosphorylation in differentiated SH-SY5Y cells: The contribution of GSK3 and ERK1/2. Environ. Toxicol. Pharmacol. 2021, 84, 103626. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Shui, X.; Mei, Y.; Xia, Y.; Lan, G.; Hu, L.; Zhang, M.; Gan, C.-L.; Li, R.; Tian, Y.; et al. miR-143-3p Inhibits Aberrant Tau Phosphorylation and Amyloidogenic Processing of APP by Directly Targeting DAPK1 in Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 7992. https://doi.org/10.3390/ijms23147992
Wang L, Shui X, Mei Y, Xia Y, Lan G, Hu L, Zhang M, Gan C-L, Li R, Tian Y, et al. miR-143-3p Inhibits Aberrant Tau Phosphorylation and Amyloidogenic Processing of APP by Directly Targeting DAPK1 in Alzheimer’s Disease. International Journal of Molecular Sciences. 2022; 23(14):7992. https://doi.org/10.3390/ijms23147992
Chicago/Turabian StyleWang, Long, Xindong Shui, Yingxue Mei, Yongfang Xia, Guihua Lan, Li Hu, Mi Zhang, Chen-Ling Gan, Ruomeng Li, Yuan Tian, and et al. 2022. "miR-143-3p Inhibits Aberrant Tau Phosphorylation and Amyloidogenic Processing of APP by Directly Targeting DAPK1 in Alzheimer’s Disease" International Journal of Molecular Sciences 23, no. 14: 7992. https://doi.org/10.3390/ijms23147992
APA StyleWang, L., Shui, X., Mei, Y., Xia, Y., Lan, G., Hu, L., Zhang, M., Gan, C.-L., Li, R., Tian, Y., Wang, Q., Gu, X., Chen, D., Zhang, T., & Lee, T. H. (2022). miR-143-3p Inhibits Aberrant Tau Phosphorylation and Amyloidogenic Processing of APP by Directly Targeting DAPK1 in Alzheimer’s Disease. International Journal of Molecular Sciences, 23(14), 7992. https://doi.org/10.3390/ijms23147992