
2-Selenouridine, a Modified Nucleoside of Bacterial tRNAs, Its Reactivity in the Presence of Oxidizing and Reducing Reagents

 , , , ,
, , , ,
Abstract
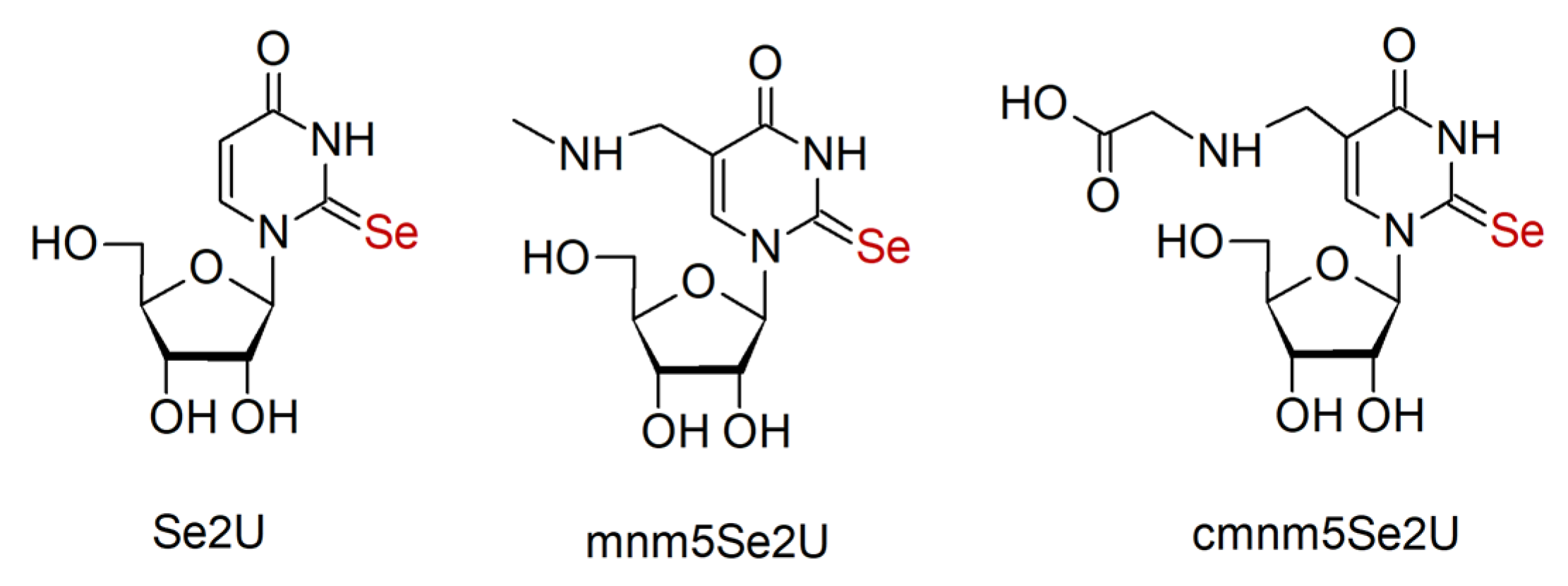
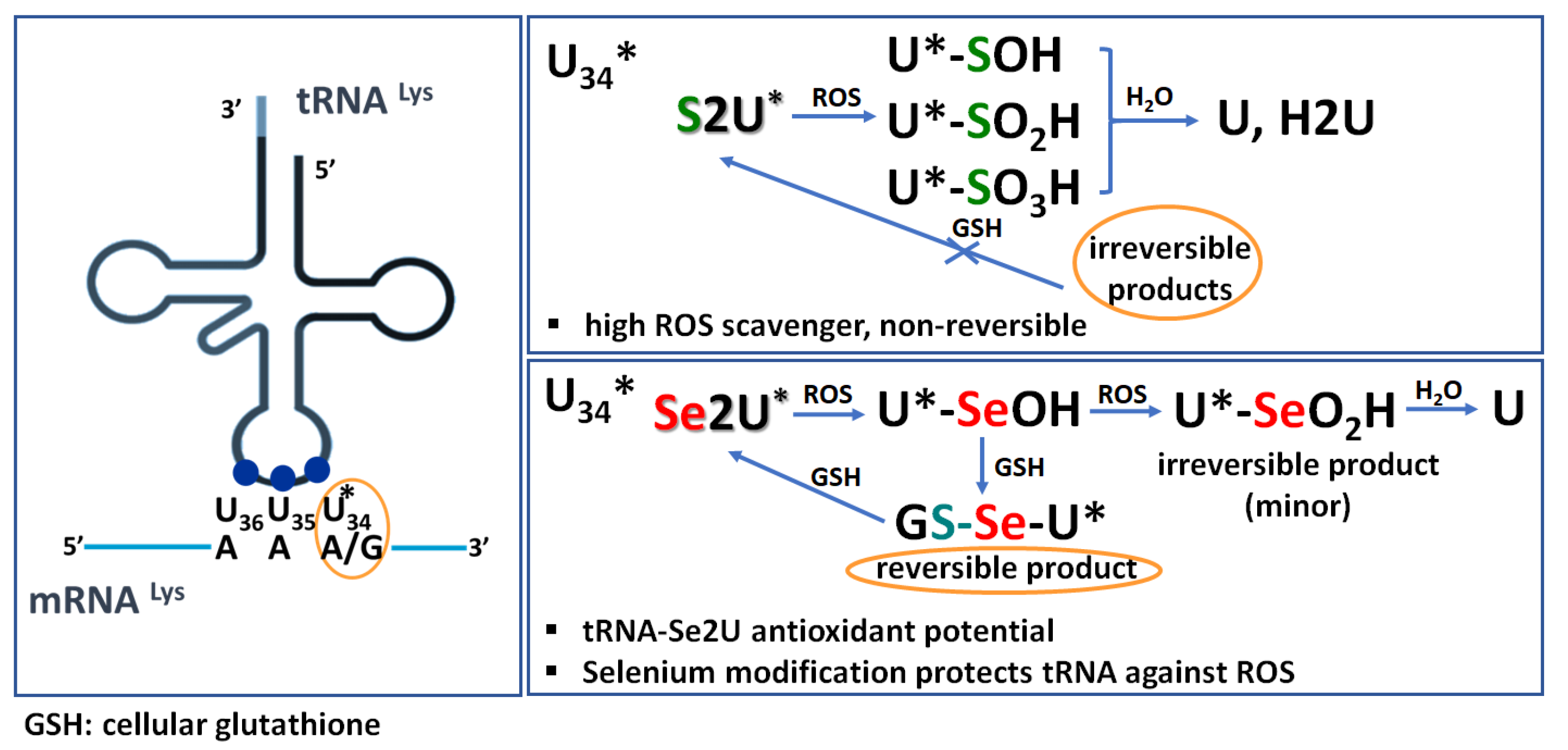
1. Introduction
2. Results
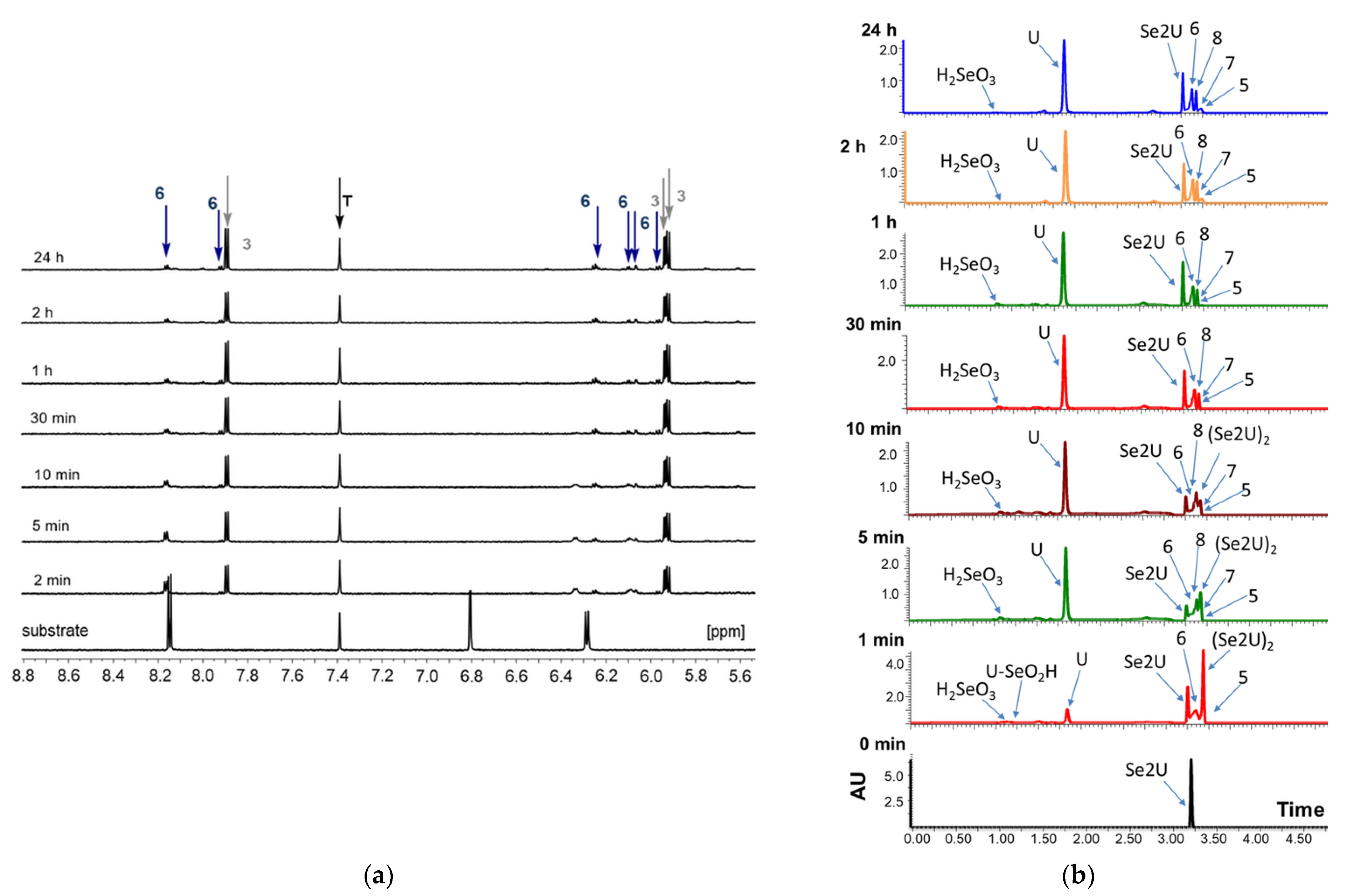
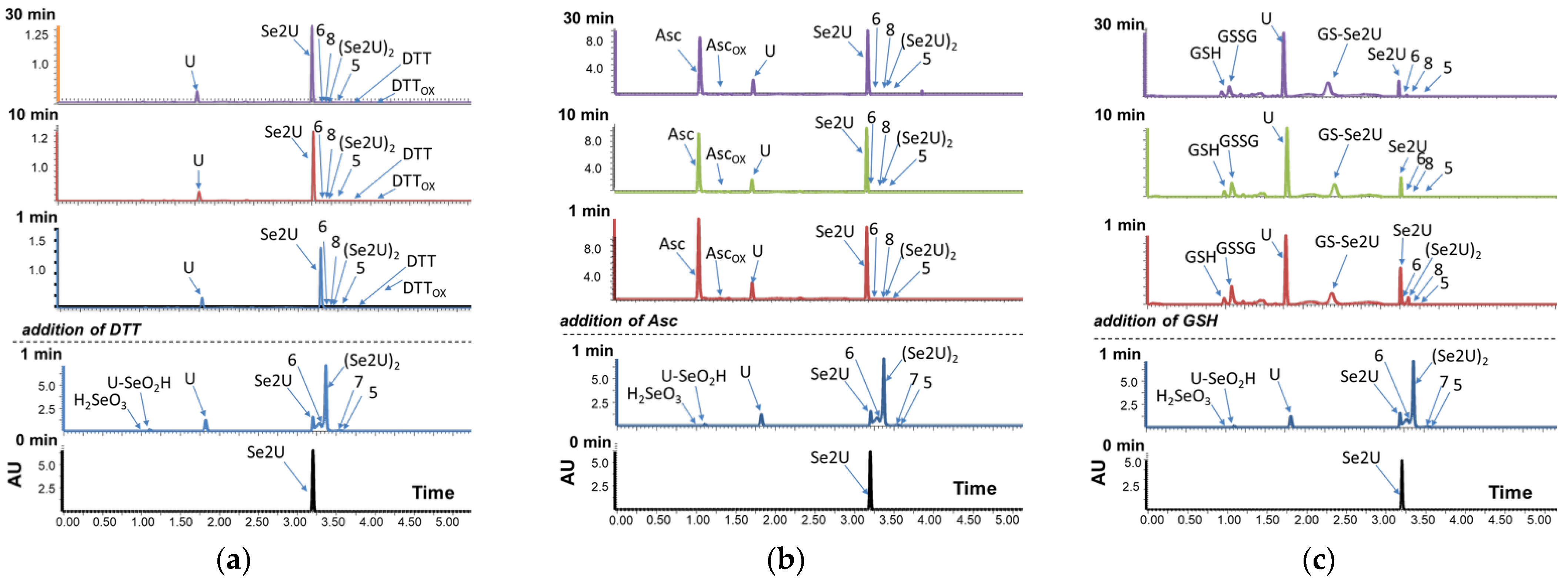
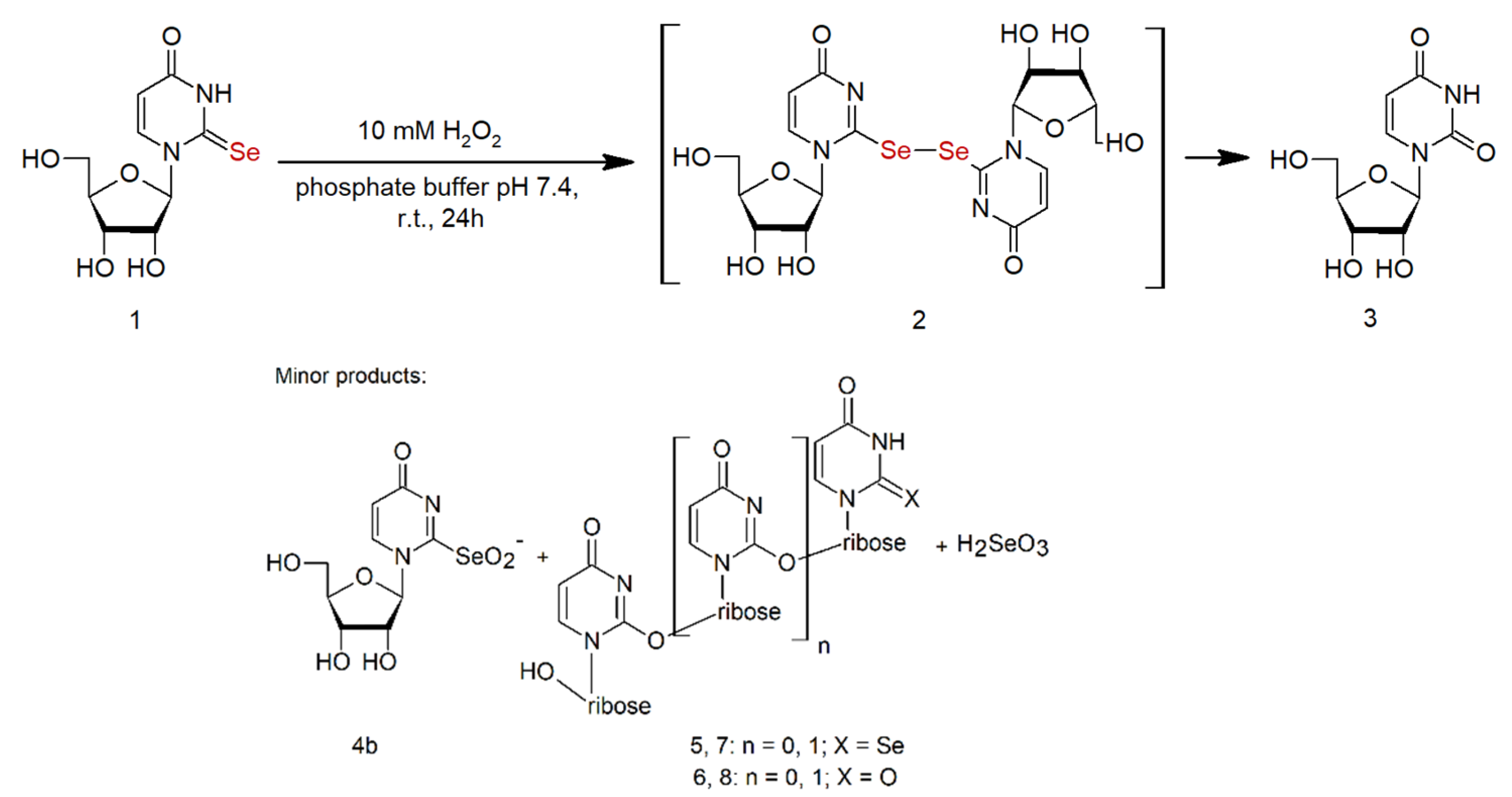
2.1. Analysis of the Oxidation Course of Se2U (1)
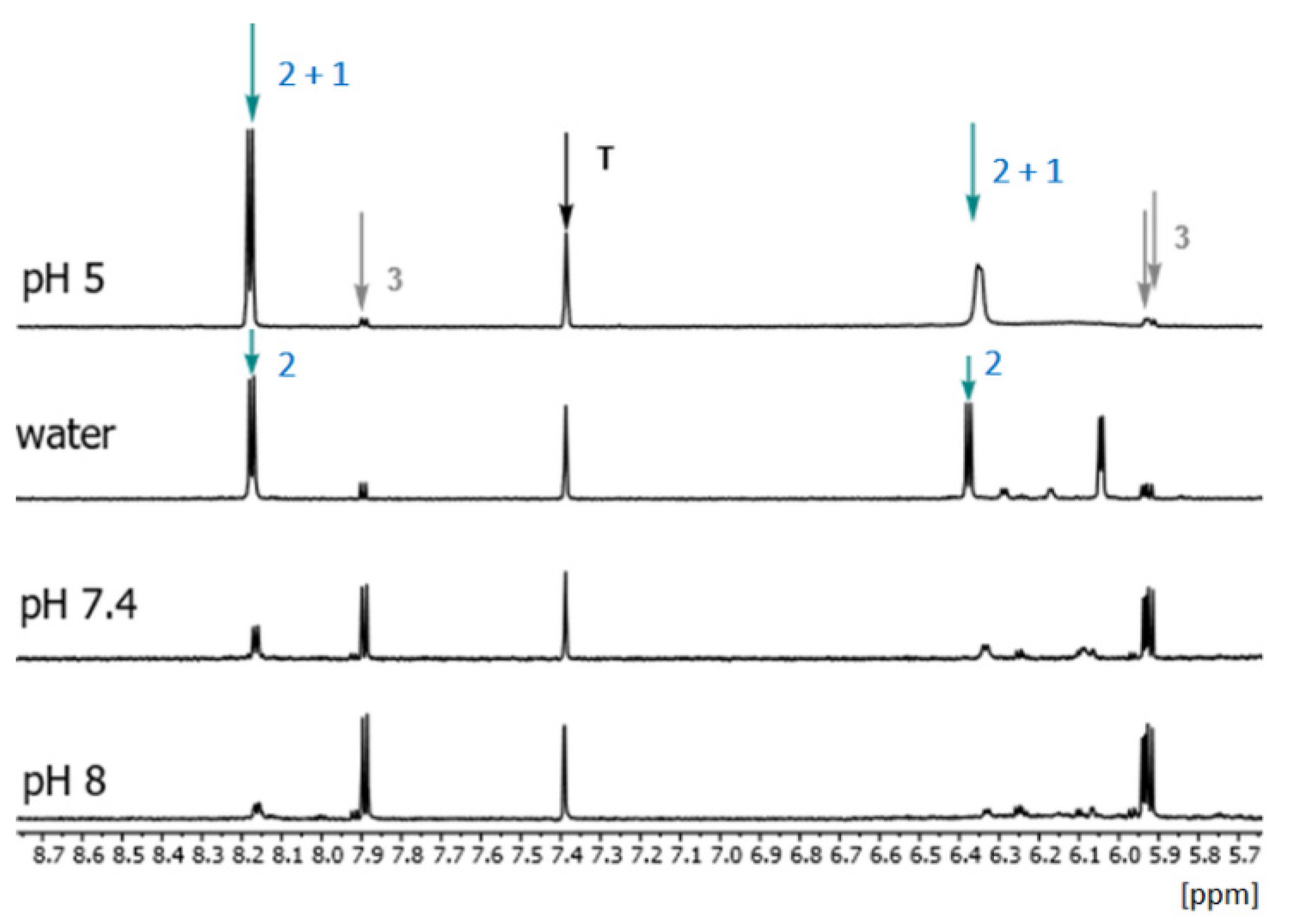
2.2. Influence of Medium pH on the Course of the Se2U Oxidation
2.3. Products with the Structure of Selenium Oxoacids in the Oxidation of Se2U
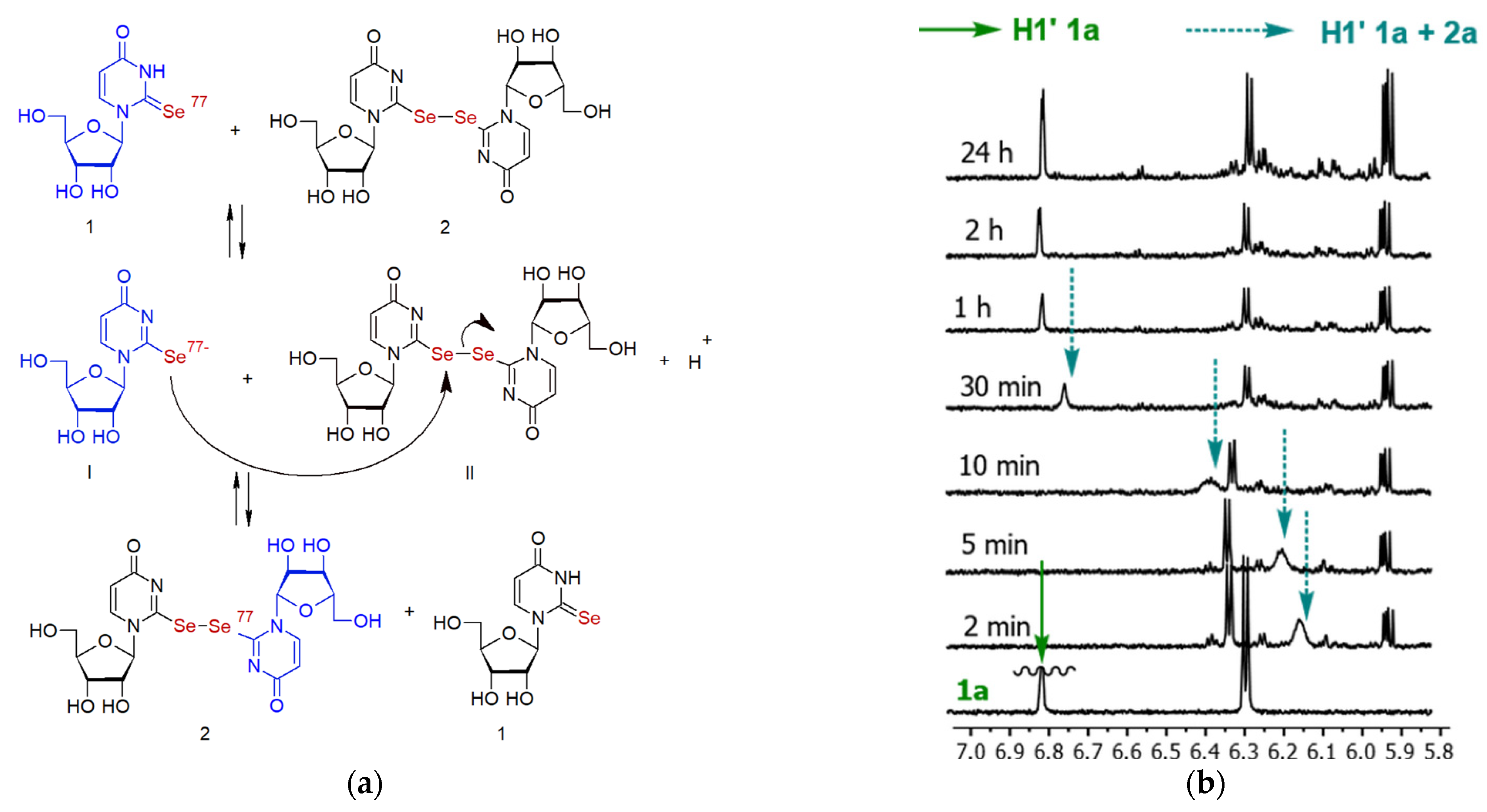
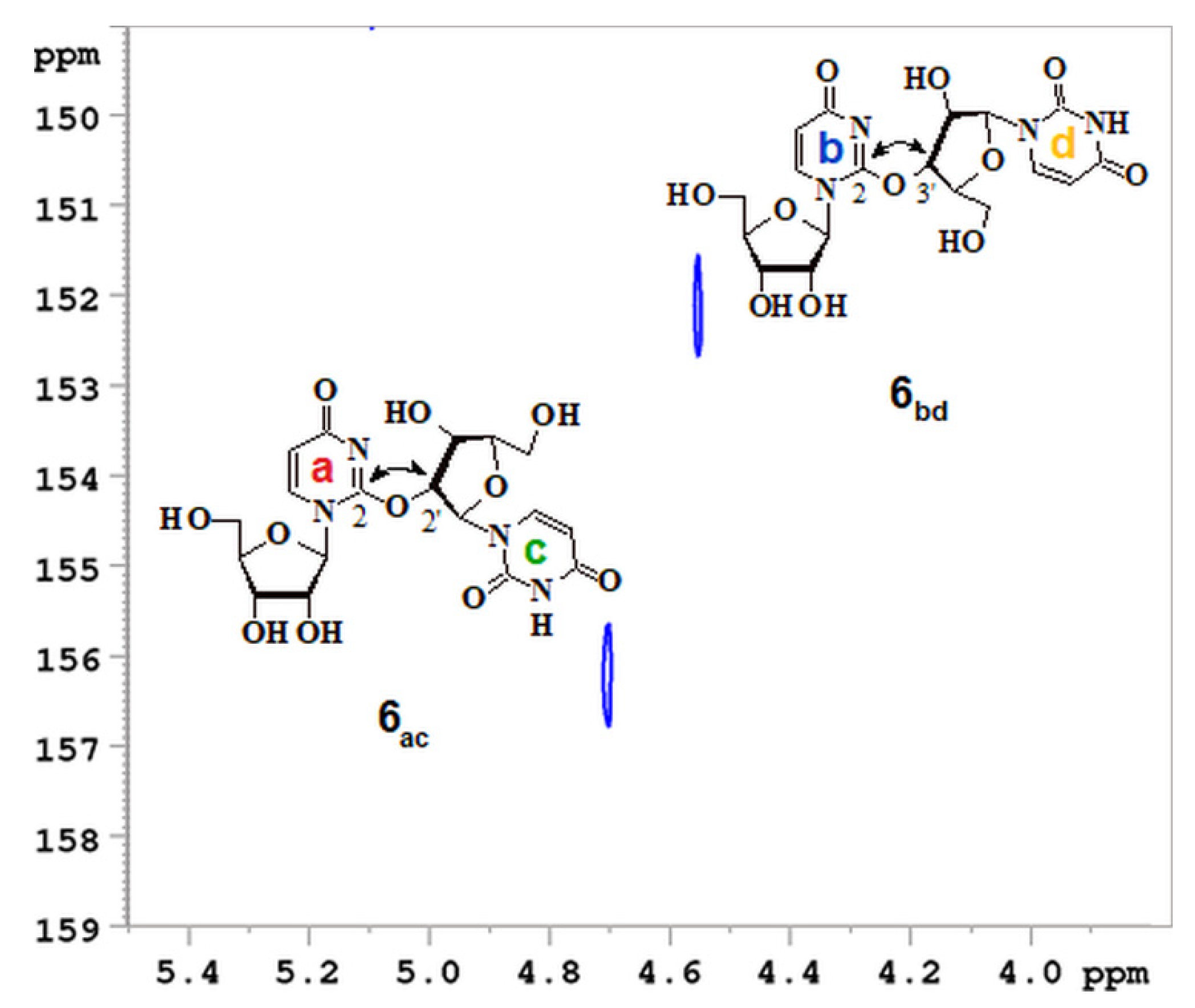
2.4. The Chemical Exchange between Se2U and (Se2U)2 in 1H NMR Time Scale
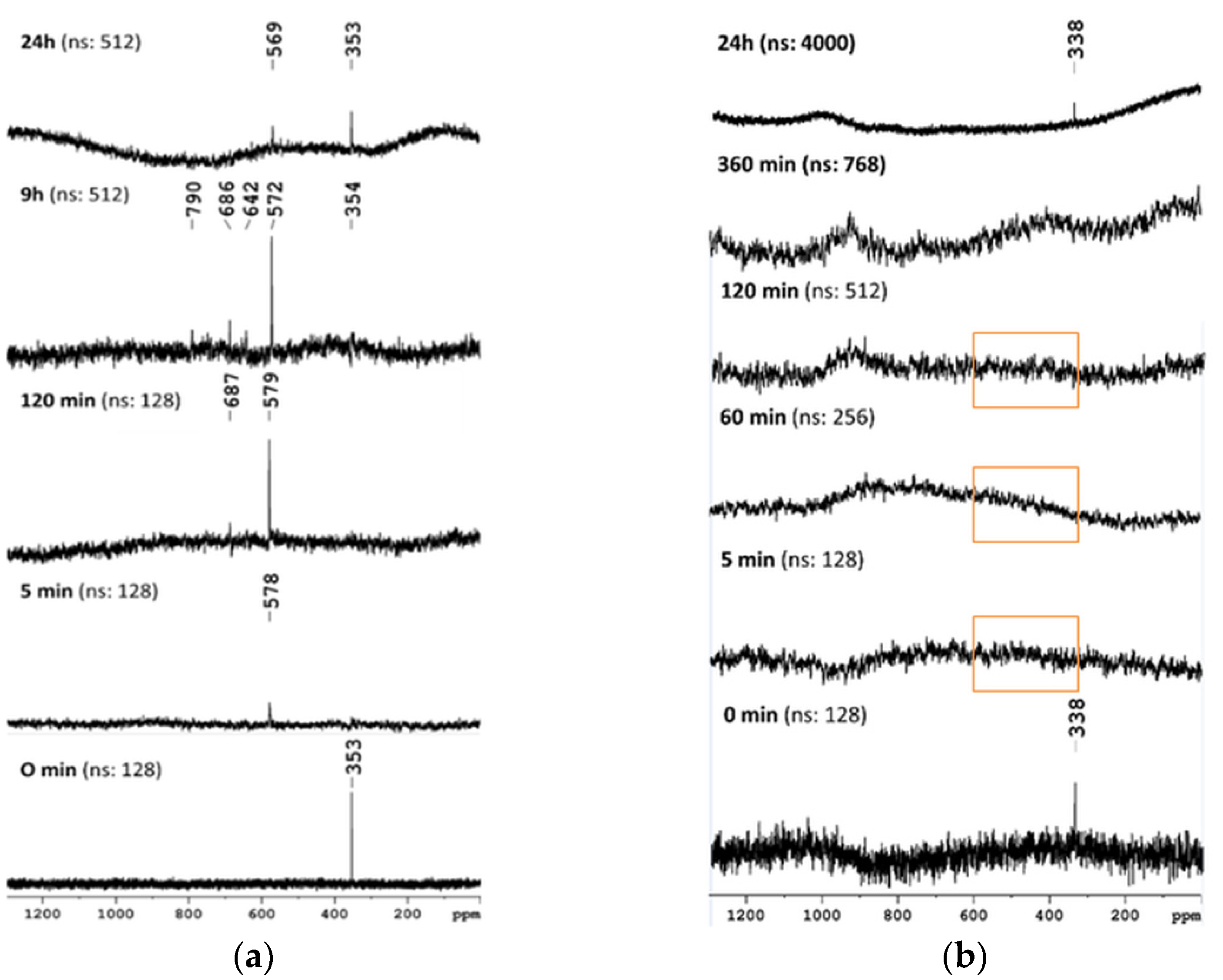
2.5. Monitoring of the Progress of Se2U Oxidation by 77Se NMR
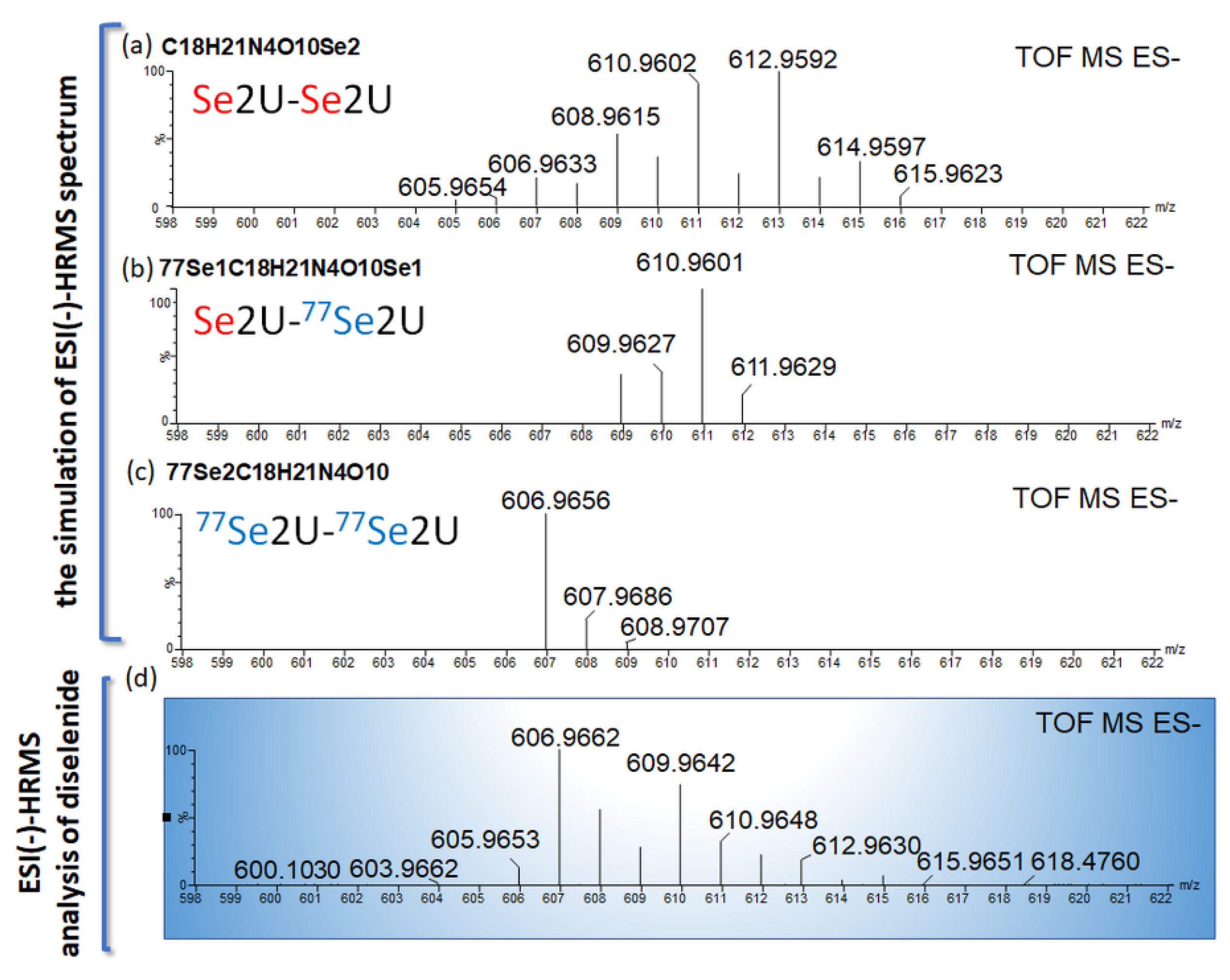
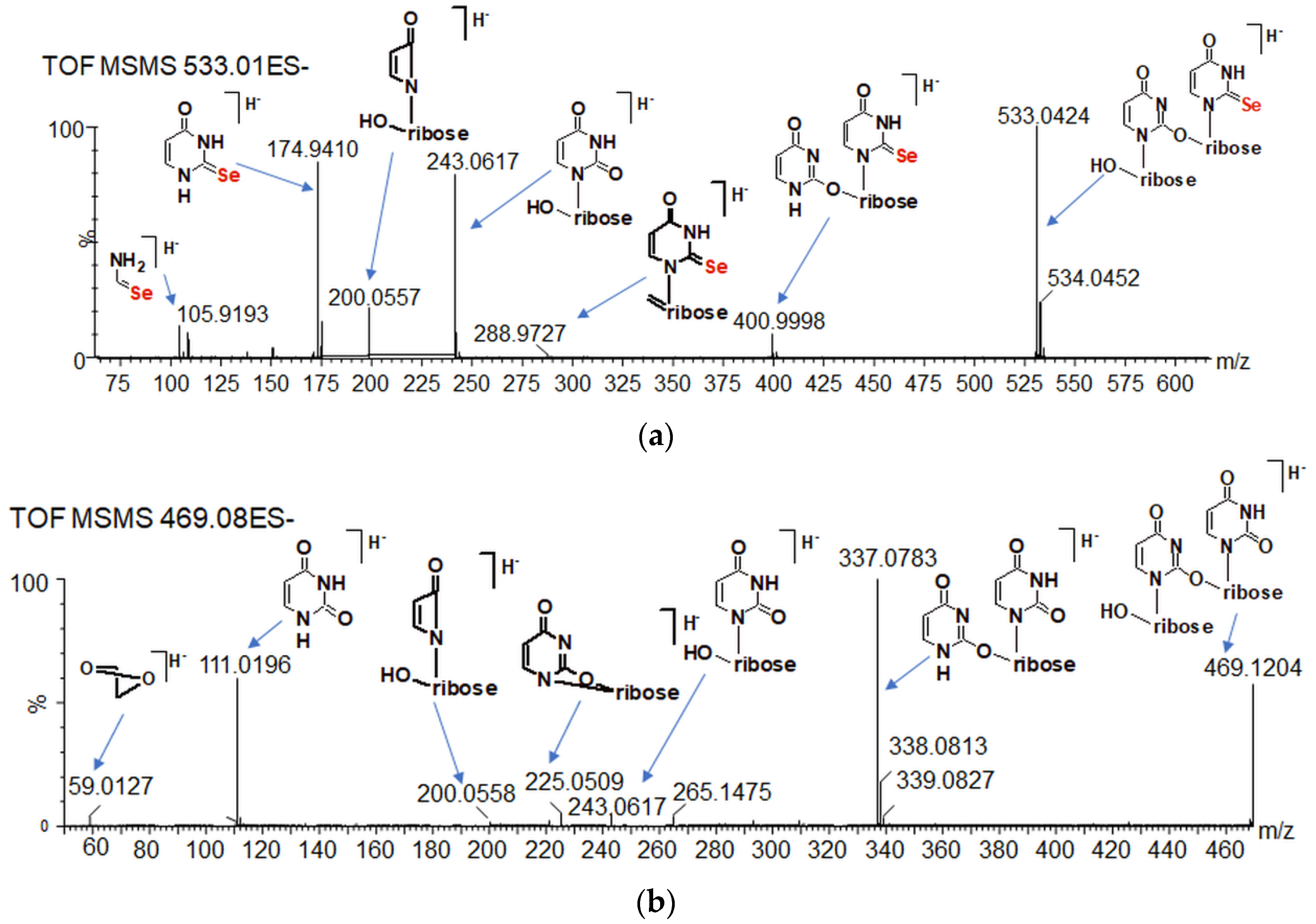
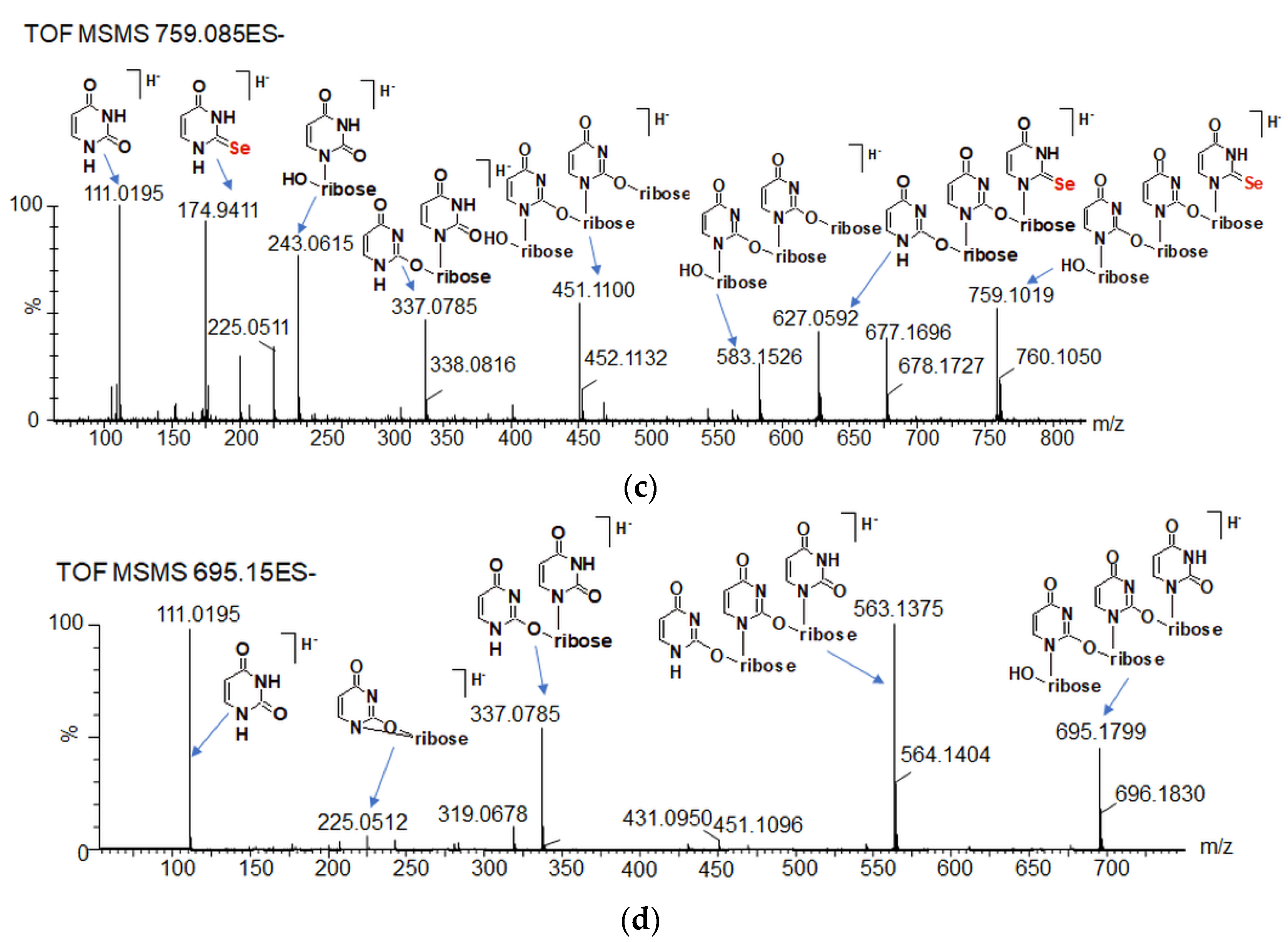
2.6. The Chemical Exchange between 77Se2U and (Se2U)2 Monitored by LC–MS
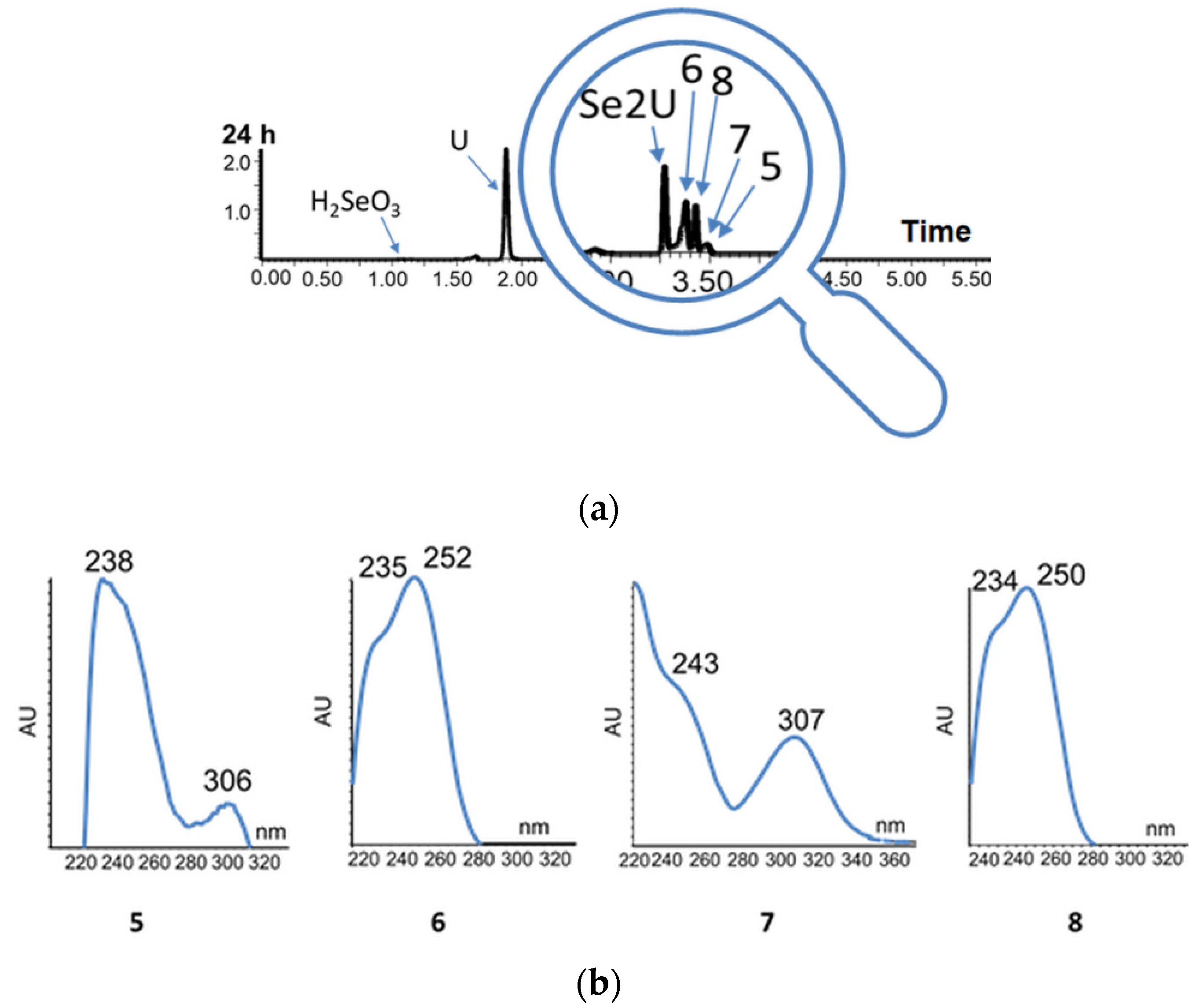
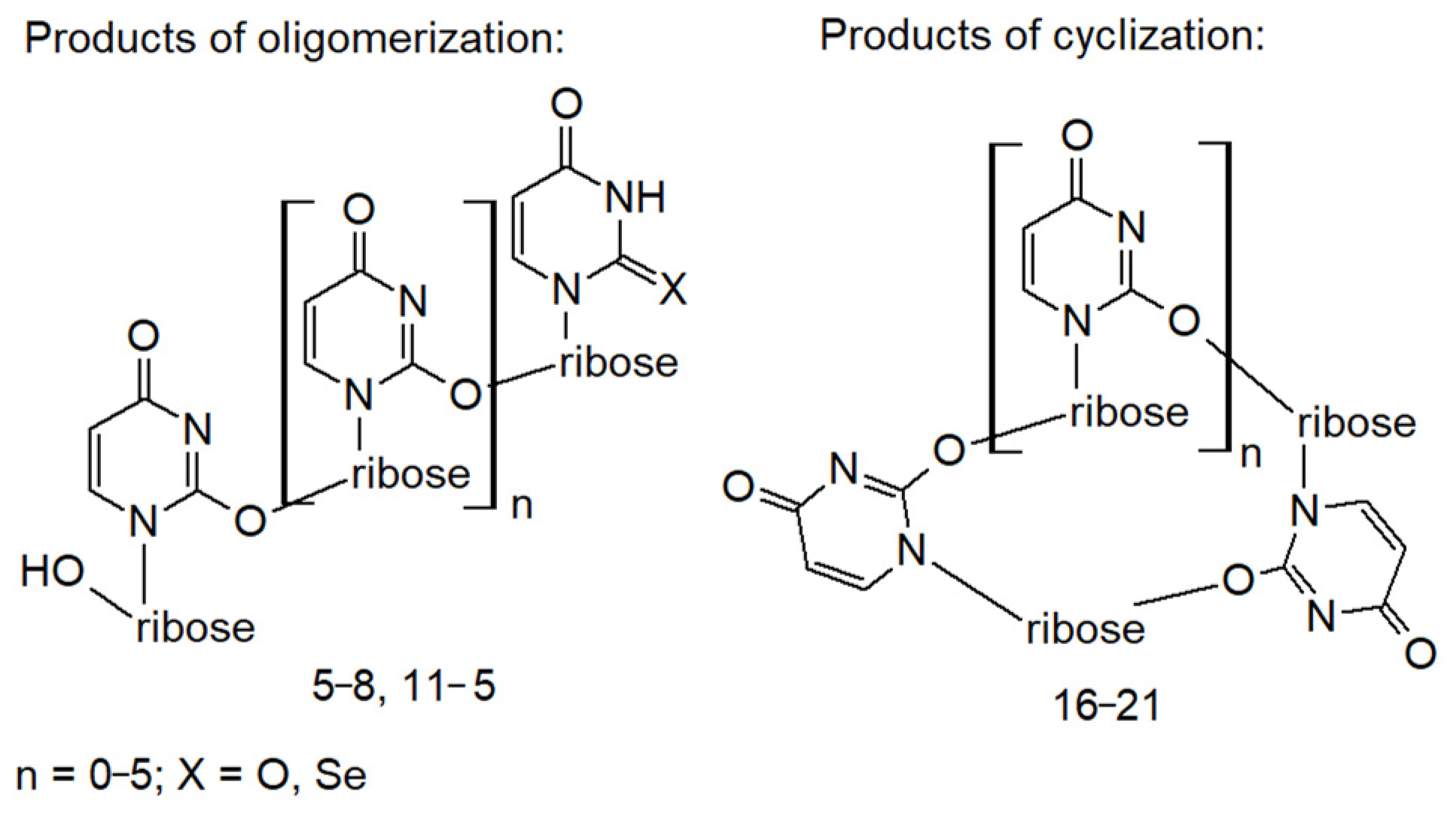
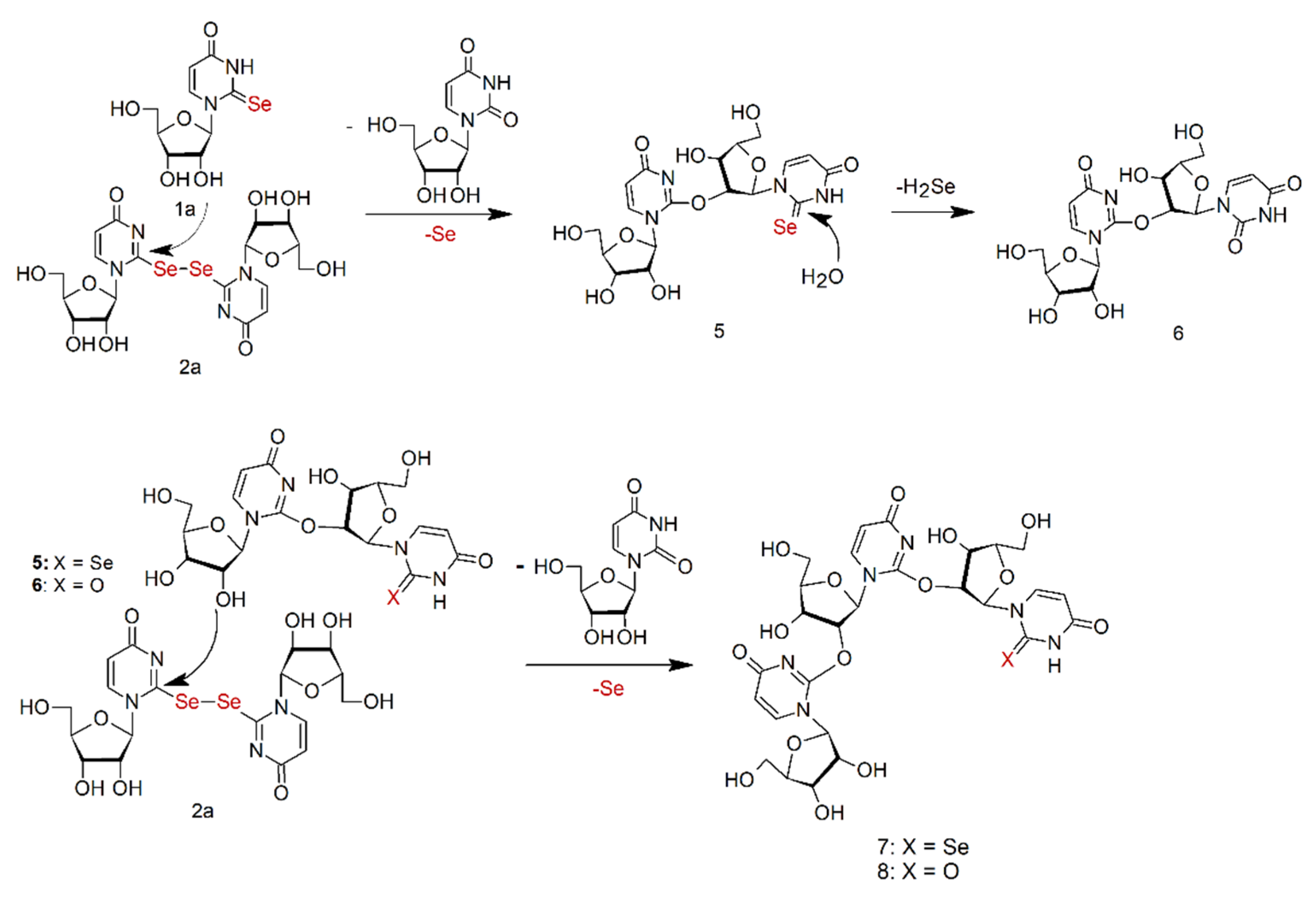
2.7. Oligomeric Side Products of the Condensation of Diselenide 2
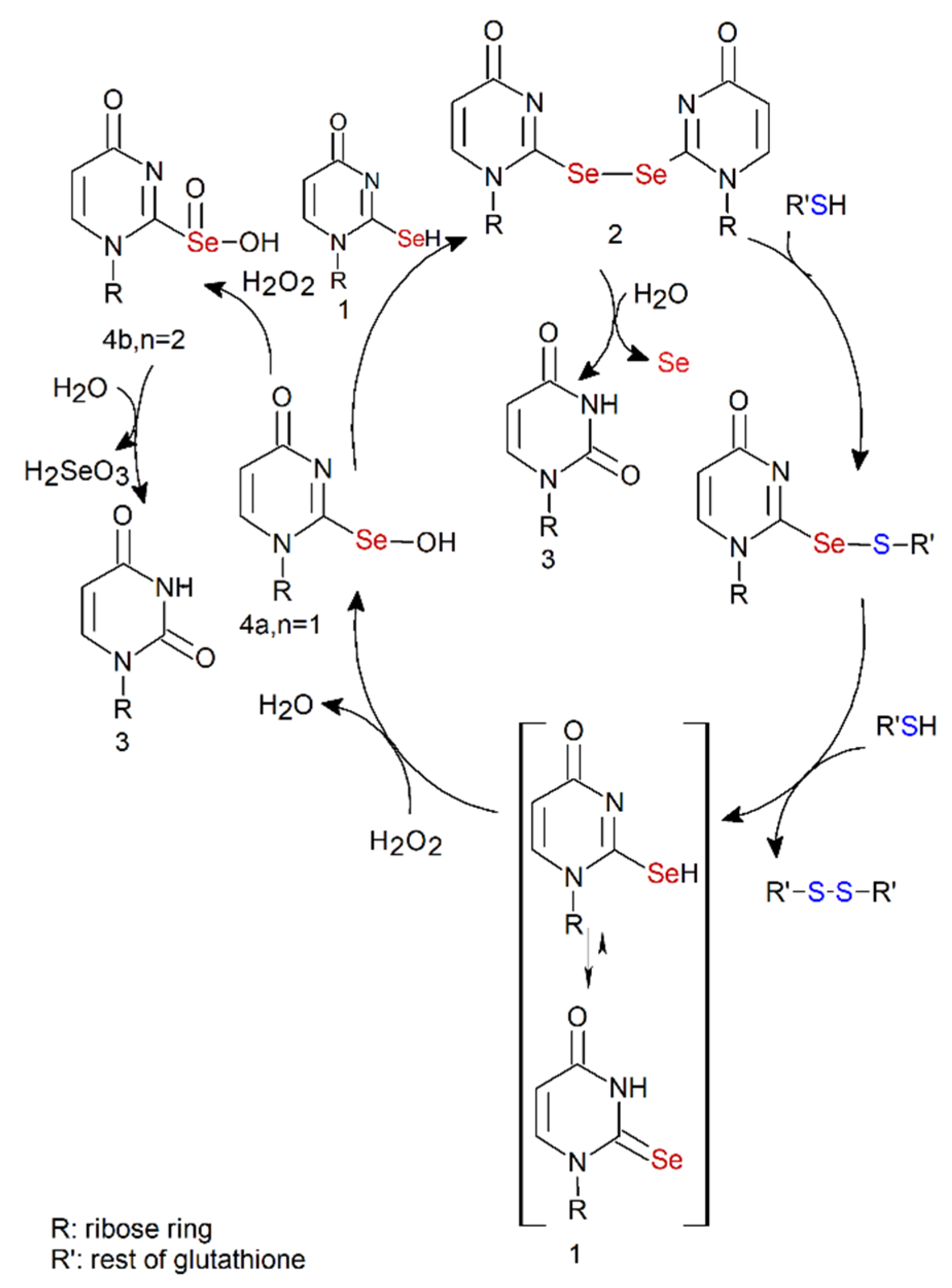
2.8. Se2U Oxidation with H2O2 and Subsequent Reduction of the Resulting Products (Rescue Assay)
3. Discussion
4. Materials and Methods
4.1. NMR Spectroscopy
4.2. Ultra-Performance Liquid Chromatography Coupled with a High-Resolution Mass Spectrometry and Photodiode Array Detection (UPLC–PDA–ESI(–)-HRMS)
4.3. Chemistry
4.3.1. Materials and Reactions
4.3.2. Synthesis of 2-selenouridine
4.3.3. H/77Se NMR Analysis of the Oxidation Experiments of 1
4.3.4. LC–MS Analysis of the Oxidation Assays of 1
4.3.5. Rescue Assay Conditions of 1 for LCMS Analysis
4.3.6. General Approach for Analysis of the Course of Oxidation of 2-selenouridine (Se2U, 1) and Identification of the Reaction Products
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Boccaletto, P.; Machnicka, M.A.; Purta, E.; Piatkowski, P.; Baginski, B.; Wirecki, T.K.; Crécy-Lagard, V.; Ross, R.; Limbach, P.A.; Kotter, A.; et al. MODOMICS: A database of RNA modification pathways. 2017 update. Nucleic. Acids Res. 2018, 46, D303–D307. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.M.; Gamper, H.; Yang, W. Post-transcriptional modifications to tRNA—A response to the genetic code degeneracy. RNA 2015, 21, 642–644. [Google Scholar] [CrossRef] [PubMed]
- Grosjean, H.; Crécy-Lagard, V.; Marck, C. Deciphering synonymous codons in the three domains of life: Co-evolution with specific tRNA modification enzymes. FEBS Lett. 2010, 584, 252–264. [Google Scholar] [CrossRef]
- Torres, A.G.; Batlle, E.; Ribas de Pouplana, L. Role of tRNA modifications in human diseases. Trends Mol. Med. 2014, 20, 306–314. [Google Scholar] [CrossRef]
- Schaffrath, R.; Leidel, S.A. Wobble uridine modifications—A reason to live, a reason to die?! RNA Biol. 2017, 14, 1209–1222. [Google Scholar] [CrossRef] [PubMed]
- Shigi, N. Biosynthesis and functions of sulfur modifications in Trna. Front. Genet. Apr. 2014, 2, 5–67. [Google Scholar] [CrossRef]
- Agris, P.F.; Eruysal, E.R.; Narendran, A.; Väre, V.Y.P.; Vangaveti, S.; Ranganathan, S.V. Celebrating wobble decoding: Half a century and still much is new. RNA Biol. 2018, 15, 537–553. [Google Scholar] [CrossRef] [PubMed]
- Rozov, A.; Demeshkina, N.; Khusainov, I.; Westhof, E.; Yusupov, M.; Yusupova, G. Novel base-pairing interactions at the tRNA wobble position crucial for accurate reading of the genetic code. Nat. Commun. 2016, 7, 10457. [Google Scholar] [CrossRef]
- Sochacka, E.; Lodyga-Chruscinska, E.; Pawlak, J.; Cypryk, M.; Bartos, P.; Ebenryter-Olbinska, K.; Leszczynska, G.; Nawrot, B. C5-substituents of uridines and 2-thiouridines present at the wobble position of tRNA determine the formation of their keto-enol or zwitterionic forms—A factor important for accuracy of reading of guanosine at the 3′-end of the mRNA codons. Nucleic. Acids Res. 2017, 45, 4825–4836. [Google Scholar] [CrossRef]
- Sierant, M.; Leszczynska, G.; Sadowska, K.; Dziergowska, A.; Rozanski, M.; Sochacka, E.; Nawrot, B. S-Geranyl-2-thiouridine wobble nucleosides of bacterial tRNAs; chemical and enzymatic synthesis of S-geranylated-RNAs and their physicochemical characterization. Nucleic. Acids Res. 2016, 44, 10986–10998. [Google Scholar] [CrossRef]
- Sochacka, E.; Szczepanowski, R.H.; Cypryk, M.; Sobczak, M.; Janicka, M.; Kraszewska, K.; Bartos, P.; Chwialkowska, A.; Nawrot, B. 2-Thiouracil deprived of thiocarbonyl function preferentially base pairs with guanine rather than adenine in RNA and DNA duplexes. Nucleic. Acids Res. 2015, 43, 2499–2512. [Google Scholar] [CrossRef] [PubMed]
- Vendeix, F.A.P.; Murphy, F.V.; Gustilo, E.; Graham, W.; Cantora, W.; Leszczynska, G.; Sproat, B.; Malkiewicz, A.; Agris, P. Human tRNALys3UUU Is Pre-Structured by Natural Modifications for Cognate and Wobble Codon Binding through Keto–Enol Tautomerism. J. Mol. Biol. 2012, 416, 467–485. [Google Scholar] [CrossRef] [PubMed]
- Murphy, F.V., IV; Ramakrishnan, V.; Malkiewicz, A.; Agris, P.F. The role of modifications in codon discrimination by tRNALysUUU. Nat. Struct. Mol. Biol. 2004, 12, 1186–1191. [Google Scholar] [CrossRef] [PubMed]
- Weixlbaumer, A.; Murphy, F.V., IV; Dziergowska, A.; Malkiewicz, A.; Vendix, F.A.P.; Agris, P.F.; Ramakrishnan, V. Mechanism for expanding the decoding capacity of transfer RNAs by modification of uridines. Nat. Struct. Mol. Biol. 2007, 14, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Ching, W.M. Characterization of selenium-containing tRNAGlu from Clostridium sticklandii. Arch. Biochem. Biophys. 1986, 244, 137–146. [Google Scholar] [CrossRef]
- Wittwer, A.J.; Tsai, L.; Ching, W.M.; Stadtman, T.C. Identification and synthesis of a naturally occurring selenonucleoside in bacterial tRNAs: 5-[(methylamino)methyl]-2-selenouridine. Biochemistry 1984, 23, 4650–4655. [Google Scholar] [CrossRef]
- Wittwer, A.J.; Stadtman, T.C. Biosynthesis of 5-methylaminomethyl-2-selenouridine, a naturally occurring nucleoside in Escherichia coli Trna. Arch. Biochem. Biophys. 1986, 248, 540–550. [Google Scholar] [CrossRef]
- Wolfe, M.D.; Ahmed, F.; Lacourciere, G.M.; Lauhon, C.T.; Stadtman, T.C.; Larson, T. Functional Diversity of the Rhodanese Homology Domain The Escherichia Coli ybbB Gene Encodes A Selenophosphate-Dependent tRNA 2-Selenouridine Synthase. J. Biol. Chem. 2004, 279, 1801–1809. [Google Scholar] [CrossRef]
- Nawrot, B.; Sochacka, E.; Duchler, M. tRNA structural and functional changes induced by oxidative stress. Cell. Mol. Life Sci. 2011, 68, 4023–4032. [Google Scholar] [CrossRef]
- Veres, Z.; Stadtman, T.C. A purified selenophosphate-dependent enzyme from Salmonella typhimurium catalyzes the replacement of sulfur in 2-thiouridine residues in tRNAs with selenium. Proc. Natl. Acad. Sci. USA 1994, 91, 8092–8096. [Google Scholar] [CrossRef]
- Jäger, G.; Chen, P.; Björk, G.R. An unmodified wobble uridine in tRNAs specific for Glutamine, Lysine, and Glutamic acid from Salmonella enterica Serovar Typhimurium results in nonviability—Due to increased missense errors? PLoS ONE 2016, 12, e0175092. [Google Scholar] [CrossRef]
- Szczupak, P.; Sierant, M.; Wielgus, E.; Radzikowska-Cieciura, E.; Kulik, K.; Krakowiak, A.; Kuwerska, P.; Leszczynska, G.; Nawrot, B. Escherichia coli tRNA 2-selenouridine synthase (SelU); elucidation of its substrate specificity for understanding the role of S-geranyl-tRNA in the conversion of 2-thio- to 2-selenouridines in bacterial tRNA. Cells 2022, 11, 1522. [Google Scholar] [CrossRef] [PubMed]
- Szczupak, P.; Radzikowska-Cieciura, E.; Kulik, K.; Madaj, R.; Sierant, M.; Krakowiak, A.; Nawrot, B. Escherichia coli tRNA 2-selenouridine synthase SelU selects its prenyl substrate to accomplish its enzymatic function. Bioorg. Chem. 2022, 122, 105739. [Google Scholar] [CrossRef]
- Sierant, M.; Leszczynska, G.; Sadowska, K.; Komar, P.; Radzikowska-Cieciura, E.; Sochacka, E.; Nawrot, B. Escherichia coli tRNA 2-selenouridine synthase (SelU) converts S2U-RNA to Se2U-RNA via S-geranylated-intermediate. FEBS Lett. 2018, 13, 2248–2258. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Sheng, J.; Hassan, A.E.; Jiang, S.; Gan, J.; Huang, Z. Novel RNA base pair with higher specificity using single selenium atom. Nucleic. Acids Res. 2012, 40, 5171–5179. [Google Scholar] [CrossRef]
- Leszczynska, G.; Cypryk, M.; Gostynski, B.; Sadowska, K.; Herman, P.; Bujacz, G.; Lodyga Chruscinska, E.; Sochacka, E.; Nawrot, B. C5-Substituted 2-Selenouridines Ensure Efficient Base Pairing with Guanosine; Consequences for Reading the NNG-3′ Synonymous mRNA Codons. Int. J. Mol. Sci. 2020, 21, 2882. [Google Scholar] [CrossRef]
- Krüger, M.K.; Pedersen, S.; Hagervall, T.G.; Sørensen, M.A. The modification of the wobble base of tRNAGlu modulates the translation rate of glutamic acid codons in vivo. J. Mol. Biol. 1998, 284, 621–631. [Google Scholar] [CrossRef]
- Hagervall, T.G.; Pomerantz, S.C.; McCloskey, J.A. Reduced misreading of asparagine codons by Escherichia coli tRNALys with hypomodified derivatives of 5-methylaminomethyl-2-thiouridine in the wobble position. J. Mol. Biol. 1998, 284, 33–42. [Google Scholar] [CrossRef]
- Wittwer, A.J.; Ching, W.M. Selenium-containing tRNA(Glu) and tRNA(Lys) from Escherichia coli: Purification, codon specificity and translational activity. Biofactors 1989, 2, 27–34. [Google Scholar]
- Atkins, J.F.; Gesteland, R.F. The twenty-first amino acid. Nature 2000, 407, 463–464. [Google Scholar] [CrossRef]
- Zoidis, E.; Seremelis, I.; Kontopoulos, N.; Danezis, G.P. Selenium-Dependent Antioxidant Enzymes: Actions and Properties of Selenoproteins. Antioxidants 2018, 7, 66. [Google Scholar] [CrossRef] [PubMed]
- Payne, N.C.; Geissler, A.; Button, A.; Sasuclark, A.R.; Schroll, A.L.; Ruggles, E.L.; Gladyshev, V.N.; Hondal, R.J. Comparison of the redox chemistry of sulfur- and selenium-containing analogs of uracil. Free Radic. Biol. Med. 2017, 104, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 3289–3303. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Glutathione and its role in cellular functions. Free Radic. Biol. Med. 1999, 27, 916–921. [Google Scholar] [CrossRef]
- Sochacka, E.; Kraszewska, K.; Sochacki, M.; Sobczak, M.; Janicka, M.; Nawrot, B. The 2-thiouridine unit in the RNA strand is desulfured predominantly to 4-pyrimidinone nucleoside under in vitro oxidative stress conditions. Chem. Commun. 2011, 47, 4914–4916. [Google Scholar] [CrossRef] [PubMed]
- Sierant, M.; Kulik, K.; Sochacka, E.; Szewczyk, R.; Sobczak, M.; Nawrot, B. Cytochrome c Catalyzes the Hydrogen Peroxide-Assisted Oxidative Desulfuration of 2-Thiouridines in Transfer RNAs. Chem. Biochem. 2018, 4, 687–695. [Google Scholar] [CrossRef]
- Bartos, P.; Ebenryter-Olbinska, K.; Sochacka, E.; Nawrot, B. The influence of the C5 substituent on the 2-thiouridine desulfuration pathway and the conformational analysis of the resulting 4-pyrimidinone products. Bioorg. Med. Chem. 2015, 23, 5587–5594. [Google Scholar] [CrossRef]
- Sochacka, E.; Bartos, P.; Kraszewska, K.; Nawrot, B. Desulfuration of 2-thiouridine with hydrogen peroxide in the physiological pH range 6.6–7.6 is pH-dependent and results in two distinct products. Bioorg. Med. Chem. Lett. 2013, 23, 5803–5805. [Google Scholar] [CrossRef]
- Lim, D.; Gründemann, D.; Seebeck, F.P. Total Synthesis and Functional Characterization of Selenoneine. Angew. Chem. Int. Ed. 2019, 58, 15026–15030. [Google Scholar] [CrossRef]
- Mangiavacchi, F.; Coelho, D.; Dias, I.F.; Di Lorenzo, I.; Grzes, P.; Palomba, M.; Rosati, O.; Bagnoli, L.; Marini, F.; Santi, C.; et al. Sweet Selenium: Synthesis and Properties of Selenium-Containing Sugars and Derivatives. Phamaceuticals 2020, 13, 211. [Google Scholar] [CrossRef]
- Sahu, P.K.; Naik, S.D.; Yu, J.; Jeong, L.S. 4-Selenonucleosides as Next-Generation Nucleosides. Eur. J. Org. Chem. 2015, 28, 6115–6124. [Google Scholar] [CrossRef]
- Jeong, L.S.; Tosh, D.K.; Choi, W.J. Development of next generation 4′-selenonucleosides. Nucleic Acids Symp. Ser. 2009, 53, 7–8. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lee, H.; Jarhad, D.B.; Yu, J.; Lee, C.; Jeong, L.S. Asymmetric Synthesis of 2′-C-Methyl-4′-selenonucleosides as Anti-Hepatitis C Virus Agents. J. Org. Chem. 2019, 84, 14414–14426. [Google Scholar] [CrossRef]
- Sancineto, L.; Piccioni, M.; De Marco, S.; Pagiotti, R.; Nascimento, V.; Braga, A.L.; Santi, C.; Pietrella, D. Diphenyl diselenide derivatives inhibitmicrobial biofilmformation involved in wound infection. BMC Microbiol. 2016, 16, 220. [Google Scholar] [CrossRef]
- Macegoniuk, K.; Grela, E.; Palus, J.; Rudzinska-Szostak, E.; Grabowiecka, A.; Biernat, M.; Berlicki, Ł. 1,2-Benzisoselenazol-3(2H)-one Derivatives As a New Class of Bacterial Urease Inhibitors. J. Med. Chem. 2016, 59, 8125–8133. [Google Scholar] [CrossRef]
- Pietrella, D. Antimicrobial Activity of Organoselenium Compounds, Organoselenium Chemistry: Between Synthesis and Biochemistry. Bentham Sci. 2014, 17, 328–344. [Google Scholar] [CrossRef][Green Version]
- Tran, P.; Kopel, J.; Ristic, B.; Marsh, H.; Fralick, J.; Reid, T. Antimicrobial seleno-organic coatings and compounds acting primarily on the plasma membrane: A review. Adv. Redox Res. 2022, 4, 100031. [Google Scholar] [CrossRef]
- Kulik, K.; Sadowska, K.; Wielgus, E.; Pacholczyk-Sienicka, B.; Sochacka, E.; Nawrot, B. Different Oxidation Pathways of 2-Selenouracil and 2-Thiouracil, Natural Components of Transfer RNA. Int. J. Mol. Sci. 2020, 21, 5956. [Google Scholar] [CrossRef]
- Landry, V.K.; Minoura, M.; Pang, K.; Buccella, D.; Kelly, B.V.; Parkin, G. Synthesis and Structural Characterization of 1-Mesityl-1,3-dihydro-imidazole-2-selone and Bis(1-mesitylimidazol-2-yl)diselenide: Experimental Evidence That the Selone Is More Stable Than the Selenol Tautomer. J. Am. Chem. Soc. 2006, 128, 12490–12497. [Google Scholar] [CrossRef]
- Prabhu, P.; Singh, B.G.; Noguchi, M.; Phadnis, P.P.; Jain, V.K.; Iwaoka, M.; Priyadarsinib, K.I. Stable selones in glutathione-peroxidase-like catalytic cycle of selenonicotinamide derivative. Org. Biomol. Chem. 2014, 12, 2404–2412. [Google Scholar] [CrossRef]
- Laube, J.; Jager, S.; Thone, C. Synthesis and Structural Studies of Pyridine-2-selenolates—Reactions with Electrophilic Phosphorus(III) Compounds and Related Complex Chemistry. Eur. J. Inorg. Chem. 2001, 8, 1983–1992. [Google Scholar] [CrossRef]
- Fenner, T.; White, J.M.; Schiesser, C.H. Preparation of 2,3-dihydroselenolo[2,3-b]pyridines and related compounds by free-radical means. Org. Biomol. Chem. 2006, 4, 466–474. [Google Scholar] [CrossRef]
- Odom, J.D.; Dawson, W.H.; Ellis, P.D. Selenium-77 relaxation time studies on compounds of biological importance: Dialkyl selenides, dialkyl diselenides, selenols, selenonium compounds, and seleno oxyacids. J. Am. Chem. Soc. 1979, 101, 5815–5822. [Google Scholar] [CrossRef]
- Shimodaira, S.; Asano, Y.; Arai, K.; Iwaoka, M. Selenoglutathione Diselenide: Unique Redox Reactions in the GPx-Like Catalytic Cycle and Repairing of Disulfide Bonds in Scrambled Protein. Biochemistry 2017, 56, 5644–5653. [Google Scholar] [CrossRef] [PubMed]
- Kraszewska, K.; Kaczynska, I.; Jankowski, S.; Karolak-Wojciechowska, J.; Sochacka, E. Desulfurization of 2-thiouracil nucleosides: Conformational studies of 4-pyrimidinone nucleosides. Bioorg. Med. Chem. 2011, 19, 2443–2449. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.; Kimball, E.; Gao, M.; Osterhout, R.; Van Dien, S.J.; Rabinowitz, J.D. Absolute Metabolite Concentrations and Implied Enzyme Active Site Occupancy in Escherichia coli. Nat. Chem. Biol. 2009, 5, 593–599. [Google Scholar] [CrossRef]
- Griffith, O.W. Biologic and pharmacologic regulation of mammalian glutathione synthesis. Free Radic. Biol. Med. 1999, 27, 922–935. [Google Scholar] [CrossRef]
- Oriana, L.; Mauri, P.; Roveric, A.; Toppoc, S.; Benazzi, L.; Bosello-Travain, V.; De Palma, A.; Maiorino, M.; Miotto, G.; Zaccarin, M.; et al. Selenocysteine oxidation in glutathione peroxidase catalysis: An MS-supported quantum mechanics study. Free Radic. Biol. Med. 2015, 87, 1–14. [Google Scholar] [CrossRef]
- Reich, H.J.; Hondal, R.J. Why Nature Chose Selenium. ACS Chem. Biol. 2016, 11, 821–841. [Google Scholar] [CrossRef]
- Vorbruggen, H.; Strehlke, P. Eine Einfache Synthese von 2-Thiopyrimidin-nucleosiden. Chem. Ber. 1973, 106, 3039–3061. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. (#) | Se2U | Molar Ratio Se2U:H2O2 | pH | T [°C] | Product Content [%] | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |||||
| 1 | Se2U 1 | 1:1 | 7.4 | rt. | - | + | 71 | + (n = 2) | + | 8 | + | + |
| 2 | 1:1 | 8.0 | rt. | - | + | 53 | - | + | 5 | + | + | |
| 3 | 1:1 | 5.0 | rt. | - | + | 100 | + (n = 1, 2) | - | - | - | - | |
| 4 | 1:1 | water | rt. | <1 | + | 97 | + (n = 1, 2) | + | 1 | + | + | |
| 5 | 1:10 | 7.4 | rt. | - | + | 100 | + (n = 2) | + | - | + | - | |
| 6 | 1:10 | water | rt. | - | + | 100 | + (n = 1, 2) | + | <1 | + | - | |
| 7 | 1:10 | water | 10 | - | + | 99 | + (n = 1, 2) | - | <1 | - | - | |
| 8 | 1:0.5 | 7.4 | rt. | 28 | + | 27 | - | + | 4 | + | + | |
| 9 | 1:0.5 | 7.4 | 10 | 19 | - | 22 | - | - | 6 | - | - | |
| 10 | 1:1 | 7.4 | 10 | - | - | 59 | - | - | 8 | - | - | |
| 11 | 1:0.5 | water | 10 | 11 | + | 87 | - | + | <1 | + | + | |
| 12 | 77Se2U | 11:0.5 | water | 10 | + | + | + | |||||
| 13 | 11:0.5 | 7.4 | 10 | + | ||||||||
| Compound | UPLC–PDA–ESI(–)-HRMS | UV | 1H NMR | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Elemental Composition | Rt [min] | m/z [M-H]− | λmax | H6 [ppm] | H5 [ppm] | H1′ [ppm] | JH6-H5 | JH1′-H2′ | |||
| Calcd | Found | ||||||||||
| 1 | Se2U | C9H12N2O5Se | 3.26 | 306.9833 | 306.9834 | 307 | 8.14 | 6.28 | 6.83 | 8.2 | 2.6 |
| 2 | (USe2U)2 | C18H22N4O10Se2 | 3.43 | 612.9592 | 612.9600 | 241 | 8.16 | 6.34 | 6.11 | 7.6 | 5.3 |
| 3 | U | C9H12N2O6 | 1.84 | 243.0617 | 243.0623 | 261 | 7.78 | 5.82 | 5.80 | 8.1 | 4.7 |
| 4a | U-SeOH | C9H12N2O6Se | 322.9782 | 322.9781 | - | 8.18 | 6.31 | 6.19 | 7.6 | 3.1 | |
| 4b | U-SeO2H | C9H12N2O7Se | 1.14 | 338.9731 | 338.9720 | 249 | 8.13 | 6.26 | 5.86 | 7.7 | 4.4 |
| 5 | C18H22N4O10Se | 3.60 | 533.0423 | 533.0418 | 238/306 | ||||||
| 6 | C18H22N4O11 | 3.37 | 469.1207 | 469.1206 | 235/252 | 8.16 | 6.25 | 6.07 | 7.6 | 3.5 | |
| 7.92 | 5.97 | 6.10 | 8.1 | 5.8 | |||||||
| 7 | C27H32N6O15Se | 3.42 | 759.1020 | 759.1020 | 243/307 | - | - | - | - | - | |
| 8 | C27H32N6O16 | 3.42 | 695.1796 | 695.1798 | 234/250 | - | - | - | - | - | |
| Conditions/Reaction Number | Content of 3 after 2 min [%] |
|---|---|
| pH 5/3 | 5 |
| Water/4 | 7 |
| pH 7.4/1 | 35 |
| pH 8/2 | 45 |
| Compound | n | X | UPLC–PDA–ESI(-)-HRMS | ||
|---|---|---|---|---|---|
| Elemental Composition | [M-H] m/z | ||||
| Calcd | Found | ||||
| Products of oligomerization | |||||
| 5 | 0 | Se | C18H21N4O10Se | 533.0423 | 533.0424 |
| 6 | 0 | O | C18H21N4O11 | 469.1207 | 469.1212 |
| 7 | 1 | Se | C27H31N6O15Se | 759.1013 | 759.1013 |
| 8 | 1 | O | C27H31N6O16 | 695.1797 | 695.1800 |
| 11 | 2 | Se | C36H41N8O20Se | 985.1602 | 985.1606 |
| 12 | 2 | O | C36H41N8O21 | 921.2386 | 921.2386 |
| 13 | 3 | O | C45H51N10O26 | 1147.2976 573.1449 a | 1147.3007 573.1464 a |
| 14 | 4 | O | C54H61N12O31 | 1373.3566 686.1744 a | 1373.3599 686.1730 a |
| 15 | 5 | O | C63H72N14O36 | 799.2039 a | 799.2028 a |
| Products of cyclization | |||||
| 16 | 0 | - | C18H19N4O10 | 451.1101 | 451.1103 |
| 17 | 1 | - | C27H29N6O15 | 677.1691 | 677.1686 |
| 18 | 2 | - | C36H39N8O20 | 903.2281 | 903.2280 |
| 19 | 3 | - | C45H49N10O25 | 1129.2870 | 1129.2883 |
| 20 | 4 | - | C54H59N12O30 | 1355.3460 | 1355.3461 |
| 21 | 5 | - | C63H69N14O35 | 790.1985 a | 790.1981 a |
Publisher′s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kulik, K.; Sadowska, K.; Wielgus, E.; Pacholczyk-Sienicka, B.; Sochacka, E.; Nawrot, B. 2-Selenouridine, a Modified Nucleoside of Bacterial tRNAs, Its Reactivity in the Presence of Oxidizing and Reducing Reagents. Int. J. Mol. Sci. 2022, 23, 7973. https://doi.org/10.3390/ijms23147973
Kulik K, Sadowska K, Wielgus E, Pacholczyk-Sienicka B, Sochacka E, Nawrot B. 2-Selenouridine, a Modified Nucleoside of Bacterial tRNAs, Its Reactivity in the Presence of Oxidizing and Reducing Reagents. International Journal of Molecular Sciences. 2022; 23(14):7973. https://doi.org/10.3390/ijms23147973
Chicago/Turabian StyleKulik, Katarzyna, Klaudia Sadowska, Ewelina Wielgus, Barbara Pacholczyk-Sienicka, Elzbieta Sochacka, and Barbara Nawrot. 2022. "2-Selenouridine, a Modified Nucleoside of Bacterial tRNAs, Its Reactivity in the Presence of Oxidizing and Reducing Reagents" International Journal of Molecular Sciences 23, no. 14: 7973. https://doi.org/10.3390/ijms23147973
APA StyleKulik, K., Sadowska, K., Wielgus, E., Pacholczyk-Sienicka, B., Sochacka, E., & Nawrot, B. (2022). 2-Selenouridine, a Modified Nucleoside of Bacterial tRNAs, Its Reactivity in the Presence of Oxidizing and Reducing Reagents. International Journal of Molecular Sciences, 23(14), 7973. https://doi.org/10.3390/ijms23147973