
Chloroquine-Induced DNA Damage Synergizes with Nonhomologous End Joining Inhibition to Cause Ovarian Cancer Cell Cytotoxicity

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
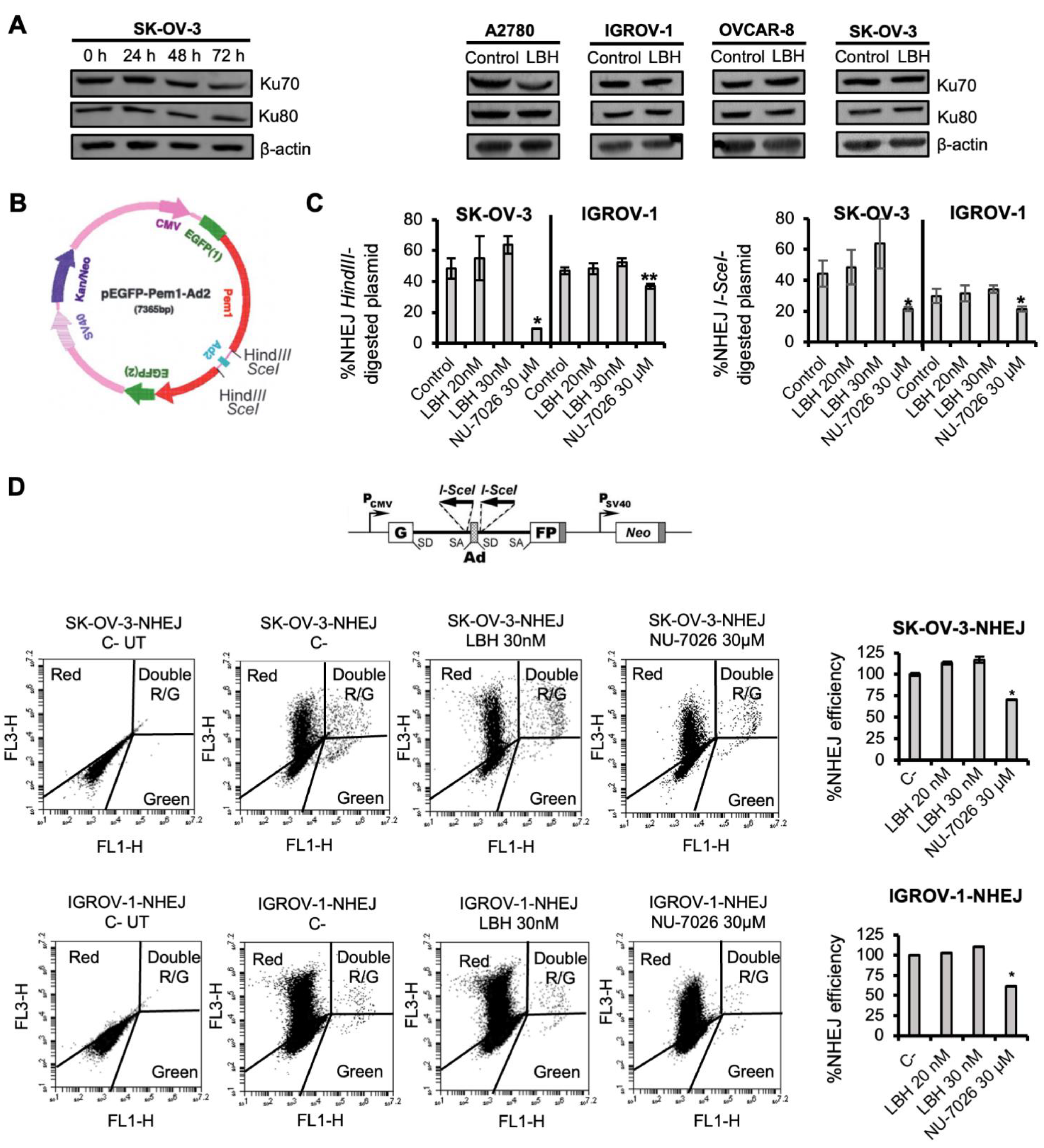
2.1. Panobinostat Does Not Affect NHEJ Efficiency in OCCLs
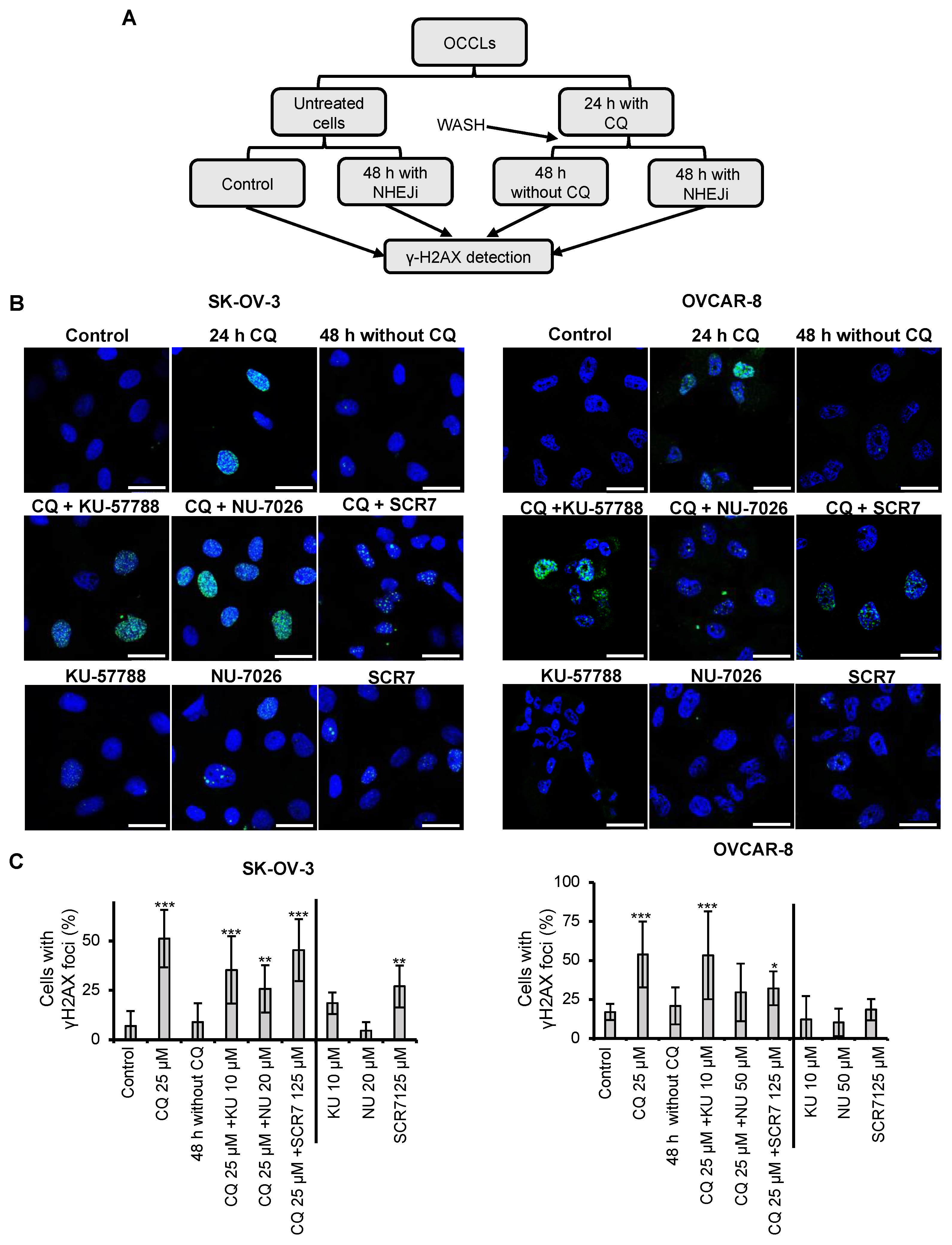
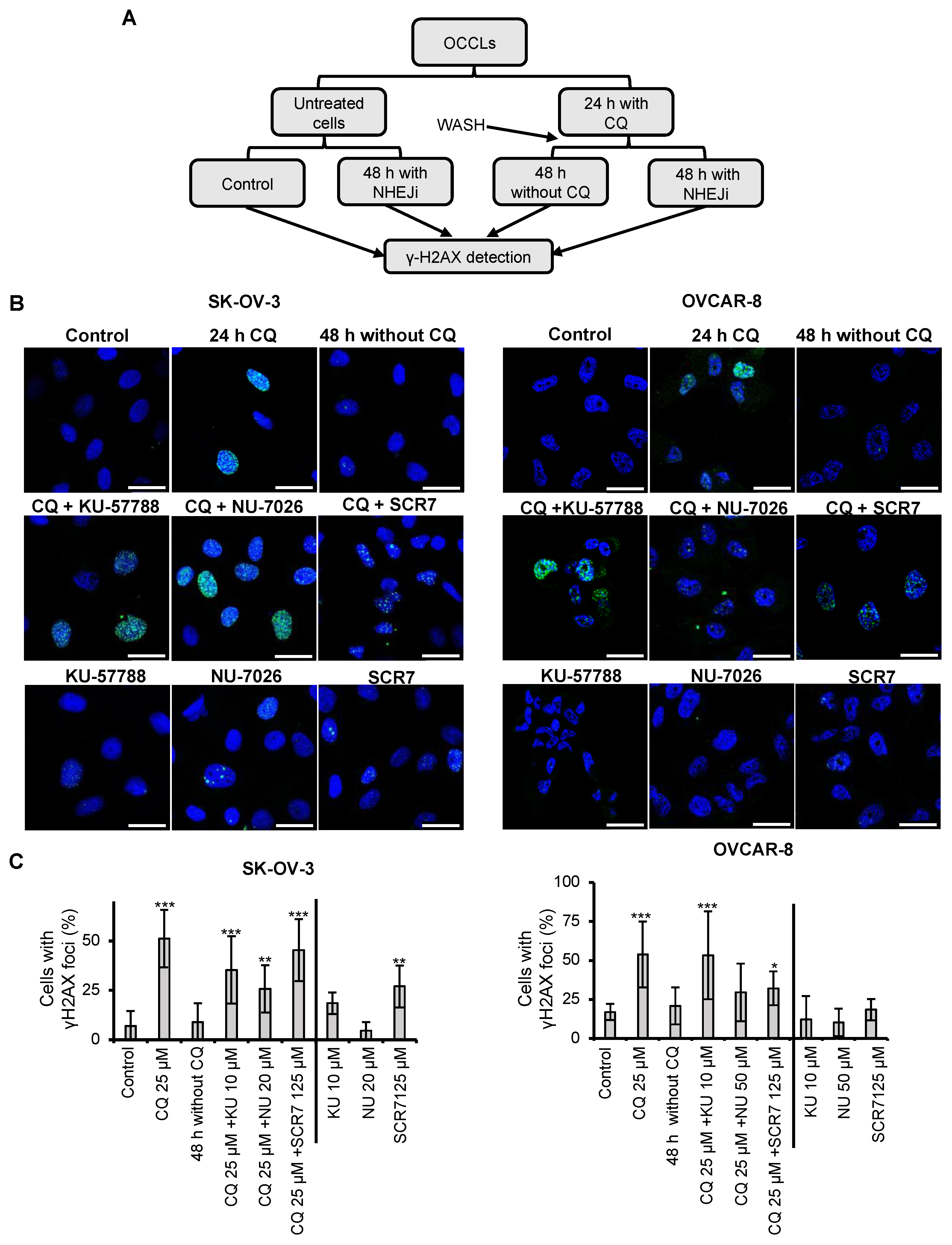
2.2. Chloroquine Induces DSBs, Which Are Repaired by NHEJ
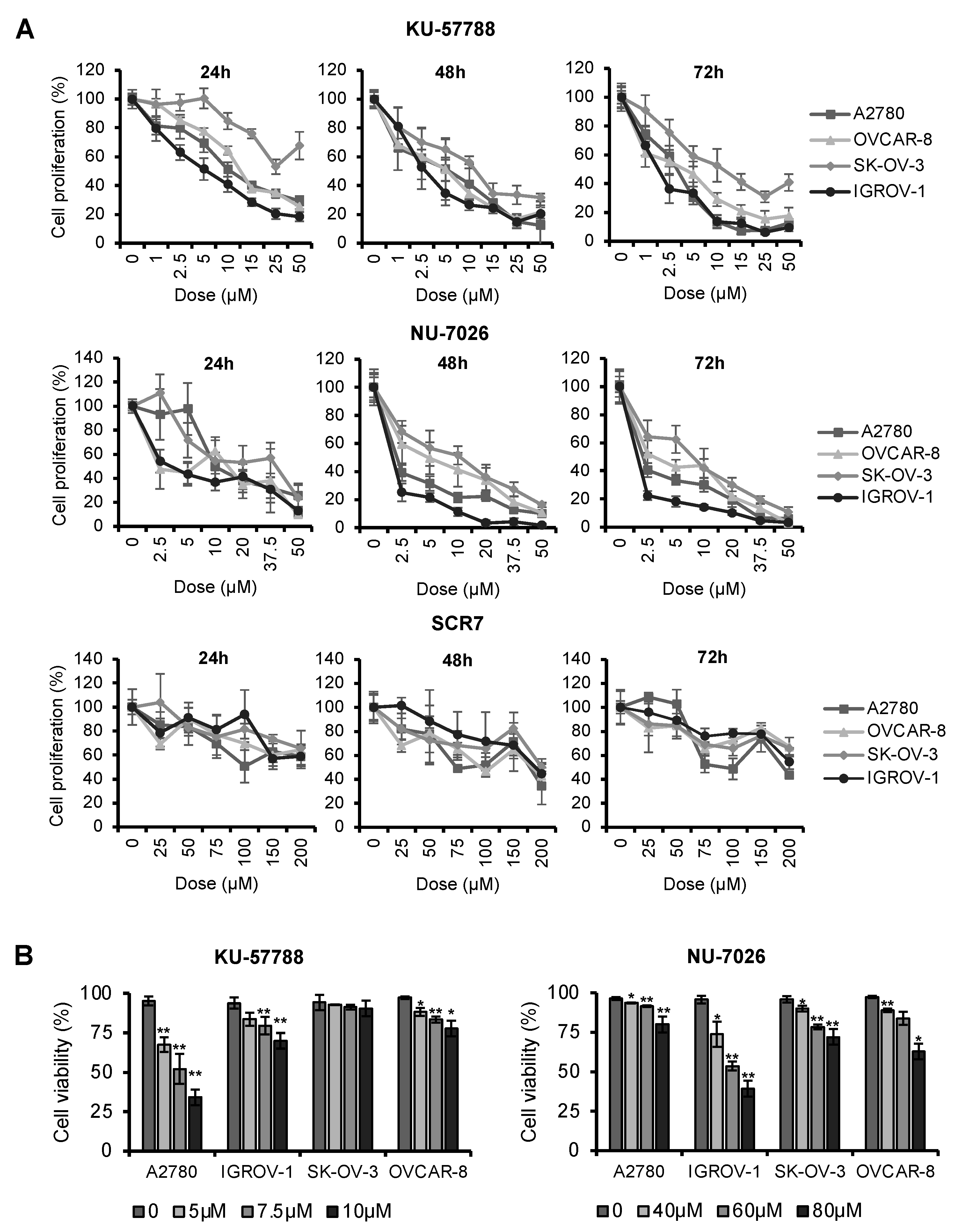
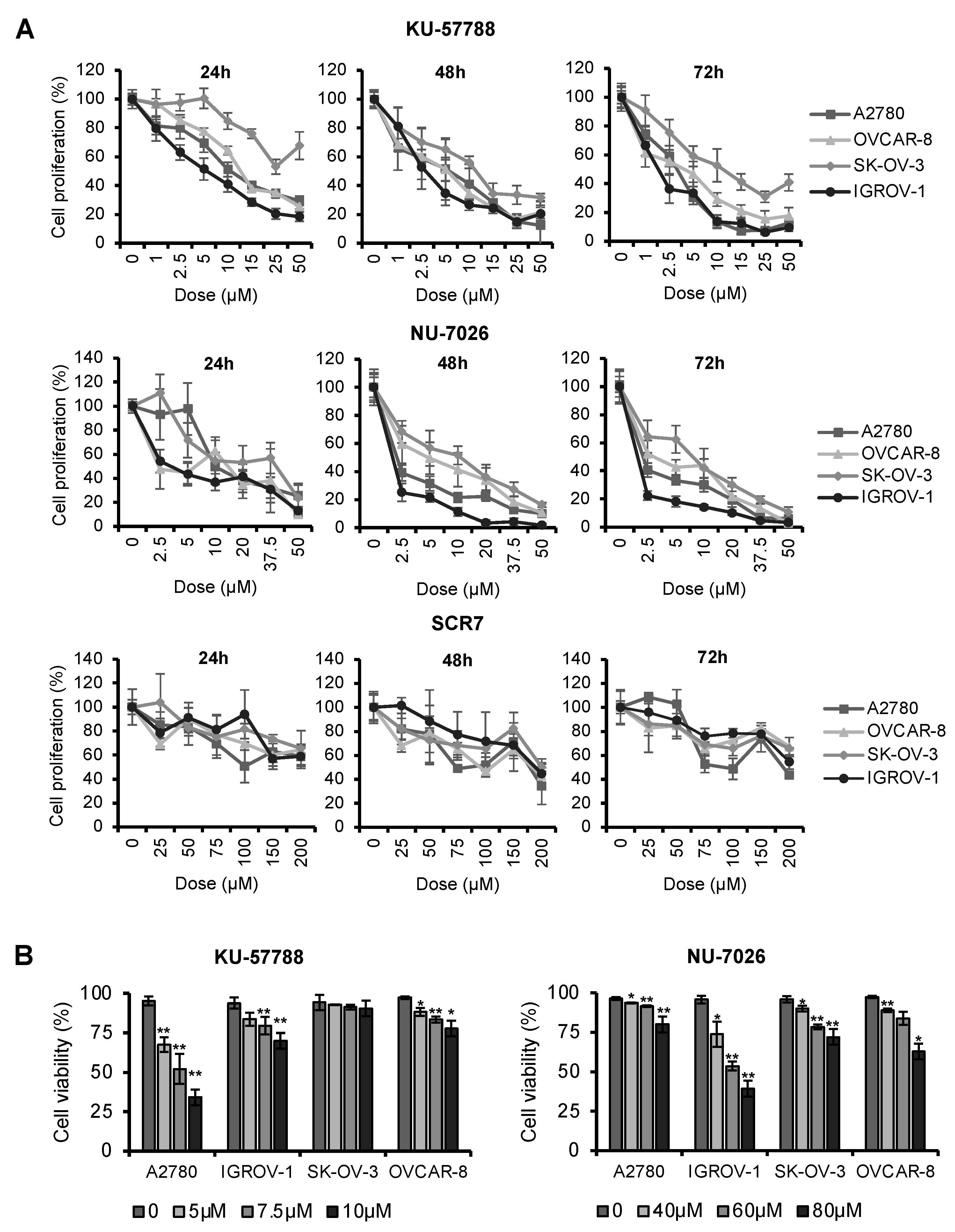
2.3. NHEJ Inhibitors Decrease Proliferation Rates and Induce Apoptosis in OCCLs
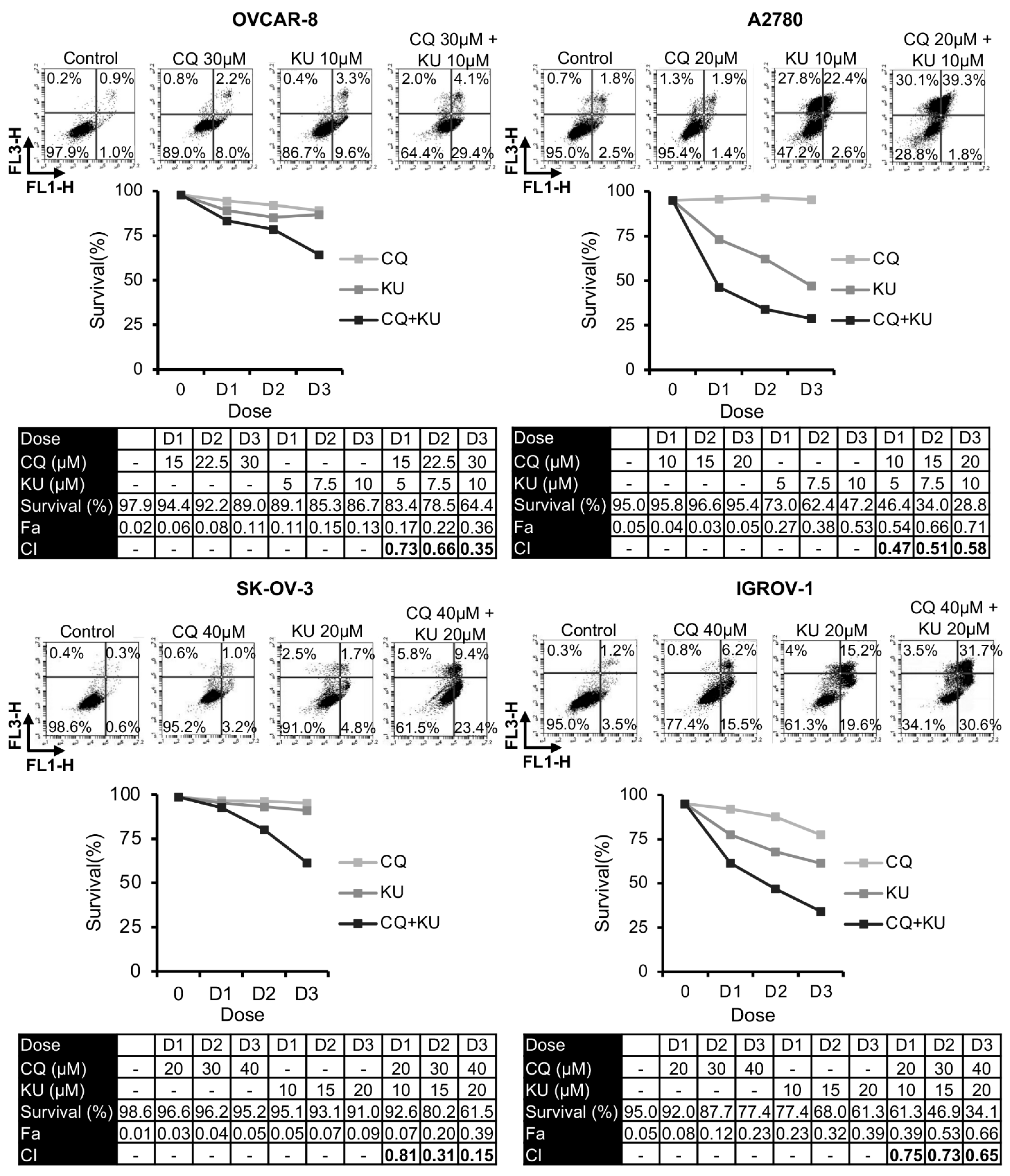
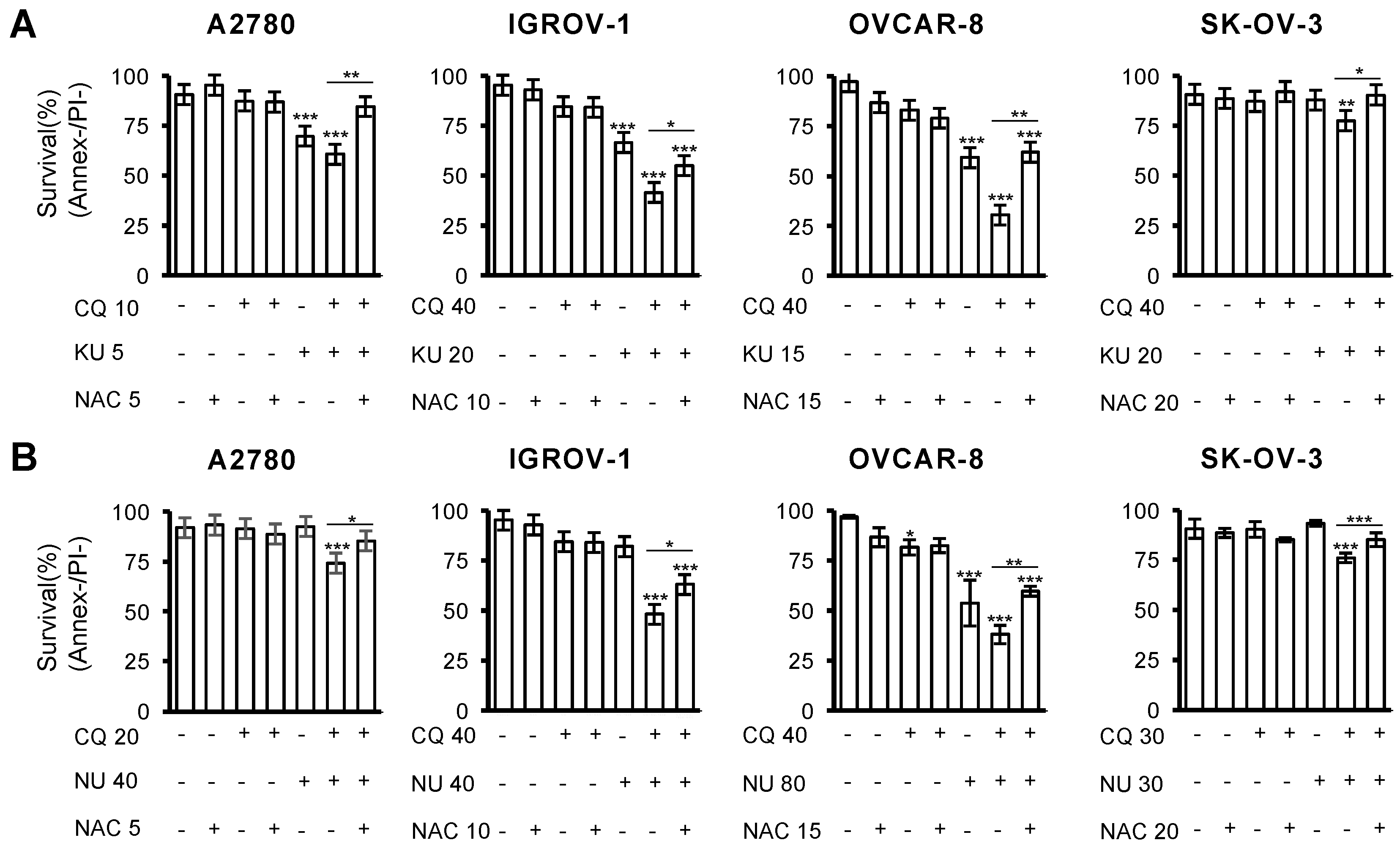
2.4. Combination of Chloroquine and NHEJ Inhibitors Synergistically Induces Cell Death in OCCLs
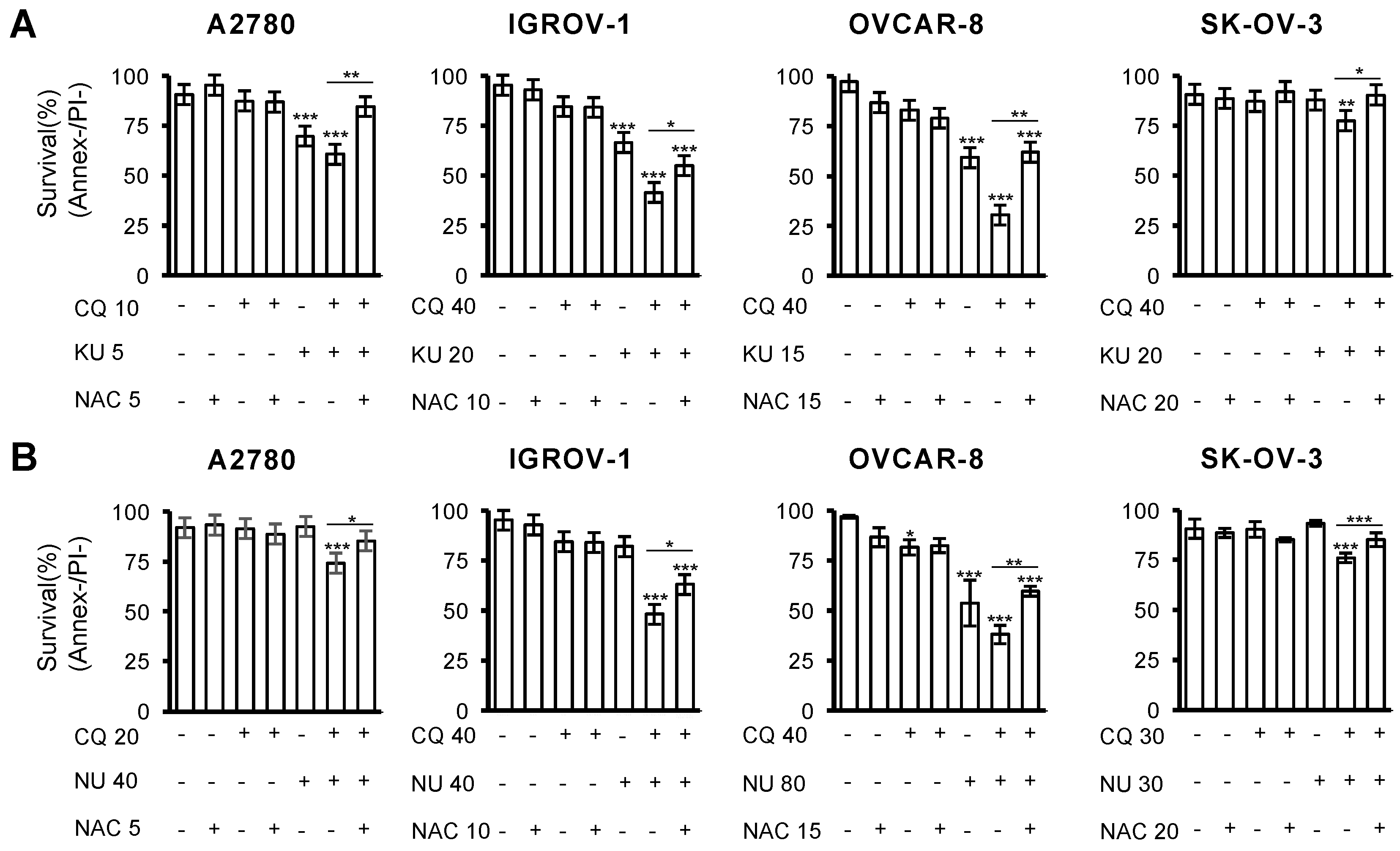
2.5. Cell Death Induced by the Combination of CQ and NHEJ Inhibitors Depends on ROS Production
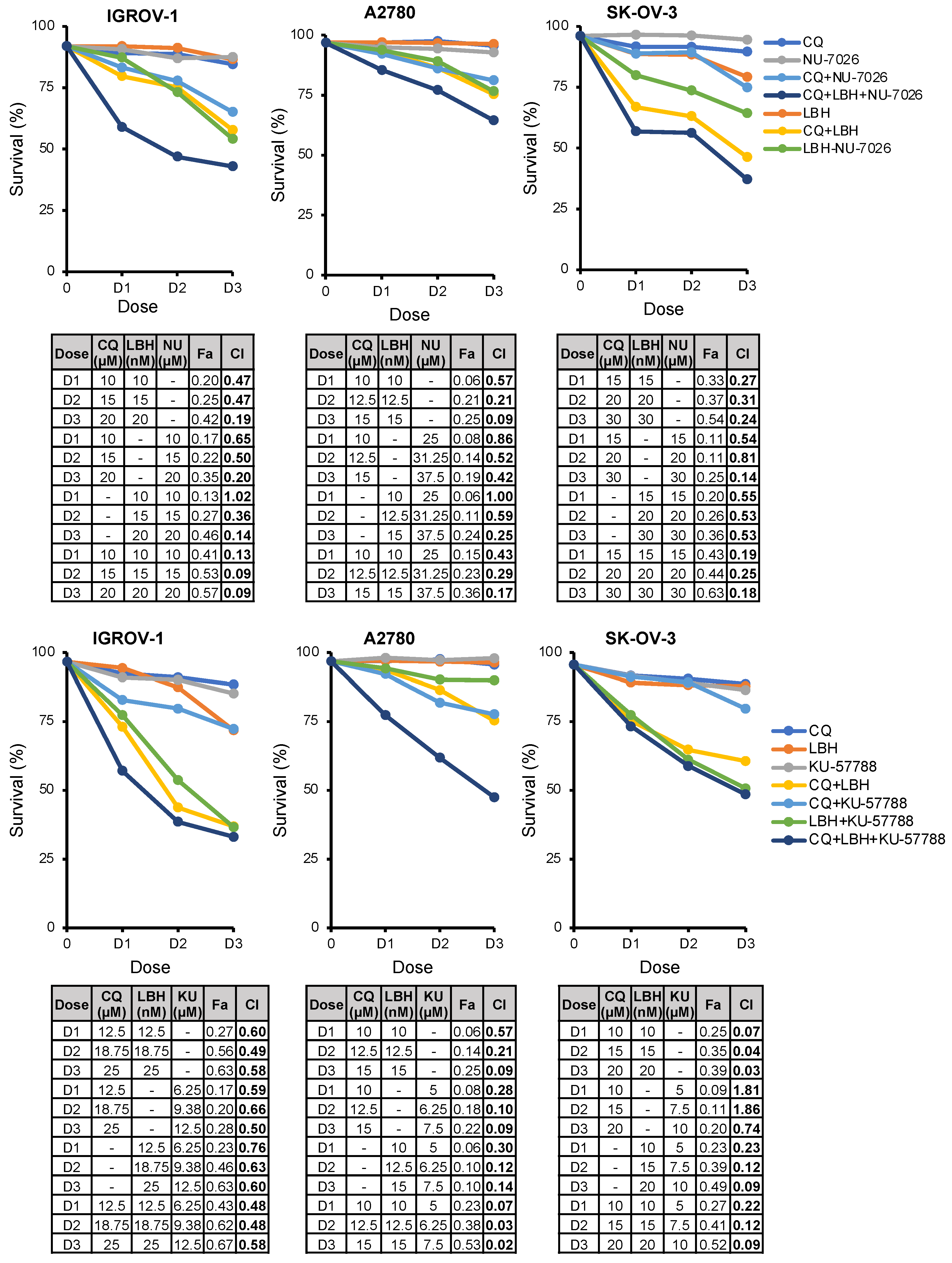
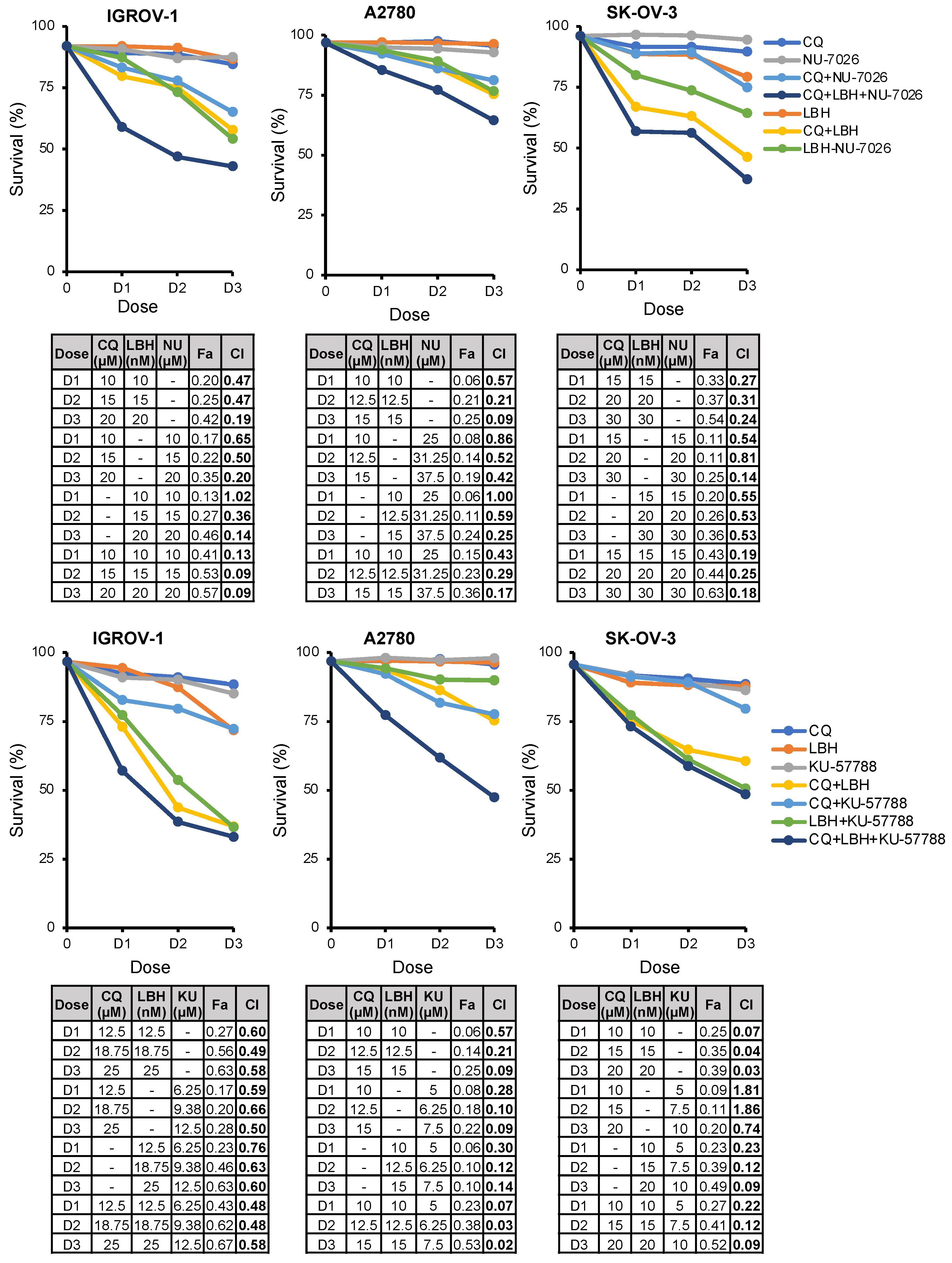
2.6. The Triple Combination CQ-NHEJi-LBH Exerts a Strong Synergy and Higher Efficacy Compared with Double Combinations
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture Conditions
4.2. Reagents
4.3. Western Blot
4.4. NHEJ Assays
4.5. Construction of Cell Lines for Detecting NHEJ Efficiency
4.6. Cell Proliferation Assay
4.7. Cell Cycle Analysis
4.8. Apoptosis Assay
4.9. Immunofluorescence
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA. Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Mancari, R.; Cutillo, G.; Bruno, V.; Vincenzoni, C.; Mancini, E.; Baiocco, E.; Bruni, S.; Vocaturo, G.; Chiofalo, B.; Vizza, E. Development of new medical treatment for epithelial ovarian cancer recurrence. Gland Surg. 2020, 9, 1149–1163. [Google Scholar] [CrossRef] [PubMed]
- Giornelli, G.H. Management of relapsed ovarian cancer: A review. Springerplus 2016, 5, 1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Han, Y.; Kim, S.I.; Kim, H.S.; Kim, S.J.; Song, Y.S. Tumor evolution and chemoresistance in ovarian cancer. NPJ Precis. Oncol. 2018, 2, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pokhriyal, R.; Hariprasad, R.; Kumar, L.; Hariprasad, G. Chemotherapy Resistance in Advanced Ovarian Cancer Patients. Biomark. Cancer 2019, 11, 1179299X19860815. [Google Scholar] [CrossRef]
- Miller, R.E.; El-Shakankery, K.H.; Lee, J.Y. PARP inhibitors in ovarian cancer: Overcoming resistance with combination strategies. J. Gynecol. Oncol. 2022, 33, e44. [Google Scholar] [CrossRef]
- Jin, C.; Yuan, M.; Bu, H.; Jin, C. Antiangiogenic Strategies in Epithelial Ovarian Cancer: Mechanism, Resistance, and Combination Therapy. J. Oncol. 2022, 2022, 4880355. [Google Scholar] [CrossRef]
- Al-Bari, A.A. Chloroquine analogues in drug discovery: New directions of uses, mechanisms of actions and toxic manifestations from malaria to multifarious diseases. J. Antimicrob. Chemother. 2014, 70, 1608–1621. [Google Scholar] [CrossRef] [Green Version]
- Varisli, L.; Cen, O.; Vlahopoulos, S. Dissecting pharmacological effects of chloroquine in cancer treatment: Interference with inflammatory signaling pathways. Immunology 2020, 159, 257–278. [Google Scholar] [CrossRef] [Green Version]
- Maes, H.; Kuchnio, A.; Peric, A.; Moens, S.; Nys, K.; DeBock, K.; Quaegebeur, A.; Schoors, S.; Georgiadou, M.; Wouters, J.; et al. Tumor vessel normalization by chloroquine independent of autophagy. Cancer Cell 2014, 26, 190–206. [Google Scholar] [CrossRef] [Green Version]
- Verbaanderd, C.; Maes, H.; Schaaf, M.B.; Sukhatme, V.P.; Pantziarka, P.; Sukhatme, V.; Agostinis, P.; Bouche, G. Repurposing drugs in oncology (ReDO)—Chloroquine and hydroxychloroquine as anti-cancer agents. Ecancermedicalscience 2017, 11, 781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.; Sun, X.; Zhang, Q.; Chen, X.; Zhao, T.; Lu, L.; Zhang, J.; Hong, Y. Histone deacetylase inhibitor trichostatin a and autophagy inhibitor chloroquine synergistically exert anti-tumor activity in H-ras transformed breast epithelial cells. Mol. Med. Rep. 2018, 17, 4345–4350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kommalapati, V.K.; Kumar, D.; Tangutur, A.D. Inhibition of JNJ-26481585-mediated autophagy induces apoptosis via ROS activation and mitochondrial membrane potential disruption in neuroblastoma cells. Mol. Cell. Biochem. 2020, 468, 21–34. [Google Scholar] [CrossRef]
- Torgersen, M.L.; Engedal, N.; Bøe, S.O.; Hokland, P.; Simonsen, A. Targeting autophagy potentiates the apoptotic effect of histone deacetylase inhibitors in t(8;21) AML cells. Blood 2013, 122, 2467–2476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, M.; Zhu, W.; Lv, X.; Yang, Q.; Liu, X.; Xie, Y.; Tang, P.; Sun, L. Encapsulation of chloroquine and doxorubicin by MPEG-PLA to enhance anticancer effects by lysosomes inhibition in ovarian cancer. Int. J. Nanomed. 2018, 13, 8231–8245. [Google Scholar] [CrossRef] [Green Version]
- Xu, R.; Ji, Z.; Xu, C.; Zhu, J. The clinical value of using chloroquine or hydroxychloroquine as autophagy inhibitors in the treatment of cancers: A systematic review and meta-analysis. Medicine 2018, 97, e12912. [Google Scholar] [CrossRef]
- Draz, H.; Goldberg, A.A.; Tomlinson Guns, E.S.; Fazli, L.; Safe, S.; Sanderson, J.T. Autophagy inhibition improves the chemotherapeutic efficacy of cruciferous vegetable-derived diindolymethane in a murine prostate cancer xenograft model. Investig. New Drugs 2018, 36, 718–725. [Google Scholar] [CrossRef]
- Wang, F.; Tang, J.; Li, P.; Si, S.; Yu, H.; Yang, X.; Tao, J.; Lv, Q.; Gu, M.; Yang, H.; et al. Chloroquine Enhances the Radiosensitivity of Bladder Cancer Cells by Inhibiting Autophagy and Activating Apoptosis. Cell. Physiol. Biochem. 2018, 45, 54–66. [Google Scholar] [CrossRef]
- Ye, H.; Chen, M.; Cao, F.; Huang, H.; Zhan, R.; Zheng, X. Chloroquine, an autophagy inhibitor, potentiates the radiosensitivity of glioma initiating cells by inhibiting autophagy and activating apoptosis. BMC Neurol. 2016, 16, 178. [Google Scholar] [CrossRef] [Green Version]
- Sehgal, A.R.; Konig, H.; Johnson, D.E.; Tang, D.; Amaravadi, R.K.; Boyiadzis, M.; Lotze, M.T. You eat what you are: Autophagy inhibition as a therapeutic strategy in leukemia. Leukemia 2015, 29, 517–525. [Google Scholar] [CrossRef]
- Carew, J.S.; Medina, E.C.; Esquivel, J.A.; Mahalingam, D.; Swords, R.; Kelly, K.; Zhang, H.; Huang, P.; Mita, A.C.; Mita, M.M.; et al. Autophagy inhibition enhances vorinostat-induced apoptosis via ubiquitinated protein accumulation. J. Cell. Mol. Med. 2010, 14, 2448–2459. [Google Scholar] [CrossRef] [PubMed]
- Carew, J.S.; Nawrocki, S.T.; Kahue, C.N.; Zhang, H.; Yang, C.; Chung, L.; Houghton, J.A.; Huang, P.; Giles, F.J.; Cleveland, J.L. Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug resistance. Blood 2007, 110, 313–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, R.; Balusu, R.; Fiskus, W.; Mudunuru, U.; Venkannagari, S.; Chauhan, L.; Smith, J.E.; Hembruff, S.L.; Ha, K.; Atadja, P.; et al. Combination of pan-histone deacetylase inhibitor and autophagy inhibitor exerts superior efficacy against triple-negative human breast cancer cells. Mol. Cancer Ther. 2012, 11, 973–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masui, A.; Hamada, M.; Kameyama, H.; Wakabayashi, K.; Takasu, A.; Imai, T.; Iwai, S.; Yura, Y. Autophagy as a Survival Mechanism for Squamous Cell Carcinoma Cells in Endonuclease G-Mediated Apoptosis. PLoS ONE 2016, 11, e0162786. [Google Scholar] [CrossRef] [Green Version]
- Tan, Q.; Wang, M.; Yu, M.; Zhang, J.; Bristow, R.G.; Hill, R.P.; Tannock, I.F. Role of Autophagy as a Survival Mechanism for Hypoxic Cells in Tumors. Neoplasia 2016, 18, 347–355. [Google Scholar] [CrossRef]
- Altman, J.K.; Szilard, A.; Goussetis, D.J.; Sassano, A.; Colamonici, M.; Gounaris, E.; Frankfurt, O.; Giles, F.J.; Eklund, E.A.; Beauchamp, E.M.; et al. Autophagy is a survival mechanism of acute myeloid leukemia precursors during dual mTORC2/mTORC1 targeting. Clin. Cancer Res. 2014, 20, 2400–2409. [Google Scholar] [CrossRef] [Green Version]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q.; et al. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4, e838. [Google Scholar] [CrossRef]
- Maycotte, P.; Aryal, S.; Cummings, C.T.; Thorburn, J.; Morgan, M.J.; Thorburn, A. Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy 2012, 8, 200–212. [Google Scholar] [CrossRef] [Green Version]
- Enzenmüller, S.; Gonzalez, P.; Debatin, K.M.; Fulda, S. Chloroquine overcomes resistance of lung carcinoma cells to the dual PI3K/mTOR inhibitor PI103 by lysosome-mediated apoptosis. Anticancer. Drugs 2013, 24, 14–19. [Google Scholar] [CrossRef]
- Choi, D.S.; Blanco, E.; Kim, Y.S.; Rodriguez, A.A.; Zhao, H.; Huang, T.H.M.; Chen, C.L.; Jin, G.; Landis, M.D.; Burey, L.A.; et al. Chloroquine eliminates cancer stem cells through deregulation of Jak2 and DNMT1. Stem Cells 2014, 32, 2309–2323. [Google Scholar] [CrossRef] [Green Version]
- Eng, C.H.; Wang, Z.; Tkach, D.; Toral-Barza, L.; Ugwonali, S.; Liu, S.; Fitzgerald, S.L.; George, E.; Frias, E.; Cochran, N.; et al. Macroautophagy is dispensable for growth of KRAS mutant tumors and chloroquine efficacy. Proc. Natl. Acad. Sci. USA 2016, 113, 182–187. [Google Scholar] [CrossRef] [Green Version]
- Ovejero-Sánchez, M.; González-Sarmiento, R.; Herrero, A.B. Synergistic effect of Chloroquine and Panobinostat in ovarian cancer through induction of DNA damage and inhibition of DNA repair. Neoplasia 2021, 23, 515–528. [Google Scholar] [CrossRef]
- Scott, S.P.; Pandita, T.K. The cellular control of DNA double-strand breaks. J. Cell. Biochem. 2006, 99, 1463–1475. [Google Scholar] [CrossRef] [Green Version]
- Robert, C.; Nagaria, P.K.; Pawar, N.; Adewuyi, A.; Gojo, I.; Meyers, D.J.; Cole, P.A.; Rassool, F.V. Histone deacetylase inhibitors decrease NHEJ both by acetylation of repair factors and trapping of PARP1 at DNA double-strand breaks in chromatin. Leuk. Res. 2016, 45, 14–23. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.; Fox, J.; Mejia, M.; Ruangpradit, W.; Saberi, A.; Kim, S.; Choi, Y.; Oh, S.; Wang, Y.; Choi, K.; et al. Histone Deacetylase Inhibitors Selectively Target Homology Dependent DNA Repair Defective Cells and Elevate Non-Homologous Endjoining Activity. PLoS ONE 2014, 9, e87203. [Google Scholar] [CrossRef]
- Slupphaug, G.; Kavli, B.; Krokan, H.E. The interacting pathways for prevention and repair of oxidative DNA damage. Mutat. Res. 2003, 531, 231–251. [Google Scholar] [CrossRef]
- Munshi, A.; Kurland, J.F.; Nishikawa, T.; Tanaka, T.; Hobbs, M.L.; Tucker, S.L.; Ismail, S.; Stevens, C.; Meyn, R.E. Histone deacetylase inhibitors radiosensitize human melanoma cells by suppressing DNA repair activity. Clin. Cancer Res. 2005, 11, 4912–4922. [Google Scholar] [CrossRef] [Green Version]
- Munshi, A.; Tanaka, T.; Hobbs, M.L.; Tucker, S.L.; Richon, V.M.; Meyn, R.E. Vorinostat, a histone deacetylase inhibitor, enhances the response of human tumor cells to ionizing radiation through prolongation of gamma-H2AX foci. Mol. Cancer Ther. 2006, 5, 1967–1974. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Zhang, T.; Teng, Z.H.; Zhang, R.; Wang, J.B.; Mei, Q.B. Sensitization to gamma-irradiation-induced cell cycle arrest and apoptosis by the histone deacetylase inhibitor trichostatin A in non-small cell lung cancer (NSCLC) cells. Cancer Biol. Ther. 2009, 8, 823–831. [Google Scholar] [CrossRef] [Green Version]
- López-Iglesias, A.A.; Herrero, A.B.; Chesi, M.; San-Segundo, L.; González-Méndez, L.; Hernández-García, S.; Misiewicz-Krzeminska, I.; Quwaider, D.; Martín-Sánchez, M.; Primo, D.; et al. Preclinical anti-myeloma activity of EDO-S101, a new bendamustine-derived molecule with added HDACi activity, through potent DNA damage induction and impairment of DNA repair. J. Hematol. Oncol. 2017, 10, 127. [Google Scholar] [CrossRef]
- Wilson, A.J.; Sarfo-Kantanka, K.; Barrack, T.; Steck, A.; Saskowski, J.; Crispens, M.A.; Khabele, D. Panobinostat sensitizes cyclin E high, homologous recombination-proficient ovarian cancer to olaparib. Gynecol. Oncol. 2016, 143, 143–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Andrade, P.V.; Andrade, A.F.; de Paula Queiroz, R.G.; Scrideli, C.A.; Tone, L.G.; Valera, E.T. The histone deacetylase inhibitor PCI-24781 as a putative radiosensitizer in pediatric glioblastoma cell lines. Cancer Cell Int. 2016, 16, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anne, M.; Sammartino, D.; Barginear, M.F.; Budman, D. Profile of panobinostat and its potential for treatment in solid tumors: An update. OncoTargets Ther. 2013, 6, 1613–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helland, Ø.; Popa, M.; Bischof, K.; Gjertsen, B.T.; McCormack, E.; Bjørge, L. The HDACi Panobinostat Shows Growth Inhibition Both In Vitro and in a Bioluminescent Orthotopic Surgical Xenograft Model of Ovarian Cancer. PLoS ONE 2016, 11, e0158208. [Google Scholar] [CrossRef] [Green Version]
- Smith, H.J.; Straughn, J.M.; Buchsbaum, D.J.; Arend, R.C. Epigenetic therapy for the treatment of epithelial ovarian cancer: A clinical review. Gynecol. Oncol. Rep. 2017, 20, 81–86. [Google Scholar] [CrossRef]
- Wilson, A.J.; Gupta, V.G.; Liu, Q.; Yull, F.; Crispens, M.A.; Khabele, D. Panobinostat enhances olaparib efficacy by modifying expression of homologous recombination repair and immune transcripts in ovarian cancer. Neoplasia 2022, 24, 63–75. [Google Scholar] [CrossRef]
- Herrero, A.B.; Gutiérrez, N.C. Targeting ongoing DNA damage in multiple myeloma: Effects of DNA damage response inhibitors on plasma cell survival. Front. Oncol. 2017, 7, 98. [Google Scholar] [CrossRef] [Green Version]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair 2008, 7, 1765–1771. [Google Scholar] [CrossRef] [Green Version]
- Pare, J.; Choi, K.; Jeong, E.; Kwon, D.; Benveniste, E.N.; Choi, C. Reactive oxygen species mediate chloroquine-induced expression of chemokines by human astroglial cells. Glia 2004, 47, 9–20. [Google Scholar] [CrossRef]
- Qu, X.; Sheng, J.; Shen, L.; Su, J.; Xu, Y.; Xie, Q.; Wu, Y.; Zhang, X.; Sun, L. Autophagy inhibitor chloroquine increases sensitivity to cisplatin in QBC939 cholangiocarcinoma cells by mitochondrial ROS. PLoS ONE 2017, 12, e0173712. [Google Scholar] [CrossRef]
- Ganguli, A.; Choudhury, D.; Datta, S.; Bhattacharya, S.; Chakrabarti, G. Inhibition of autophagy by chloroquine potentiates synergistically anti-cancer property of artemisinin by promoting ROS dependent apoptosis. Biochimie 2014, 107, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Park, D.; Lee, Y. Biphasic Activity of Chloroquine in Human Colorectal Cancer Cells. Dev. Reprod. 2014, 18, 225–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandsma, I.; Gent, D.C. Pathway choice in DNA double strand break repair: Observations of a balancing act. Genome Integr. 2012, 3, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Liang, S.; Ochi, T.; Chirgadze, D.Y.; Huiskonen, J.T.; Blundell, T.L. Understanding the structure and role of DNA-PK in NHEJ: How X-ray diffraction and cryo-EM contribute in complementary ways. Prog. Biophys. Mol. Biol. 2019, 147, 26–32. [Google Scholar] [CrossRef]
- Ciszewski, W.M.; Tavecchio, M.; Dastych, J.; Curtin, N.J. DNA-PK inhibition by NU7441 sensitizes breast cancer cells to ionizing radiation and doxorubicin. Breast Cancer Res. Treat. 2014, 143, 47–55. [Google Scholar] [CrossRef]
- Shaheen, F.S.; Znojek, P.; Fisher, A.; Webster, M.; Plummer, R.; Gaughan, L.; Smith, G.C.M.; Leung, H.Y.; Curtin, N.J.; Robson, C.N. Targeting the dna double strand break repair machinery in prostate cancer. PLoS ONE 2011, 6, e20311. [Google Scholar] [CrossRef]
- Alikarami, F.; Safa, M.; Faranoush, M.; Hayat, P.; Kazemi, A. Inhibition of DNA-PK enhances chemosensitivity of B-cell precursor acute lymphoblastic leukemia cells to doxorubicin. Biomed. Pharmacother. 2017, 94, 1077–1093. [Google Scholar] [CrossRef]
- Tichý, A.; Novotná, E.; Durisová, K.; Salovská, B.; Sedlaríková, R.; Pejchal, J.; Zárybnická, L.; Vávrová, J.; Sinkorová, Z.; Rezácová, M. Radio-sensitization of human leukaemic molt-4 cells by DNA-dependent protein kinase inhibitor, NU7026. Acta Med. (Hradec Králové) 2012, 55, 66–73. [Google Scholar] [CrossRef] [Green Version]
- Baker, N.C.; Ekins, S.; Williams, A.J.; Tropsha, A. A bibliometric review of drug repurposing. Drug Discov. Today 2018, 23, 661–672. [Google Scholar] [CrossRef]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Chen, D.; Xie, J.; Fiskesund, R.; Dong, W.; Liang, X.; Lv, J.; Jin, X.; Liu, J.; Mo, S.; Zhang, T.; et al. Chloroquine modulates antitumor immune response by resetting tumor-associated macrophages toward M1 phenotype. Nat. Commun. 2018, 9, 873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, T.; Li, P.; Luo, Z.; Chen, X.; Zhang, J.; Wang, C.; Chen, P.; Dong, Z. Chloroquine inhibits hepatocellular carcinoma cell growth in vitro and in vivo. Oncol. Rep. 2016, 35, 43–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farombi, E.O. Genotoxicity of chloroquine in rat liver cells: Protective role of free radical scavengers. Cell Biol. Toxicol. 2006, 22, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Poillet-Perez, L.; Despouy, G.; Delage-Mourroux, R.; Boyer-Guittaut, M. Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biol. 2015, 4, 184–192. [Google Scholar] [CrossRef] [Green Version]
- Liang, D.H.; Choi, D.S.; Ensor, J.E.; Kaipparettu, B.A.; Bass, B.L.; Chang, J.C. The autophagy inhibitor chloroquine targets cancer stem cells in triple negative breast cancer by inducing mitochondrial damage and impairing DNA break repair. Cancer Lett. 2016, 376, 249–258. [Google Scholar] [CrossRef] [Green Version]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, M.; Raghavan, S.C. DNA Double-Strand Break Repair Inhibitors as Cancer Therapeutics. Chem. Biol. 2015, 22, 17–29. [Google Scholar] [CrossRef] [Green Version]
- Restle, A.; Färber, M.; Baumann, C.; Böhringer, M.; Scheidtmann, K.H.; Müller-Tidow, C.; Wiesmüller, L. Dissecting the role of p53 phosphorylation in homologous recombination provides new clues for gain-of-function mutants. Nucleic Acids Res. 2008, 36, 5362–5375. [Google Scholar] [CrossRef]
- Kumar, R.J.; Chao, H.X.; Simpson, D.A.; Feng, W.; Cho, M.-G.; Roberts, V.R.; Sullivan, A.R.; Shah, S.J.; Wozny, A.-S.; Fagan-Solis, K.; et al. Dual inhibition of DNA-PK and DNA polymerase theta overcomes radiation resistance induced by p53 deficiency. NAR Cancer 2020, 2, zcaa038. [Google Scholar] [CrossRef]
- Willmore, E.; De Caux, S.; Sunter, N.J.; Tilby, M.J.; Jackson, G.H.; Austin, C.A.; Durkacz, B.W. A novel DNA-dependent protein kinase inhibitor, NU7026, potentiates the cytotoxicity of topoisomerase II poisons used in the treatment of leukemia. Blood 2004, 103, 4659–4665. [Google Scholar] [CrossRef]
- Davidson, D.; Coulombe, Y.; Martinez-Marignac, V.L.; Amrein, L.; Grenier, J.; Hodkinson, K.; Masson, J.Y.; Aloyz, R.; Panasci, L. Irinotecan and DNA-PKcs inhibitors synergize in killing of colon cancer cells. Investig. New Drugs 2012, 30, 1248–1256. [Google Scholar] [CrossRef] [PubMed]
- Veuger, S.J.; Curtin, N.J.; Richardson, C.J.; Smith, G.C.M.; Durkacz, B.W. Radiosensitization and DNA repair inhibition by the combined use of novel inhibitors of DNA-dependent protein kinase and poly(ADP-ribose) polymerase-1. Cancer Res. 2003, 63, 6008–6015. [Google Scholar] [PubMed]
- Lee, T.W.; Wong, W.W.; Dickson, B.D.; Lipert, B.; Cheng, G.J.; Hunter, F.W.; Hay, M.P.; Wilson, W.R. Radiosensitization of head and neck squamous cell carcinoma lines by DNA-PK inhibitors is more effective than PARP-1 inhibition and is enhanced by SLFN11 and hypoxia. Int. J. Radiat. Biol. 2019, 95, 1597–1612. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Thomas, H.D.; Batey, M.A.; Cowell, I.G.; Richardson, C.J.; Griffin, R.J.; Calvert, A.H.; Newell, D.R.; Smith, G.C.M.; Curtin, N.J. Preclinical evaluation of a potent novel DNA-dependent protein kinase inhibitor NU7441. Cancer Res. 2006, 66, 5354–5362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medová, M.; Medo, M.; Hovhannisyan, L.; Muñoz-Maldonado, C.; Aebersold, D.M.; Zimmer, Y. DNA-PK in human malignant disorders: Mechanisms and implications for pharmacological interventions. Pharmacol. Ther. 2020, 215, 107617. [Google Scholar] [CrossRef] [PubMed]
- Gordhandas, S.B.; Manning-Geist, B.; Henson, C.; Iyer, G.; Gardner, G.J.; Sonoda, Y.; Moore, K.N.; Aghajanian, C.; Chui, M.H.; Grisham, R.N. Pre-clinical activity of the oral DNA-PK inhibitor, peposertib (M3814), combined with radiation in xenograft models of cervical cancer. Sci. Rep. 2022, 12, 974. [Google Scholar] [CrossRef]
- Wise, H.C.; Iyer, G.V.; Moore, K.; Temkin, S.M.; Gordon, S.; Aghajanian, C.; Grisham, R.N. Activity of M3814, an Oral DNA-PK Inhibitor, In Combination with Topoisomerase II Inhibitors in Ovarian Cancer Models. Sci. Rep. 2019, 9, 18882. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, M.; Nambiar, M.; Sharma, S.; Karki, S.S.; Goldsmith, G.; Hegde, M.; Kumar, S.; Pandey, M.; Singh, R.K.; Ray, P.; et al. An inhibitor of nonhomologous end-joining abrogates double-strand break repair and impedes cancer progression. Cell 2012, 151, 1474–1487. [Google Scholar] [CrossRef] [Green Version]
- Bartek, J. DNA damage response, genetic instability and cancer: From mechanistic insights to personalized treatment. Mol. Oncol. 2011, 5, 303–307. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Lian, G. ROS and diseases: Role in metabolism and energy supply. Mol. Cell. Biochem. 2020, 467, 1–12. [Google Scholar] [CrossRef]
- Fok, J.H.L.; Ramos-Montoya, A.; Vazquez-Chantada, M.; Wijnhoven, P.W.G.; Follia, V.; James, N.; Farrington, P.M.; Karmokar, A.; Willis, S.E.; Cairns, J.; et al. AZD7648 is a potent and selective DNA-PK inhibitor that enhances radiation, chemotherapy and olaparib activity. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.X.; Ni, X.L.; Zhang, H.; Wu, M.; Liu, J.; Xu, S.; Yang, L.L.; Fu, S.Z.; Wu, J. Preparation, characterization, in vitro and in vivo anti-tumor effect of thalidomide nanoparticles on lung cancer. Int. J. Nanomed. 2018, 13, 2463–2476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, X.; Lara, H.; Wagner, K.T.; Saripalli, S.; Hyder, S.N.; Foote, M.; Sethi, M.; Wang, E.; Caster, J.M.; Zhang, L.; et al. Improving DNA double-strand repair inhibitor KU55933 therapeutic index in cancer radiotherapy using nanoparticle drug delivery. Nanoscale 2015, 7, 20211–20219. [Google Scholar] [CrossRef] [Green Version]
- Belhadj, Z.; Zhan, C.; Ying, M.; Wei, X.; Xie, C.; Yan, Z.; Lu, W. Multifunctional targeted liposomal drug delivery for efficient glioblastoma treatment. Oncotarget 2017, 8, 66889–66900. [Google Scholar] [CrossRef] [Green Version]
- Seluanov, A.; Mittelman, D.; Pereira-Smith, O.M.; Wilson, J.H.; Gorbunova, V. DNA end joining becomes less efficient and more error-prone during cellular senescence. Proc. Natl. Acad. Sci. USA 2004, 101, 7624–7629. [Google Scholar] [CrossRef] [Green Version]
- Chicaybam, L.; Barcelos, C.; Peixoto, B.; Carneiro, M.; Limia, C.G.; Redondo, P.; Lira, C.; Paraguassú-Braga, F.; De Vasconcelos, Z.F.M.; Barros, L.; et al. An Efficient Electroporation Protocol for the Genetic Modification of Mammalian Cells. Front. Bioeng. Biotechnol. 2017, 4, 99. [Google Scholar] [CrossRef] [Green Version]
- Chou, T.C. Drug combination studies and their synergy quantification using the chou-talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [Green Version]
- The Jamovi Project. Jamovi (Version 2.2) [Computer Software]. 2021. Available online: https://www.jamovi.org (accessed on 25 April 2022).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | A2780 | IGROV-1 | OVCAR-8 | SK-OV-3 |
|---|---|---|---|---|
| Inhibitors | IC50 (95% CI) | IC50 (95% CI) | IC50 (95% CI) | IC50 (95% CI) |
| KU-57788 | 2.56 µM (2.25–2.89 µM) | 1.86 µM (1.62–2.12 µM) | 3.42 µM (2.81–4.14 µM) | 11.07 µM (9.07–13.51 µM) |
| NU-7026 | 2.51 µM (2.10–2.97 µM) | 0.96 µM (0.81–1.12 µM) | 4.27 µM (3.45–5.26 µM) | 7.30 µM (6.38–8.36 µM) |
| SCR7 | 204.5 µM (129.7–348.8 µM) | 329.8 µM (253.1–446.9 µM) | 305.4 µM (223.2–439.4 µM) | 269.5 µM (202.3–373.1 µM) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ovejero-Sánchez, M.; Rubio-Heras, J.; Vicente de la Peña, M.d.C.; San-Segundo, L.; Pérez-Losada, J.; González-Sarmiento, R.; Herrero, A.B. Chloroquine-Induced DNA Damage Synergizes with Nonhomologous End Joining Inhibition to Cause Ovarian Cancer Cell Cytotoxicity. Int. J. Mol. Sci. 2022, 23, 7518. https://doi.org/10.3390/ijms23147518
Ovejero-Sánchez M, Rubio-Heras J, Vicente de la Peña MdC, San-Segundo L, Pérez-Losada J, González-Sarmiento R, Herrero AB. Chloroquine-Induced DNA Damage Synergizes with Nonhomologous End Joining Inhibition to Cause Ovarian Cancer Cell Cytotoxicity. International Journal of Molecular Sciences. 2022; 23(14):7518. https://doi.org/10.3390/ijms23147518
Chicago/Turabian StyleOvejero-Sánchez, María, Jorge Rubio-Heras, María del Carmen Vicente de la Peña, Laura San-Segundo, Jesús Pérez-Losada, Rogelio González-Sarmiento, and Ana Belén Herrero. 2022. "Chloroquine-Induced DNA Damage Synergizes with Nonhomologous End Joining Inhibition to Cause Ovarian Cancer Cell Cytotoxicity" International Journal of Molecular Sciences 23, no. 14: 7518. https://doi.org/10.3390/ijms23147518
APA StyleOvejero-Sánchez, M., Rubio-Heras, J., Vicente de la Peña, M. d. C., San-Segundo, L., Pérez-Losada, J., González-Sarmiento, R., & Herrero, A. B. (2022). Chloroquine-Induced DNA Damage Synergizes with Nonhomologous End Joining Inhibition to Cause Ovarian Cancer Cell Cytotoxicity. International Journal of Molecular Sciences, 23(14), 7518. https://doi.org/10.3390/ijms23147518