Abstract
Some prevention strategies, including vaccines and antibiotic alternatives, have been developed to reduce enterotoxigenic Escherichia coli proliferation in animal production. In this study, a wild-type strain of BE311 with a virulent heat-stable enterotoxin gene identical to E. coli K99 was isolated for its high potential for gene expression ability. The whole genome of E. coli BE311 was sequenced for gene analyses and editing. Subsequently, the fluorescent gene mCherry was successfully knocked into the genome of E. coli BE311 by CRISPR/Cas9. The E. coli BE311–mCherry strain was precisely quantified through the fluorescence intensity and red colony counting. The inflammatory factors in different intestinal tissues all increased significantly after an E. coli BE311–mCherry challenge in Sprague–Dawley rats (p < 0.05). The heat-stable enterotoxin gene of E. coli BE311 was knocked out, and an attenuated vaccine host E. coli BE311-STKO was constructed. Flow cytometry showed apoptotic cell numbers were lower following a challenge of IPEC-J2 cells with E. coli BE311-STKO than with E. coli BE311. Therefore, the E. coli BE311–mCherry and E. coli BE311-STKO strains that were successfully constructed based on the gene knock-in and knock-out technology could be used as ideal candidates in ETEC challenge models and for the development of attenuated vaccines.
1. Introduction
Enterotoxigenic Escherichia coli (ETEC) is one of the main causes of diarrhea in farm animals, including piglets, calves, and lambs [1], and infections by ETEC in animal husbandry and aquaculture result in severe economic losses due to high animal morbidity and mortality [2]. Generally, ETEC in an exogenous environment enters animals orally and is subsequently recognized by specific receptors in the small intestine. It attaches to the intestinal epithelium using a colonizing factor antigen (also called adhesin) and then colonizes the small intestine [3]. After this colonization, ETEC begins rapid proliferation, reaching 109 colony forming units (CFU) per gram of tissue. This ETEC infection directly leads to intestinal and systemic inflammation, gut barrier dysfunction, and the subsequent growth failure of infected animals [4].
The expression of virulence factors, such as adhesin and enterotoxins, by ETEC is responsible for its pathogenicity. During the process of infection, ETEC binds to small intestinal enterocytes by adhesin and then secretes heat-labile (LT) and/or heat-stable (ST) enterotoxins, which disrupt the intestinal barrier function and cause watery diarrhea [5]. For many years, antibiotics have been widely used in animal production for the prevention and control of animal diarrhea by targeting ETEC. However, the abuse of antibiotics greatly increases safety risks by promoting antibiotic resistance. Some alternative prevention strategies, including vaccines and antibiotic alternatives [6,7,8,9], are required to reduce ETEC infection in animal production.
The ETEC strains can be modified by DNA recombinant technology to retain the desired immunogenicity while selectively eliminating toxicity, resulting in a safer and more effective vaccine suitable for commercial production [10]. The colonization factors of CS6 fimbriae have been targeted for recombinant engineered expression as potential vaccines and have provided protection against ETEC infection in the Aotus nancymaae diarrheal challenge model [11]. Live attenuated vaccines are also being actively pursued in vaccine research. For example, the live-attenuated ACE527 is a live, oral, multivalent vaccine that consists of three genetically attenuated and engineered strains of ETEC and contains antigens covering a wide range of ETEC surface colonization factors (CFA/I, CFA/II, and CFA/IV) as well as LT-B, the binding subunit of the LT toxin [12,13,14].
Live ETEC with toxin-free mutations can also be effective oral vaccines and can be used as recipient strains for the development of multivalent vaccines [15,16,17]. For example, the spontaneous mutant E1392/75-2A, which was engineered for the loss of the ST and LT toxins but continued to express CFA/II, provided 75% protection against a challenge with an LT+, ST+, and CFA/II+ ETEC strain [18]. Similarly, re-inoculation with an avirulent strain expressing adhesive fimbriae and a non-toxic form of LT provided significant short-term protection in young piglets from a challenge with a virulent ETEC strain expressing the same fimbrial adhesin and enterotoxin [19]. Therefore, ETEC-based engineered modifications are of special importance for ETEC vaccine development and enterotoxin analyses.
The evaluation of the protective efficiency of ETEC vaccine candidates requires a suitable animal infection model [11,18,19,20] that is also suitable for the development of antibiotic alternatives for in vivo anti-ETEC activities [21,22,23,24]. Reported ETEC infection models have typically used methods such as plate counting, quantitative PCR for unique virulence genes, and an enzyme-linked immunosorbent assay (ELISA) to detect and quantify ETEC [25,26]. However, ETEC is also widespread in the environment, such as in food, water, and bioaerosols [27,28,29], which renders the quantification of ETEC less specific and poorly targeted. A sensitive, selective, and rapid platform for ETEC detection is, therefore, urgently needed for challenge models.
Recently, the heterologous luciferase firefly and fluorescent protein genes have been transferred into recipient bacterial strains to provide the rapid, accurate, and visual quantification of bacteria [30,31]. Fluorescent protein genes have an advantage over reporter genes such as luciferase because the protein products spontaneously fluoresce without the addition of any substrate or cofactor [32]. The genome integration of heterologous genes has also been demonstrated to be better than plasmid-borne overexpression in terms of an increased stability and lower metabolic burden [33]. It is worth mentioning that the CRISPR/Cas9 system is one of the most powerful and revolutionary genome editing tools used to precisely manipulate the genome of various organisms [34]. Organisms of interest can now be labeled using specific-site knock-in and the integrated expression of exogenous fluorescent proteins in hosts based on CRISPR/Cas9. In the case of the ETEC infection, the underlying mechanism can be better understood by various gene modifications, including overexpression, gene deletion, or the precise regulation of virulence factors, to aid in uncovering their functions [35]. A CRISPR-Cas9 system-based continual genome editing strategy, including gene insertions and knock-outs of both single and multiple (up to three) targets in the E. coli genome, has been developed and is now widely applied [36,37].
In this work, our aim was to use the CRISPR-Cas9 system technology to directly modify ETEC for specific fluorescence labeling and enterotoxin analyses, based on the ability of ETEC strains to perform heterologous gene expression. The resulting modified strain was expected to serve as an attenuated host for vaccine development and as a general platform for ETEC-specific detection in ETEC challenge models.
2. Results
2.1. Identification and Characterization of E. coli BE311
The recipient strains and expression vectors that showed the confirmed expression of exogenous genes were specifically selected for further gene editing. Table 1 shows that pQE30-GFP was the most effective expression vector in the E. coli strains. The E. coli Top10, K99, and BE311 showed the strongest fluorescence as recipient strains after transformation with pQE30-GFP. The fluorescence images of colonies showed a similarly strong GFP fluorescence intensity in E. coli BE311 to that in E. coli K99 and Top10 (Figure 1A).

Table 1.
The GFP-expressing ability of different E. coli strains and vectors.
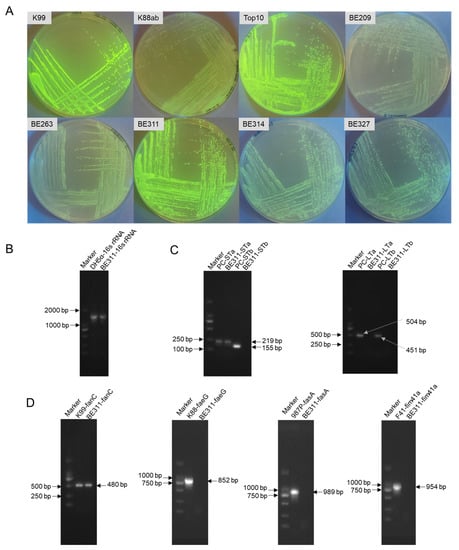
Figure 1.
Identification and characterization of E. coli BE311. (A), The expression of pQE30-GFP in different E. coil strains. (B), PCR amplification of the 16S rRNA of E. coli BE311. (C), Identification of the enterotoxins in E. coli BE311 by agarose gel electrophoresis (AGE). (D), Identification of the adhesions in E. coli BE311 by AGE.
The 16S rRNA amplification (Figure 1B) and sequencing confirmed that the BE311 strain belonged to Escherichia coli. The extracted genomic DNA of BE311 was used as a template to identify the virulence factors contained in that strain. As shown in Figure 1C, the type of enterotoxins harbored in E. coli BE311 was tested, and the strains containing STa (219 bp), STb (155 bp), LTa (504 bp), and LTb (451 bp) were used as positive controls. Only the STa enterotoxin gene was present in BE311 (Figure 1C). The K99 (480 bp), K88 (852 bp), 987P (989 bp), and F41 (954 bp) adhesins were successively amplified and served as positive controls. The same primers were used to amplify the BE311 strain adhesins, and the results showed that E. coli BE311 contained a fanC adhesin identical to the one from E. coli K99. This confirmed the successful isolation of a wild-type ETEC, designated the BE311 strain, that harbored K99 (F5) fimbriae. The BE311 strain was deposited in the China Center for Type Culture Collection (Wuhan, China) and named E. coli ETEC BE311 (CCTCC NO: M2019733).
2.2. Whole Genome Sequencing of E. coli BE311
The whole genome sequencing showed that the chromosome of E. coli BE311 was 4,840,702 bp (50.74% G + C content) long and encoded 4622 genes (Figure 2A) annotated to Refseq. The whole genome sequence of the E. coli BE311 chromosome was deposited in GenBank with the Accession Number SUB11437696. The complete sequence of E. coli BE311 was compared to the VFDB database to identify potential virulence factors. The E. coli BE311 chromosome encodes a variety of potential virulence factors (Supplementary Table S1), including adhesions, toxins, and invasion proteins. Overall, 1364 candidate pseudogenes were identified in the E. coli BE311 genome, and the total length of the predicted pseudogenes was 323,478 bp (Supplementary Figure S1).
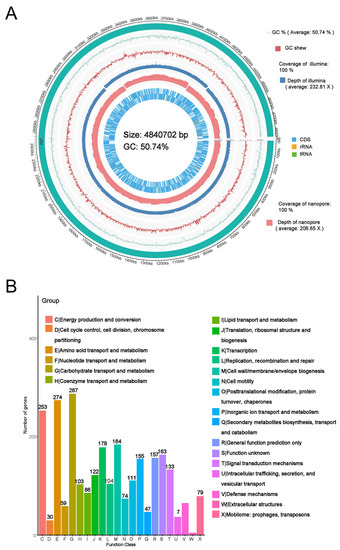
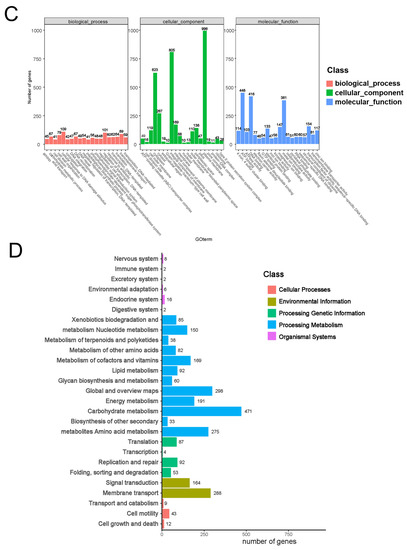
Figure 2.
Whole genome sequencing of E. coli BE311. (A), The circus plot of E. coli BE311 genome. (B), The COG analysis of E. coli BE311 genome. (C), GO analysis of E. coli BE311 genome. (D), KEGG analysis of E. coli BE311 genome.
Bias in the functional content distribution across the replicons in the genome was analyzed using Clusters of Orthologous Groups (COG) of proteins. The analysis identified a total of 2706 annotated putative sequences to COG functional categories and was used to predict their role in metabolism in the E. coli BE311 strain. Figure 2B shows that the annotation sequences of E. coli BE311 were subdivided into 22 classification items. In addition to some general function sequences and unknown function sequences, the top three protein functional classification items were carbohydrate transport and metabolism (287, 10.6%), amino acid transport and metabolism (274, 10.13%), and energy production and conversion (253, 9.35%). A total of 15,480 gene annotation sequences from E. coli BE311 were classified into three classifications of the biological process, cellular component, and molecular function, with the number and proportion of annotation sequences being 5054 (32.65%), 3863 (24.95%), and 6563 (42.40%), respectively (Figure 2C). Most of the gene sequences were involved in the cellular component classification. In addition, E. coli BE311 was predicted to contain 14 gene islands (Supplementary Table S2) and two CRISPR sequences (Supplementary Table S3). The Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis annotated a total of 3175 genes to different pathways. The genes were mainly enriched in carbohydrate metabolism, global and overview maps, and amino acid metabolism (Figure 2D).
2.3. Expression and Analysis of mCherry in E. coli BE311
A labeled, stably expressed and detectable ETEC strain for use in the ETEC challenge model was obtained by the knock-in of the mCherry gene into the E. coli BE311 genome by CRISPR/Cas9, based on the information obtained by whole genome sequencing. Figure 3A shows a graphical representation of the process. The mCherry gene was amplified from the genome of the edited strains by a pair of primers for the original yheO. As shown in Figure 3B, the amplified sequence size of the positive edited strain with the mCherry gene knock-in was 1650 bp, while the size of the wild-type E. coli BE311 was 814 bp. The successful knock-in of the mCherry gene into the E. coli BE311 was verified by agarose gel electrophoresis.
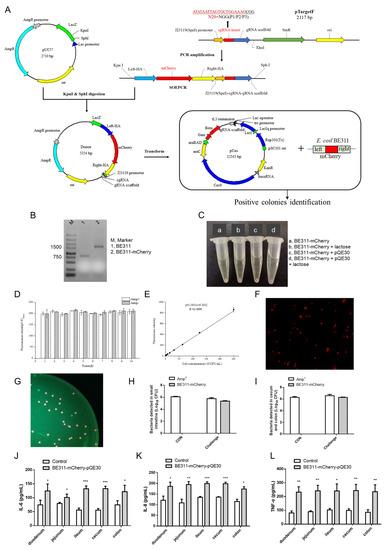
Figure 3.
Expression and analysis of mCherry in BE311. (A), The process of knocking mCherry into BE311 based on CRISPR/Cas9. (B), Agarose gel electrophoresis detection of the mCherry in BE311 genome. (C), Red fluorescence expression in different strains with or without lactose inducement. (D), Stability analysis of BE311–mCherry–pQE30 with or without ampicillin. (E), Construction of the correlation curve between fluorescence intensity and concentration of BE311–mCherry–pQE30. (F), The fluorescence of E. coli BE311–mCherry–pQE30 captured under fluorescence microscope. (G–I), The gut microbiota of E. coli BE311–mCherry–pQE30-challenged mice was obtained and cultured in LB plate for 24 h. The fluorescence of colonies and bacteria in intestines (H,I) was identified under blue exciting light and fluorescence microscope. (J–L), The expression of IL-6, IL-8, and TNF-α in different intestines of E. coli BE311–mCherry–pQE30-challenged SD rats. Statistical significance was determined using Students’ t test. * p < 0.05, ** p < 0.01, *** p < 0.001.
The edited E. coli BE311 strain had no resistance gene and could not be recovered specifically. The expression of the mCherry gene also had to be induced by lactose or IPTG for sufficient red brightness to be visibly distinguished. The pQE30 vector was transfected into the edited E. coli BE311 for antibiotic resistance with the aim of ETEC recovery and quantification in the challenge model. Interestingly, the edited E. coli BE311 with pQE30 not only showed ampicillin resistance, but also showed red fluorescence without lactose induction, and the fluorescence was as strong as the induced fluorescence (Figure 3C). The stability of the E. coli BE311–mCherry containing the pQE30 plasmid was confirmed, and the strain was passaged 10 times with or without ampicillin. The results indicated that the fluorescence intensity of E. coli BE311 was still strong even after several generations of culture and even when cultured without ampicillin (Figure 3D). Figure 3E shows that the concentration of BE311–mCherry was highly correlated with the fluorescence intensity (R2 = 0.9999), indicating a relationship between the fluorescence intensity and concentration of E. coli BE311–mCherry–pQE30. The E. coli BE311–mCherry transformed with pQE30 showed obvious red fluorescence and a short bar shape under the fluorescence microscope (Figure 3F).
The E. coli BE311–mCherry containing pQE30 (named BE311–mCherry–pQE30) was used to challenge Sprague–Dawley rats, and the bacteria in different intestinal segments were recovered and spread on ampicillin agar plates. The resulting BE311–mCherry–pQE30 colonies showed a red brightness that could be clearly distinguished under green light excitation (Figure 3G). The ampicillin-resistant bacteria were detected in both the small and large intestines (Figure 3H,I), and the E. coli BE311–mCherry–pQE30 strain was readily identifiable as red colonies (Figure 3G). The examination of different intestinal segments revealed significantly increased levels of inflammatory factors (including IL-6, IL-8, and TNF-α) in the duodenum, jejunum, ileum, cecum, and colon (p < 0.05) after the E. coli BE311–mCherry challenge (Figure 3J–L).
2.4. Construction and Functional Analysis of STKO-BE311
The virulence ST gene of BE311 was knocked out by CRISPR/Cas9, according to the process shown in Figure 4A. The genomes of wild-type E. coli BE311 and BE311-STKO were isolated and amplified for the ST gene. The agarose gel electrophoresis results revealed a DNA band between 100 and 250 bp in wild-type E. coli BE311, but it was absent in BE311-STKO, indicating that ST had been successfully removed from the E. coli BE311 genome (Figure 4B). This knock-out of the ST gene did not interfere with the growth of E. coli BE311-STKO, since the growth curve was similar to that of wild-type E. coli BE311 (Figure 4C). A comparison of the virulence and infection effects of E. coli BE311 and BE311-STKO cocultured with IPEC-J2 cells (Figure 4D) revealed that the BE311 challenge obviously increased apoptosis in IPEC-J2 cells (Figure 4E, p < 0.01), while BE311-STKO did not. Nevertheless, the deletion of ST in BE311 significantly decreased the total apoptosis rate in IPEC-J2 cells compared with wild-type BE311 (Figure 4E, p < 0.01).
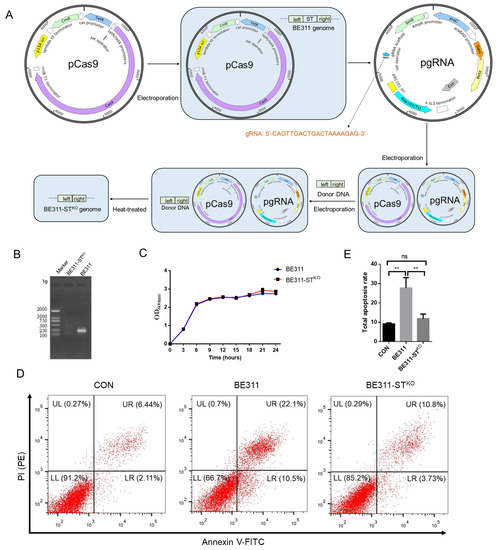
Figure 4.
Construction and analysis of BE311-STKO. (A), The knock-out process of ST gene in BE311. (B), Amplification of ST gene in wild-type and ST-knock-out BE311 strains. (C), The growth curve of BE311 and BE311-STKO strains within 24 h. (D), IPEC-J2 cells were challenged with BE311 or BE311-STKO for 6 h. Then the cellular apoptosis was examined by flow cytometry. (E), The total apoptosis rate in IPEC-J2 cells challenged with BE311 or BE311-STKO for 6 h. ** p < 0.01, ns, no significant difference.
3. Discussion
ETEC has a high incidence, rapid onset, and high mortality in the global pig farming industry. The diarrhea in piglets caused mainly by ETEC infection seriously affects piglet growth and production performance and is one of the major technology challenges in modern swine production [38]. The long-term use and abuse of antibiotics have resulted in a number of issues, including dysbiosis of the animals’ intestinal microbiota, the accumulation of drug residues, and environmental pollution by drugs [39]. As a result, an urgent need exists in the breeding industry for the development of effective vaccines or antibiotic alternatives against ETEC [10,13].
The targeted transformation of wild bacteria depends on the ability of the recipient strains to express exogenous genes, and this is closely related to the chosen expression vectors [40,41,42]. Green fluorescent protein is often used as an indicator to monitor heterologous protein expression in bacteria or transgenic plants [43,44]. In the present study, the recipient strains of E. coli were screened for their potential use as live vaccines or as indictors in challenge models. Therefore, strains with a high heterologous gene expression ability were preferentially selected for gene editing by CRISPR/Cas9 CRISPR/Cas9. As shown in Figure 1A and Table 2, both the recipient strains and the expression vectors contributed to the expression of GFP. Interestingly, the wild-type ETEC, including K99 and BE311, showed strong fluorescence protein expression, close to that of E. coli Top10, whereas the ability of E. coli K88 to express exogenous proteins was inferior to that of E. coli K99 and BE311. E. coli BE311 shared a homologous adhesin of STI to that found in E. coli K99. This result revealed that E. coli BE311 was a superior wild-type ETEC host strain for expressing exogenous genes. As a wild-type strain, the E. coli BE311 isolated and identified in the present study could be successfully employed in further gene editing.
Table 2.
Primers used for PCR.
The rapid development of microbial genomics has led to the use of whole genome sequencing technology for the sequencing of microbial genomic DNA on a large scale and identification of its key regulatory factors [45]. To our knowledge, no reference sequences have been reported in public databases for the whole genome sequences of K99 or F5-fimbriae-harboring ETEC. In the present study, whole genome sequencing was carried out for E. coli BE311 with the aim of determining the gene sequences and gene functions of E. coli BE311 and to discover novel virulence factors. The total length of the BE311 genome was 4,840,702 bp encoding 4622 genes (Figure 2A), which was similar to the E. coli strains identified in previous studies, with 5.2 to 5.5 Mbp genomes and 4633 to 5150 coding sequences (CDSs) [46,47,48]. We found that the virulence genes, ST, and fimbriae sequences of BE311 were identical to those of K99 (data not shown), suggesting that BE311 was an F5-like fimbria ETEC strain. The whole genome sequence identified in the present study will provide a novel reference for further studies on E. coli K99.
A study on enterohemorrhagic Escherichia coli (EHEC) O157:H7 has shown that the identified proteins were grouped into three important functional groups: metabolism, information storage and processing, cellular processes, and signaling [49]. Correspondingly, the BE311 genes were mainly annotated to cellular components and metabolism pathways (Figure 2A). These crucial enriched proteins may contribute to the pathogenesis of the bacterium; however, the specific gene functions of BE311 require further clarification based on more rigorous experiments. Furthermore, based on the whole genome sequence, the gRNA and the editing sites in E. coli BE311 could be determined for further gene knock-in and knock-out modifications by specific CRISPR/Cas9 technology [50].
In searching for inhibitors against ETEC as antibiotic alternatives, some infection experimental models are often established based on the ETEC infection [51,52,53]. The decreased colonization and proliferation of ETEC in the gastrointestinal tract is a key aim in the control of pathogen infection, making in vivo ETEC tracking particularly important. Conventional selective medium-based colony counting methods are now widely supplemented by PCR techniques, but this involves a complicated series of steps, including RNA extraction from tissues or cells, amplification, and the detection of specific virulence gene expression [54,55]. In early reports, GFP-tagged E. coli were constructed and successfully used to track the fate of ETEC distributed in the gut based on fluorescence signals [56,57]. These assays based on GFP-tagged E. coli provide a rapid and specific method for the determination of the ETEC load in challenge models. Red fluorescent proteins (RFPs) with longer emission wavelengths are more penetrating and sensitive than the green fluorescent proteins, especially for the testing of the direct imaging [58]. Therefore, mCherry was selected as the RFP in the present study for expression in E. coli BE311 (Figure 3A).
In previous studies, most of the bioluminescent reporter genes, such as lux, luc, ruc, and gfp, were expressed in the recipient hosts through plasmid transformation methods [31,59,60,61]. The reporter genes were, therefore, not integrated into the host chromosome and were easily lost, even without selection pressure [62]. In the present study, we constructed E. coli BE311–mCherry by knocking mCherry into the E. coli BE311 genome using CRISPR/Cas9 (Figure 3B). The red fluorescent protein could then be expressed after lactose induction (Figure 3C). We also increased the selectivity of E. coli BE311 in plate counting by transforming a vacant pQE30 plasmid to carry ampicillin resistance. Interestingly, the E. coli BE311–mCherry strain containing the vacant plasmid pQE30 (named E. coli BE311–mCherry–pQE30) showed a stable and efficient expression of the fluoresce signal even without lactose induction (Figure 3C,D). The pQE-30 vector contained a double lacO operator repression module downstream of the T5 promoter. The possible reason for the observed spontaneous fluorescence was that most of the lac repressors expressed by E. coli BE311 were combined into the lacO operator of pQE-30 so that the transcription and expression of mCherry was no longer repressed. This would also explain the fact that the fluorescence intensity of E. coli BE311–mCherry–pQE30 was not significantly enhanced after lactose induction (Figure 3C). However, we have not tested whether this effect can be achieved by incorporating a resistance cassette with the chromosomal DNA, together with the fluorescence gene. This would be a better option if the bacteria could both express fluorescence and harbor resistance, and the chromosomal mutation would be more rigorous and convincing as a theoretical control to the wild-type strain. The relative experiments will be completed in our future work.
The pathogenicity of E. coli BE311–mCherry–pQE30 was verified in the rat model, since the inflammatory factors were significantly increased in all intestinal segments after administration of the RFP-tagged ETEC. This result indicated that the insertion of mCherry did not affect the pathogenicity of the ETEC. Moreover, the RFP-tagged ETEC was easily recovered and counted on ampicillin agar plates. The ampicillin-resistant strains, except for E. coli BE311–mCherry–pQE30, could also be detected on the agar plates, and the RFP label helped to identify the administered ETEC, as those colonies were significantly different from the endogenous bacterial colonies (Figure 3H,I). Therefore, the E. coli BE311–mCherry–pQE30 constructed using CRISPR/Cas9 gene editing was an ideal tool strain that will be suitable for detecting the load of ETEC in challenge models.
Virulence factor-deficient hosts are helpful tools for ETEC vaccine exploration [63]. Heat-stable enterotoxins (STs) do not lose their activity when heated at 100 °C for 30 min, and their small size and 3D structure make them more heat resistant [64]. We recognized that the presence of ST in E. coli BE311 might affect its safety as a vaccine or vaccine vector; therefore, we constructed an ST knockout strain, again using CRISPR/Cas9 gene editing (Figure 4A). At the gene level, the ST gene knockout was verified, as shown in Figure 4B. The decreased apoptosis induced by E. coli BE311-STKO in IPEC-J2 cells indicated that the presence of ST could disrupt the function of intestinal epithelial cells to some extent, and the toxicity of E. coli BE311 was weakened by the knockout of the ST gene. The ETEC infection mechanism is very complicated [65]; however, the E. coli BE311-STKO strain constructed by CRISPR/Cas9 gene editing helped in the exploration of ST functions and could provide attenuated ETEC strains for vaccine development.
In summary, based on the screening of ETEC for its gene expression ability, we isolated a wild-type enterotoxigenic E. coli BE311, whose ST gene was the same as that of E. coli K99. The whole genome of E. coli BE311 was sequenced for gene editing based on CRISPR/Cas9 technology. Two strains, E. coli BE311–mCherry–pQE30 and E. coli BE311-STKO, were successfully constructed based on a gene knock-in and knock-out, respectively. The two strains could represent ideal candidates for ETEC challenge model studies and attenuated vaccine development.
4. Materials and Methods
4.1. Bacterial Strains and Plasmids
Three standard ETEC strains, E. coli K88ab (CVCC C83901), E. coli K99 (CVCC C83922), and E. coli 987P (CVCC1523), were purchased from the China Institute of Veterinary Drug Control (Beijing, China). Four standard E. coli expression host strains, E. coli BL21, E. coli Rosetta, E. coli TG1, and E. coli Top10, and three expression vectors, pQE30, pET9a, and pET28a, were purchased from Solarbio Life Science Co., Ltd. (Beijing, China). Five ETEC strains were isolated from the feces of piglets with diarrhea by sorbitol MacConkey agar enrichment streaks, single colony morphology identification, 16S rRNA sequencing, and PCR identification of specific virulence genes, and kept in our laboratory. The fluorescent plasmids pUC-GFP and pUC-mCherry were kindly gifted by Dr. Xiaosheng Leung of the College of Life Science, South-Central Minzu University.
The E. coli strains were cultured in Luria–Bertani (LB) broth (1% tryptone, 0.5% yeast extract, and 1% NaCl) or on LB-agar plates. Bacteria harboring the pQE30 plasmid were cultured in LB medium with 100 μg/mL ampicillin. E. coli strains harboring the pET28a and pET9a plasmids were cultured in LB medium with 100 μg/mL kanamycin. The supplier of pCas9 (#62655) and gRNA (#62656) was Addgene (Watertown, MA, USA).
4.2. Screening of ETEC for Heterologous Gene Expression and Identification of Virulence Factors
The pUC-GFP plasmid was digested with Nde I/BamH I to obtain the gfp gene. The gfp DNA fragments (about 700 bp) were purified with a DNA gel extraction kit (Takara, Japan). The gfp fragments were then inserted into identical sites in three vectors (pET-28a, pET-9a, and pQE-30) and transformed into E. coli Top10 cells. Restriction enzyme identification, PCR, and sequence analysis were then used to obtain the recombinant 3 plasmids (pET28a-GFP, pET9a-GFP, and pQE30-GFP), which were then transformed into E. coli expression host strains and ETEC strains. On the second day, the recombinant and non-recombinant colonies were identified by directly observing their fluorescence under blue light excitation. The wild-type E. coli BE311 strain was selected for further CRISPR/Cas9 modification based on its strongest green fluorescence.
E. coli BE311 genomic DNA was extracted with a BacteriaGen DNA Kit (Cwbiotech, China), and the 16s rRNA sequence was amplified and analyzed by BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi) (accessed on 6 March 2019) and agarose electrophoresis. The known virulence factors of ETEC and BE311 were amplified using the primers shown in Table 2, according to the reference sequences. The PCR products were verified by agarose gel electrophoresis.
4.3. Whole-Genome Sequencing (WGS)
4.3.1. DNA Preparation
The bacterial genomic DNA of the screened E. coli BE311 was extracted using a DNA miniprep kit (Qiagen, Dusseldorf, Germany) according to the manufacturer’s protocol. The amounts of DNA in the samples were quantified according to Illumina sequencing sample requirements. The DNA samples were dissolved in 10 mM Tris buffer to give a 10 nM concentration in a minimum 10 μL volume. The DNA purity was determined by calculating A260/280 and A260/230 ratios in a Nanodrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, USA). The quantified DNA was used to prepare genomic DNA libraries using the SQK-LSK109 ligation kit (ONT, Oxford, UK) and sequenced on a PromethION sequencer (Oxford Nanopore Technologies, Oxford, UK) and the Illumina platform (San Diego, CA, USA) using standard protocols.
4.3.2. Analysis of DNA Sequence Data
The filtered reads from Hiseq were assembled with Assembler1.2. The genome was annotated and open reading frames (ORFs) identified using Prokka and/or PATRIC 3.6.5 with default settings. The WGS data sets were analyzed using an open-access bioinformatics webtool available at the Center for Genomic Epidemiology (http://www.genomicepidemiology.org/ (accessed on 6 March 2019) ). Serotype Finder 2.0 was used for in silico typing based on the WGS of the assembled genomes/contigs in FASTA format, with a threshold of 90% identity and a 60% total serotype gene length. Virulence genes were identified using Virulence Finder 2.0 with thresholds of 90% identity and 60% minimum length. Virulence genes were aligned to the VFDB database (http://www.mgc.ac.cn/VFs/main.htm). Clustered regularly interspaced short palindromic repeats (CRISPRs), genomics islands (GIs), and pseudogene candidates and prophages were determined by CRISPRfinder (https://crispr.i2bc.paris-saclay.fr/), IslandViewer 4 (http://www.pathogenomics.sfu.ca/islandviewer/), and Pseudofinder, respectively (All the websites in this section were accessed on 3–8 April 2021). The functions of the identified genes were divided into three categories: cell composition, molecular function, and biological process. The top 20 enriched GO slims with the most genes under each category were selected for plotting. After KEGG annotation, the genes were classified according to their associated KEGG metabolic pathways.
4.4. Construction of mCherry-Labeled E. coli Based on CRISPR/Cas9
4.4.1. Plasmid Construction
The CRISPR/Cas9 vector was constructed based on the yheO gene sequence published in Genbank (GenBank accession number NC_000913.3). Primers yheO-genomic-F and yheO-genomic-R were used to amplify the yheO gene from E. coli BE311 genomic DNA. The guide sequence yheO-gRNA was designed according to Cas9 target design principles. The CRISPR/Cas9 plasmids containing sgRNA encoding sequences were constructed using plasmid pTargetF as a template. The primers P1, P2, and P3 were designed to construct the recombinant plasmid pTargetF-sgRNA. The T5 promoter with the mCherry gene was obtained using primers mCherry-F and mCherry-R. The primers Left-HA-F, Left-HA-R, Right-HA-F, and Right-HA-R were used to amplify the yehO-Left-HA and yehO-Right-HA genes from E. coli BE311. The fragment J23119(Spe I)-sgRNA-gRNA scaffold was amplified by the primers SSG-F and SSG-R. These four fragments (Left-HA + mCherry + Right-HA + J23119 (Spe I)-sgRNA-gRNA) were fused by ligating the upstream and downstream editing templates using overlapping PCR. The editing template containing the Left-HA/mCherry/Right-HA fused sequences and the fragment J23119 (Spe I)-sgRNA-gRNA scaffold was digested with Kpn I and Sph I, and the target templates were inserted into the pUC57 vector. The mCherry gene was cloned from the plasmid pUC-mCherry. The donor DNA was obtained by mixing the yehO gene upstream and downstream editing templates with the mCherry gene and sgRNA, and then using the mixed fragment as a template to amplify the final donor DNA by overlapping PCR with double enzyme digestion. All these primers are listed in Table 3.
Table 3.
Primers used in the construction of mCherry-labeled E. coli BE311.
4.4.2. Genome Editing
The pCas plasmid was transformed into E. coli BE311 competent cells, and the transformed cells were plated on LB agar containing kanamycin and cultured at 30 °C. E. coli BE311 colonies harboring the pCas plasmid were selected and used to prepare competent cells. For electroporation, 50 μL of the E. coli BE311 competent cells was mixed with 100 ng pTargetT series DNA and 400 ng donor DNA and subjected to electroporation in a 2 mm Gene Pulser cuvette (#1652082EDU, Bio-Rad, Hercules, CA, USA) at 2.5 kV. The product was suspended immediately in 1 mL ice-cold LB medium. The cells were allowed to recover at 30 °C for 1 h before plating on LB agar containing kanamycin (50 mg/L) and spectinomycin (50 mg/L), and incubating overnight at 30 °C. The transformants were identified by colony PCR and DNA sequencing.
The pTarget series was cured by inoculating the edited colony harboring both pCas and pTarget series into 2 mL LB medium containing kanamycin (50 mg/L) and isopropyl-β-D-thiogalactopyranoside (IPTG; 0.5 mM). The culture was incubated for 16 h, diluted, and plated on LB agar containing kanamycin (50 mg/L). The colonies were confirmed as cured by determining their sensitivity to spectinomycin (50 mg/L) and were then used in a second round of genome editing. The pCas series was cured by growing the colonies nonselectively overnight at 37 °C [66].
Figure 3A outlines the process used for the construction of the E. coli BE311 expression system with mCherry fluorescent protein (named E. coli BE311–mCherry) as the reporter gene. A blank vector pQE30 was transformed into the E. coli BE311–mCherry to test expression with or without inducement. The strains were cultured in 37 °C in LB for 12 h and then induced by addition of 5 g/L lactose and incubation at 18 °C for 10 h. The fluorescence of the different strains was observed in daylight after centrifugation.
4.4.3. Verification of the E. coli BE311–mCherry Strain
The effects of mCherry labeling in E. coli BE311 were tested by PCR using primers 5′-GTAGAAGCCGGTAAAGGCGA-3′ and 5′-AGAAACGCTGCTATCCGCTC-3′. The genomic DNA of E. coli BE311 and recombinant strain E. coli BE311–mCherry were used as templates. The final PCR reaction volume was 25 μL, and included 0.5 mmol/L primers, 1.5 mmol/L Mg2+, 200 mmol/L dNTPs, 50 ng of template DNA, and appropriate ddH2O. The PCR reaction program began with initial denaturation at 98 °C for 3 min, followed by 35 cycles of denaturation at 98 °C for 10 s, annealing at 60 °C for 20 s, and extension at 72 °C for 7 s. The final extension was held at 72 °C for 5 min. The resulting PCR products were separated on 1% agarose gel and subjected to DNA sequencing. The PCR product size of BE311 genome was 814 bp, and the product size amplified from the mCherry-positive genome was 1650 bp.
4.4.4. Fluorescence Intensity Determination of E. coli BE311–mCherry
The E. coli BE311–mCherry from the fermentation broth was harvested for analysis by centrifugation (6000× g for 5 min), washed twice, and suspended in physiological saline. The suspensions of E. coli BE311–mCherry with the initial optical density were diluted about tenfold at a gradient of 2 times and the fluorescence intensity was determined. The suspensions were plated for counting, and the linear correlation between strain concentrations (CFU/mL) and the fluorescence intensity (AU) was established.
A fresh bacterial solution of E. coli BE311–mCherry was washed twice with sterile double distilled water, and the excitation and emission wavelengths of mCherry at 587 nm/610 nm, respectively, were used to determine the fluorescence intensity of E. coli BE311–mCherry in an F-7000 fluorescence spectrophotometer (Hitachi, Ltd., Tokyo, Japan). A sample of the bacterial solution was evenly coated on a clean glass slide, and the fluorescence was observed under a fluorescence microscope (Nikon Eclipse C1, Minato, Japan).
4.5. Construction of an ST-Knock-Out E. coli BE311 Strain Based on CRISPR/Cas9
4.5.1. Plasmid and Editing Template DNA Construction
The guide RNA and editing template were designed based on the whole genome sequence of E. coli BE311 obtained in this study. The ST gene sequence was used to design the corresponding gRNA in the open website (http://www.rgenome.net/cas-offinder/) (accessed on 8 June 2021); its sequence was 5′-CAGTTGACTGACTAAAAGAG-3′. The gRNA was synthesized and linked to the plasmid pgRNA. The sequence of E. coli BE311 was then used to insert the sequence of the left and right homologous arms of the ST gene into pUC57 to obtain the editing template. Primers 5′-ATGAAAAAGCTAATGTTGGCAATTT-3′ and 5′-TTAATAACATCCAGCACAGGCAGGA-3′ were used to amplify the homologous arm sequences from pUC57-ST. The PCR products were used as the donor DNA.
4.5.2. Genome Editing with Chemical Transformation
E. coli BE311 competent cells containing pCas were prepared and electroporated by mixing 100 μL competent cells with 100 ng pgRNA and 400 ng donor DNA, and electroporating in a 2 mm Gene Pulser cuvette (Bio-Rad Korea, Seoul, Korea) at 2.5 kV. The product was suspended immediately in 700 μL ice-cold LB medium. The cells were allowed to recover at 30 °C for 1 h before plating onto LB agar containing kanamycin (50 mg/L) and ampicillin (50 mg/L), and incubation overnight at 30 °C. Positive clones were identified by colony PCR. The positive strains were heat treated to cure pCas and pgRNA.
4.5.3. Verification of the Editing Efficiency
The genomic DNAs of wild-type and engineered E. coli BE311 (named BE311-STKO) were prepared from 4 mL scale overnight cultures in LB medium using a bacterial genomic DNA extraction kit (Cwbiotech, Taizhou, China). The ST gene was amplified using the primers 5′-CGAACAAGAAAAGGATAAAACCA-3′ and 5′-TATAAAAGCGTTGTTTTTTATTA-3′.
4.6. Stability Detection of E. coli BE311–mCherry and BE311-STKO
Overnight cultures of E. coli BE311–mCherry and E. coli BE311-STKO were transferred to fresh LB medium at an inoculum volume of 1% and incubated at 37 °C, 200 rpm/min. The bacterial solution was passaged, continuously, 10 times, each time at an interval of 24 h, diluted, and the OD600 nm absorbance and fluorescence intensity were measured.
4.7. E. coli BE311–mCherry–pQE30 Challenge In Vivo
Twelve male Sprague–Dawley rats were randomly divided into two groups: a control group and the ETEC challenge group. Rats in the ETEC group were intragastrically administered with 1 mL (106 CFU) E. coli BE311–mCherry–pQE30 daily via oral gavage for 7 days. The rats in the control group were orally gavaged with 1 mL isopycnic phosphate buffered saline (PBS) daily for 7 days. All rats were sacrificed at day 8, and the intestines were separated into five gastrointestinal segments, including the duodenum, jejunum, ileum, cecum, and colon. Each intestinal sample was completely suspended in PBS, and then homogenized, filtered through sterile gauze, and diluted 10-fold with normal saline. The numbers of anti-Amp+ and BE311–mCherry bacteria were calculated by colony counting on LB agar plates containing 50 mg/L ampicillin. The expression of IL-6, IL-8, and TNF-α in different intestinal segments was tested with an ELISA kit (CAMILO Biological, Naijing, China), according to the kit instructions. The animal experiment was approved by Scientific Ethics and Safety Committee of South-Central Minzu University (Ethical permission number: 2020-SCUEC-006).
4.8. Flow Cytometry Analysis of Apoptosis in Cells Infected with E. coli BE311-STKO
Apoptosis was assessed using an Annexin V-FITC/PI double-stain assay kit (Beyotime, Haimen, China), according to the manufacturer’s instructions. Briefly, the IPEC-J2 cells were seeded in 60 mm dishes and cultured with PBS or E. coli BE311 and BE311-STKO for 6 h. The cells were then collected and stained in binding buffer with 5 μL polyimide (PI) solution and 10 μL FITC-conjugated annexin V for 15 min in the dark at room temperature. Apoptotic cells were detected with a CytoFLEX flow cytometer (BD, Franklin Lakes, NJ, USA), and the data were analyzed with the FlowJo software program (Version 10, FlowJo, Ashland, OR, USA).
4.9. Statistical Analysis
Student’s t test and one-way ANOVA statistical analysis were used for statistical comparisons of differences between two or more than two groups, respectively. Column charts were constructed from the mean ± SD values using GraphPad Prism 6.0 (Graphpad Inc., San Diego, CA, USA). Values of p < 0.05 were deemed statistically significant. All experiments were performed as at least three independent replicates.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms23147502/s1.
Author Contributions
Conceptualization, X.G. and S.L.; Methodology, L.Z.; Software, S.L.; Validation, T.T. and S.L.; Formal Analysis, X.L.; Investigation, T.T., Y.S., J.H. and G.W.; Resources, X.G.; Data Curation, S.L. and L.Z.; Writing—Original Draft Preparation, S.L.; Writing—Review and Editing, X.G.; Visualization, X.G.; Supervision, X.G.; Project Administration, X.G.; Funding Acquisition, X.G. and S.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (No. 32072767 and 31672455), the Knowledge Innovation Program of the Wuhan-Shuguang Project (No. 2022020801020409), and the Fundamental Research Funds for the Central Universities, South-Central MinZu University (Grant Number: CZP22003 and CZQ22012).
Institutional Review Board Statement
The animal study protocol was approved by the Scientific Ethics and Safety Committee of South-Central Minzu University (protocol code: 2020-SCUEC-006, 3 April 2020).
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are openly available in Genebank at SUB11437696.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Francis, D.H. Enterotoxigenic Escherichia coli infection in pigs and its diagnosis. J. Swine Health Prod. 2002, 10, 171–175. [Google Scholar]
- Kim, N.; Gu, M.J.; Kye, Y.C.; Ju, Y.J.; Hong, R.; Ju, D.B.; Pyung, Y.J.; Han, S.H.; Park, B.C.; Yun, C.H. Bacteriophage EK99P-1 alleviates enterotoxigenic Escherichia coli K99-induced barrier dysfunction and inflammation. Sci. Rep. 2022, 12, 941. [Google Scholar] [CrossRef]
- Dubreuil, J.D.; Isaacson, R.E.; Schifferli, D.M. Animal enterotoxigenic Escherichia coli. EcoSal Plus 2016, 7, 1128. [Google Scholar] [CrossRef]
- Brubaker, J.; Zhang, X.; Bourgeois, A.L.; Harro, C.; Sack, D.A.; Chakraborty, S. Intestinal and systemic inflammation induced by symptomatic and asymptomatic enterotoxigenic E. coli infection and impact on intestinal colonization and ETEC specific immune responses in an experimental human challenge model. Gut Microbes. 2021, 13, 1891852. [Google Scholar] [CrossRef]
- Roussel, C.; Sivignon, A.; de Vallée, A.; Garrait, G.; Denis, S.; Tsilia, V.; Ballet, N.; Vandekerckove, P.; Van de Wiele, T.; Barnich, N.; et al. Anti-infectious properties of the probiotic Saccharomyces cerevisiae CNCM I-3856 on enterotoxigenic E. coli (ETEC) strain H10407. Appl. Microbiol. Biotechnol. 2018, 102, 6175–6189. [Google Scholar] [CrossRef]
- Laird, T.J.; Abraham, S.; Jordan, D.; Pluske, J.R.; Hampson, D.J.; Trott, D.J.; O’Dea, M. Porcine enterotoxigenic Escherichia coli: Antimicrobial resistance and development of microbial-based alternative control strategies. Vet. Microbiol. 2021, 258, 109117. [Google Scholar] [CrossRef]
- Luo, Y.; Liu, L.; Chen, D.; Yu, B.; Zheng, P.; Mao, X.; Huang, Z.; Yu, J.; Luo, J.; Yan, H.; et al. Dietary supplementation of fructooligosaccharides alleviates enterotoxigenic E. coli-induced disruption of intestinal epithelium in a weaned piglet model. Br. J. Nutr. 2021, 1–27. [Google Scholar] [CrossRef]
- Mirhoseini, A.; Amani, J.; Nazarian, S. Review on pathogenicity mechanism of enterotoxigenic Escherichia coli and vaccines against it. Microb. Pathog. 2018, 117, 162–169. [Google Scholar] [CrossRef]
- Wang, W.; Ma, H.; Yu, H.; Qin, G.; Tan, Z.; Wang, Y.; Pang, H. Screening of Lactobacillus plantarum Subsp. plantarum with potential probiotic activities for inhibiting ETEC K88 in weaned piglets. Molecules 2020, 25, 4481. [Google Scholar] [CrossRef]
- Hur, J.; Lee, J.H. Comparative evaluation of a vaccine candidate expressing enterotoxigenic Escherichia coli (ETEC) adhesins for colibacillosis with a commercial vaccine using a pig model. Vaccine 2012, 30, 3829–3833. [Google Scholar] [CrossRef]
- Ramakrishnan, A.; Joseph, S.S.; Reynolds, N.D.; Poncet, D.; Maciel, M.; Nunez, G.; Espinoza, N.; Nieto, M.; Castillo, R.; Royal, J.M.; et al. Evaluation of the immunogenicity and protective efficacy of a recombinant CS6-based ETEC vaccine in an Aotus nancymaae CS6+ ETEC challenge model. Vaccine 2021, 39, 487–494. [Google Scholar] [CrossRef]
- Chakraborty, S.; Randall, A.; Vickers, T.J.; Molina, D.; Harro, C.D.; DeNearing, B.; Brubaker, J.; Sack, D.A.; Bourgeois, A.L.; Felgner, P.L. Interrogation of a live-attenuated enterotoxigenic Escherichia coli vaccine highlights features unique to wild-type infection. NPJ Vaccines 2019, 4, 37. [Google Scholar] [CrossRef]
- Harro, C.; Bourgeois, A.L.; Sack, D.; Walker, R.; DeNearing, B.; Brubaker, J.; Maier, N.; Fix, A.; Dally, L.; Chakraborty, S. Live attenuated enterotoxigenic Escherichia coli (ETEC) vaccine with dmLT adjuvant protects human volunteers against virulent experimental ETEC challenge. Vaccine 2019, 37, 1978–1986. [Google Scholar] [CrossRef]
- Darsley, M.J.; Chakraborty, S.; DeNearing, B.; Sack, D.A.; Feller, A.; Buchwaldt, C.; Bourgeois, A.L.; Walker, R.; Harro, C.D. The oral, live attenuated enterotoxigenic Escherichia coli vaccine ACE527 reduces the incidence and severity of diarrhea in a human challenge model of diarrheal disease. Clin. Vaccine Immunol. 2012, 19, 1921–1931. [Google Scholar] [CrossRef]
- Holmgren, J.; Bourgeois, L.; Carlin, N.; Clements, J.; Gustafsson, B.; Lundgren, A.; Nygren, E.; Tobias, J.; Walker, R.; Svennerholm, A.M. Development and preclinical evaluation of safety and immunogenicity of an oral ETEC vaccine containing inactivated E. coli bacteria overexpressing colonization factors CFA/I, CS3, CS5 and CS6 combined with a hybrid LT/CT B subunit antigen, administered alone and together with dmLT adjuvant. Vaccine 2013, 31, 2457–2464. [Google Scholar]
- Harutyunyan, S.; Neuhauser, I.; Mayer, A.; Aichinger, M.; Szijártó, V.; Nagy, G.; Nagy, E.; Girardi, P.; Malinoski, F.J.; Henics, T. Characterization of shigetec, a novel live attenuated combined vaccine against shigellae and etec. Vaccines 2020, 8, 689. [Google Scholar] [CrossRef]
- McKenzie, R.; Darsley, M.; Thomas, N.; Randall, R.; Carpenter, C.; Forbes, E.; Finucane, M.; Sack, R.B.; Hall, E.; Bourgeois, A.L. A double-blind, placebo-controlled trial to evaluate the efficacy of PTL-003, an attenuated enterotoxigenic E. coli (ETEC) vaccine strain, in protecting against challenge with virulent ETEC. Vaccine 2008, 26, 4731–4739. [Google Scholar] [CrossRef]
- McKenzie, R.; Bourgeois, A.L.; Engstrom, F.; Hall, E.; Chang, H.S.; Gomes, J.G.; Kyle, J.L.; Cassels, F.; Turner, A.K.; Randall, R. Comparative safety and immunogenicity of two attenuated enterotoxigenic Escherichia coli vaccine strains in healthy adults. Infect. Immun. 2006, 74, 994–1000. [Google Scholar] [CrossRef][Green Version]
- Santiago-Mateo, K.; Zhao, M.; Lin, J.; Zhang, W.; Francis, D.H. Avirulent K88 (F4)+ Escherichia coli strains constructed to express modified enterotoxins protect young piglets from challenge with a virulent enterotoxigenic Escherichia coli strain that expresses the same adhesion and enterotoxins. Vet. Microbiol. 2012, 159, 337–342. [Google Scholar] [CrossRef]
- Zhang, H.; Xu, Y.; Zhang, Z.; You, J.; Yang, Y.; Li, X. Protective immunity of a multivalent vaccine candidate against piglet diarrhea caused by enterotoxigenic Escherichia coli (ETEC) in a pig model. Vaccine 2018, 36, 723–728. [Google Scholar] [CrossRef]
- Chen, S.; Wu, X.; Xia, Y.; Wang, M.; Liao, S.; Li, F.; Yin, J.; Ren, W.; Tan, B.; Yin, Y. Effects of dietary gamma-aminobutyric acid supplementation on amino acid profile, intestinal immunity, and microbiota in ETEC-challenged piglets. Food Funct. 2020, 11, 9067–9074. [Google Scholar] [CrossRef]
- Yang, K.; Jiang, Z.; Zheng, C.; Wang, L.; Yang, X. Effect of Lactobacillus plantarum on diarrhea and intestinal barrier function of young piglets challenged with enterotoxigenic Escherichia coli K88. J. Anim. Sci. 2014, 92, 1496–1503. [Google Scholar] [CrossRef]
- Peng, X.; Wang, R.; Hu, L.; Zhou, Q.; Liu, Y.; Yang, M.; Fang, Z.; Lin, Y.; Xu, S.; Feng, B. Enterococcus faecium NCIMB 10415 administration improves the intestinal health and immunity in neonatal piglets infected by enterotoxigenic Escherichia coli K88. J. Anim. Sci. Biotechnol. 2019, 10, 72. [Google Scholar] [CrossRef]
- Spitzer, F.; Vahjen, W.; Pieper, R.; Martinez-Vallespin, B.; Zentek, J. A standardised challenge model with an enterotoxigenic F4+ Escherichia coli strain in piglets assessing clinical traits and faecal shedding of fae and est-II toxin genes. Arch. Anim. Nutr. 2014, 68, 448–459. [Google Scholar] [CrossRef]
- Ghapoutsa, R.N.; Boda, M.; Gautam, R.; Ndze, V.N.; Mugyia, A.E.; Etoa, F.X.; Bowen, M.D.; Esona, M.D. Detection of diarrhoea associated rotavirus and co-infection with diarrhoeagenic pathogens in the littoral region of Cameroon using ELISA, RT-PCR and Luminex xTAG GPP assays. BMC Infect. Dis. 2021, 21, 614. [Google Scholar] [CrossRef]
- Rocha, L.B.; Ozaki, C.Y.; Horton, D.S.; Menezes, C.A.; Silva, A.; Fernandes, I.; Magnoli, F.C.; Vaz, T.M.; Guth, B.E.; Piazza, R.M. Different assay conditions for detecting the production and release of heat-labile and heat-stable toxins in enterotoxigenic Escherichia coli isolates. Toxins 2013, 5, 2384–2402. [Google Scholar] [CrossRef]
- Gonzales-Siles, L.; Sjöling, Å. The different ecological niches of enterotoxigenic Escherichia coli. Environ. Microbiol. 2016, 18, 741–751. [Google Scholar] [CrossRef]
- Farling, S.; Rogers, T.; Knee, J.S.; Tilley, E.A.; Brown, J.; Deshusses, M.A. Bioaerosol emissions associated with pit latrine emptying operations. Sci. Total Environ. 2019, 648, 1082–1086. [Google Scholar] [CrossRef]
- Ginn, O.; Rocha-Melogno, L.; Bivins, A.; Lowry, S.; Cardelino, M.; Nichols, D.; Tripathi, S.N.; Soria, F.; Andrade, M.; Bergin, M.; et al. Detection and quantification of enteric pathogens in aerosols near open wastewater canals in cities with poor sanitation. Environ. Sci. Technol. 2021, 55, 14758–14771. [Google Scholar] [CrossRef]
- Billard, P.; DuBow, M.S. Bioluminescence-based assays for detection and characterization of bacteria and chemicals in clinical laboratories. Clin. Biochem. 1998, 31, 1–14. [Google Scholar] [CrossRef]
- Wu, C.M.; Chung, T.C. Mice protected by oral immunization with Lactobacillus reuteri secreting fusion protein of Escherichia coli enterotoxin subunit protein. FEMS Immunol. Med. Microbiol. 2007, 50, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, M. Green fluorescent protein (GFP): Applications, structure, and related photophysical behavior. Chem. Rev. 2002, 102, 759–782. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lin, Z.; Huang, C.; Zhang, Y.; Wang, Z.; Tang, Y.J.; Chen, T.; Zhao, X. Metabolic engineering of Escherichia coli using CRISPR-Cas9 meditated genome editing. Metab. Eng. 2015, 31, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.E.; Yeh, I.H.; Sung, L.Y.; Wu, M.Y.; Chao, Y.P.; Ng, I.S.; Hu, Y.C. Enhanced integration of large DNA into E. coli chromosome by CRISPR/Cas9. Biotechnol. Bioeng. 2017, 114, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Hou, M.; Sun, S.; Feng, Q.; Dong, X.; Zhang, P.; Shi, B.; Liu, J.; Shi, D. Genetic editing of the virulence gene of Escherichia coli using the CRISPR system. PeerJ 2020, 8, e8881. [Google Scholar] [CrossRef]
- Jiang, Y.; Chen, B.; Duan, C.; Sun, B.; Yang, J.; Yang, S. Multigene editing in the Escherichia coli genome via the CRISPR-Cas9 system. Appl. Environ. Microbiol. 2015, 81, 2506–2514. [Google Scholar] [CrossRef]
- Wan, P.; Cui, S.; Ma, Z.; Chen, L.; Li, X.; Zhao, R.; Xiong, W.; Zeng, Z. Reversal of mcr-1-mediated colistin resistance in Escherichia coli by CRISPR-Cas9 system. Infect. Drug Resist. 2020, 13, 1171–1178. [Google Scholar] [CrossRef]
- Gresse, R.; Chaucheyras-Durand, F.; Fleury, M.A.; Van de Wiele, T.; Forano, E.; Blanquet-Diot, S. Gut microbiota dysbiosis in postweaning piglets: Understanding the keys to health. Trends Microbiol. 2017, 25, 851–873. [Google Scholar] [CrossRef]
- Kraemer, S.A.; Ramachandran, A.; Perron, G.G. Antibiotic pollution in the environment: From microbial ecology to public policy. Microorganisms 2019, 7, 180. [Google Scholar] [CrossRef]
- Busso, D.; Delagoutte-Busso, B.; Moras, D. Construction of a set Gateway-based destination vectors for high-throughput cloning and expression screening in Escherichia coli. Anal. Biochem. 2005, 343, 313–321. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, X.; Yu, L.; Tian, Y.; Li, S.; Leng, F.; Ma, J.; Chen, J. Effects of Sr2+ on the preparation of Escherichia coli DH5α competent cells and plasmid transformation. PeerJ 2020, 8, e9480. [Google Scholar] [CrossRef]
- Kang, D.; Kim, Y.; Cha, H. Comparison of green fluorescent protein expression in two industrial Escherichia coli strains, BL21 and W3110, under co-expression of bacterial hemoglobin. Appl. Microbiol. Biotechnol. 2002, 59, 523–528. [Google Scholar] [PubMed]
- Drew, D.E.; Von Heijne, G.; Nordlund, P.; de Gier, J.-W.L. Green fluorescent protein as an indicator to monitor membrane protein overexpression in Escherichia coli. FEBS Lett. 2001, 507, 220–224. [Google Scholar] [CrossRef]
- Richards, H.; Halfhill, M.; Millwood, R.; Stewart, C. Quantitative GFP fluorescence as an indicator of recombinant protein synthesis in transgenic plants. Plant. Cell Rep. 2003, 22, 117–121. [Google Scholar] [CrossRef]
- Robins-Browne, R.M.; Holt, K.E.; Ingle, D.J.; Hocking, D.M.; Yang, J.; Tauschek, M. Are Escherichia coli pathotypes still relevant in the era of whole-genome sequencing? Front. Cell Infect. Microbiol. 2016, 6, 141. [Google Scholar] [CrossRef]
- Ji, X.; Liang, B.; Sun, Y.; Zhu, L.; Zhou, B.; Guo, X.; Liu, J. An Extended-spectrum beta-lactamase-producing hybrid Shiga-toxigenic and enterotoxigenic Escherichia coli strain isolated from a piglet with diarrheal disease in northeast China. Foodborne Pathog. Dis. 2020, 17, 382–387. [Google Scholar] [CrossRef]
- Morita, S.; Sato, S.; Maruyama, S.; Nagasaka, M.; Murakami, K.; Inada, K.; Uchiumi, M.; Yokoyama, E.; Asakura, H.; Sugiyama, H.; et al. Whole-genome sequence analysis of Shiga toxin-producing Escherichia coli O157 strains isolated from wild deer and boar in Japan. J. Vet. Med. Sci. 2021, 83, 1860–1868. [Google Scholar] [CrossRef]
- García, V.; Gambino, M.; Pedersen, K.; Haugegaard, S.; Olsen, J.E.; Herrero-Fresno, A. F4- and F18-positive enterotoxigenic Escherichia coli isolates from diarrhea of postweaning pigs: Genomic characterization. Appl. Environ. Microbiol. 2020, 86, e01913-20. [Google Scholar] [CrossRef]
- Da Silva, W.M.; Bei, J.; Amigo, N.; Valacco, M.P.; Amadio, A.; Zhang, Q.; Wu, X.; Yu, T.; Larzabal, M.; Chen, Z.; et al. Quantification of enterohemorrhagic Escherichia coli O157:H7 protein abundance by high-throughput proteome. PLoS ONE 2018, 13, e0208520. [Google Scholar] [CrossRef]
- von Mentzer, A.; Tobias, J.; Wiklund, G.; Nordqvist, S.; Aslett, M.; Dougan, G.; Sjöling, Å.; Svennerholm, A.M. Identification and characterization of the novel colonization factor CS30 based on whole genome sequencing in enterotoxigenic Escherichia coli (ETEC). Sci. Rep. 2017, 7, 12514. [Google Scholar] [CrossRef]
- Han, X.; Ding, S.; Ma, Y.; Fang, J.; Jiang, H.; Li, Y.; Liu, G. Lactobacillus plantarum and Lactobacillus brevis alleviate intestinal inflammation and microbial disorder induced by ETEC in a murine model. Oxid. Med. Cell Longev. 2021, 2021, 6867962. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zhang, Y.; Zhang, D.; Bai, Y.; Liu, G.; Li, P.; Li, J.; Li, Y. Immunogenicity and protective efficacy of enterotoxigenic Escherichia coli (ETEC) total RNA against ETEC challenge in a mouse model. Sci. Rep. 2020, 10, 20530. [Google Scholar] [CrossRef] [PubMed]
- Vermeire, B.; Gonzalez, L.M.; Jansens, R.J.J.; Cox, E.; Devriendt, B. Porcine small intestinal organoids as a model to explore ETEC-host interactions in the gut. Vet. Res. 2021, 52, 94. [Google Scholar] [CrossRef]
- Bussalleu, E.; Pinart, E.; Yeste, M.; Briz, M.; Sancho, S.; Torner, E.; Bonet, S. A PCR technique to detect enterotoxigenic and verotoxigenic Escherichia coli in boar semen samples. Res. Vet. Sci. 2012, 93, 31–33. [Google Scholar] [CrossRef] [PubMed]
- Prieto, A.; López-Novo, C.; Díaz, P.; Díaz-Cao, J.M.; López-Lorenzo, G.; Antón, C.; Remesar, S.; García-Dios, D.; López, C.; Panadero, R.; et al. Antimicrobial susceptibility of enterotoxigenic Escherichia coli from diarrhoeic neonatal calves in spain. Animals 2022, 12, 264. [Google Scholar] [CrossRef]
- Matsuo, T.; Okoda, Y.; Badgar, B.; Inoue, N.; Xuan, X.; Taylor, D.; Fujisaki, K. Fate of GFP-expressing Escherichia coli in the midgut and response to ingestion in a tick, Ornithodoros moubata (Acari: Argasidae). Exp. Parasitol. 2004, 108, 67–73. [Google Scholar] [CrossRef]
- Park, P.-G.; Cho, M.-H.; Rhie G-e Jeong, H.; Youn, H.; Hong, K.-J. GFP-tagged E. coli shows bacterial distribution in mouse organs: Pathogen tracking using fluorescence signal. Clin. Exp. Vaccine Res. 2012, 1, 83–87. [Google Scholar] [CrossRef][Green Version]
- Barolo, S.; Castro, B.; Posakony, J.W. New Drosophila transgenic reporters: Insulated P-element vectors expressing fast-maturing RFP. Biotechniques 2004, 36, 436–442. [Google Scholar] [CrossRef]
- Hua, Z.; Wu, C.; Fan, G.; Tang, Z.; Cao, F. The antibacterial activity and mechanism of ginkgolic acid C15:1. BMC Biotechnol. 2017, 17, 5. [Google Scholar] [CrossRef]
- Rocha, E.M.; Marinotti, O.; Serrão, D.M.; Correa, L.V.; Katak, R.M.; de Oliveira, J.C.; Muniz, V.A.; de Oliveira, M.R.; do Nascimento Neto, J.F.; Pessoa, M.C.F.; et al. Culturable bacteria associated with Anopheles darlingi and their paratransgenesis potential. Malar. J. 2021, 20, 40. [Google Scholar] [CrossRef]
- Ferris, R.A.; McCue, P.M.; Borlee, G.I.; Glapa, K.E.; Martin, K.H.; Mangalea, M.R.; Hennet, M.L.; Wolfe, L.M.; Broeckling, C.D.; Borlee, B.R. Model of chronic equine endometritis involving a pseudomonas aeruginosa biofilm. Infect. Immun. 2017, 85, e00332-17. [Google Scholar] [CrossRef] [PubMed]
- Douglas, G.L.; Klaenhammer, T.R. Directed chromosomal integration and expression of the reporter gene gusA3 in Lactobacillus acidophilus NCFM. Appl. Environ. Microbiol. 2011, 77, 7365–7371. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Xu, Y.; Li, X.; Li, G.; Zhao, H.; Wang, L. Expression and purification of a recombinant enterotoxin protein using different E. coli host strains and expression vectors. Protein J. 2021, 40, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhong, Z.; Luo, Y.; Cox, E.; Devriendt, B. Heat-stable enterotoxins of enterotoxigenic Escherichia coli and their impact on host immunity. Toxins 2019, 11, 24. [Google Scholar] [CrossRef] [PubMed]
- Fleckenstein, J.M.; Hardwidge, P.R.; Munson, G.P.; Rasko, D.A.; Sommerfelt, H.; Steinsland, H. Molecular mechanisms of enterotoxigenic Escherichia coli infection. Microbes Infect. 2010, 12, 89–98. [Google Scholar] [CrossRef]
- Wang, X.; He, J.; Le, K. Making point mutations in Escherichia coli BL21 genome using the CRISPR-Cas9 system. FEMS Microbiol. Lett. 2018, 365, fny060. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).