
Hepatic Expression of the Na+-Taurocholate Cotransporting Polypeptide Is Independent from Genetic Variation

 ,
,  , , , , ,
, , , , ,  , ,
, , 
Abstract
1. Introduction
2. Results
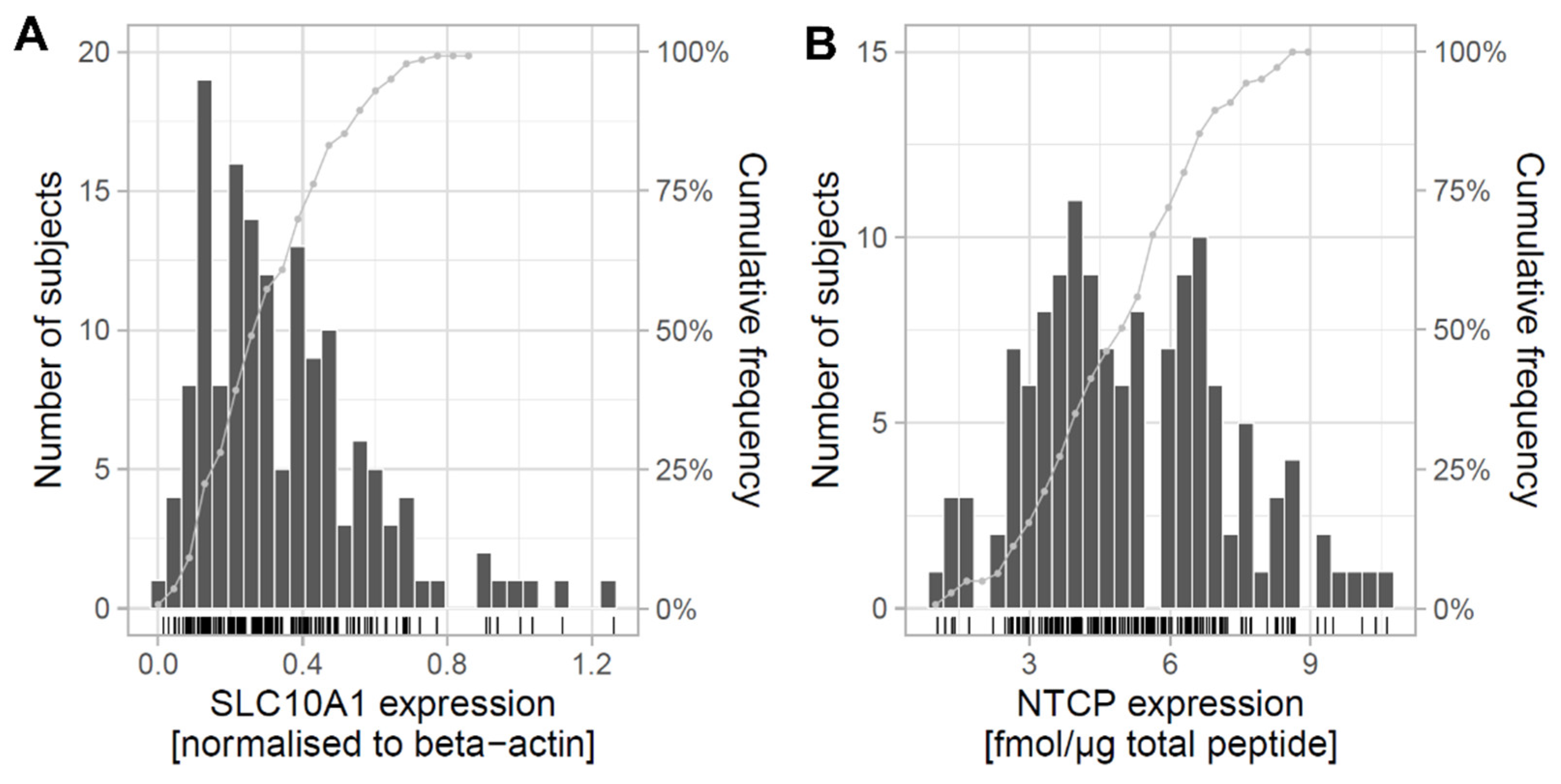
2.1. Expression of SLC10A1/NTCP in Human Liver and Correlation with Clinical Data
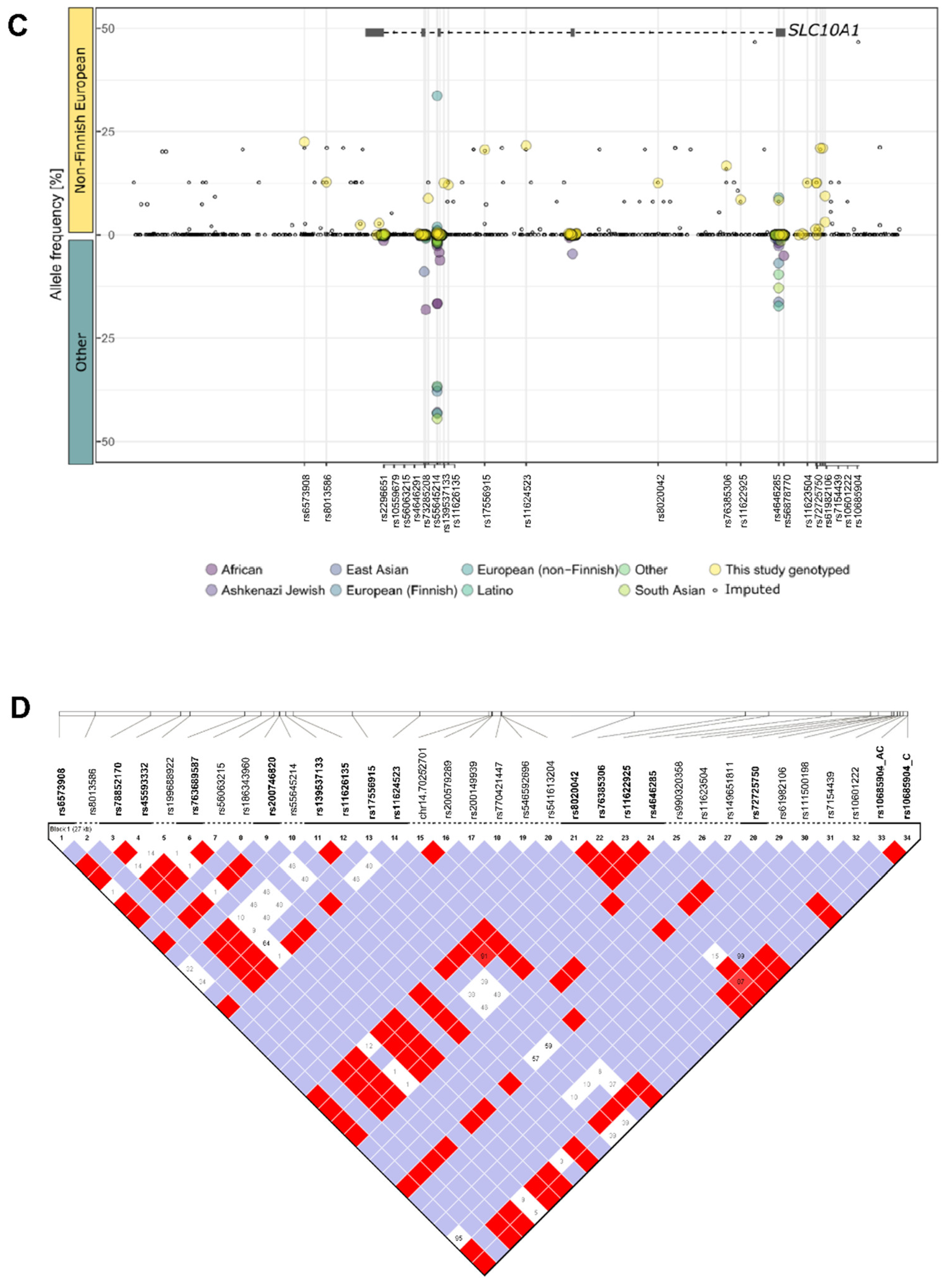
2.2. Genetic Variability of SLC10A1 in Human Liver
2.3. Impact of SLC10A1 Genetics on SLC10A1/NTCP Expression
2.4. Genetic Variability and Expression of SLC10A1 in Human Livers of the Cancer Genome Atlas
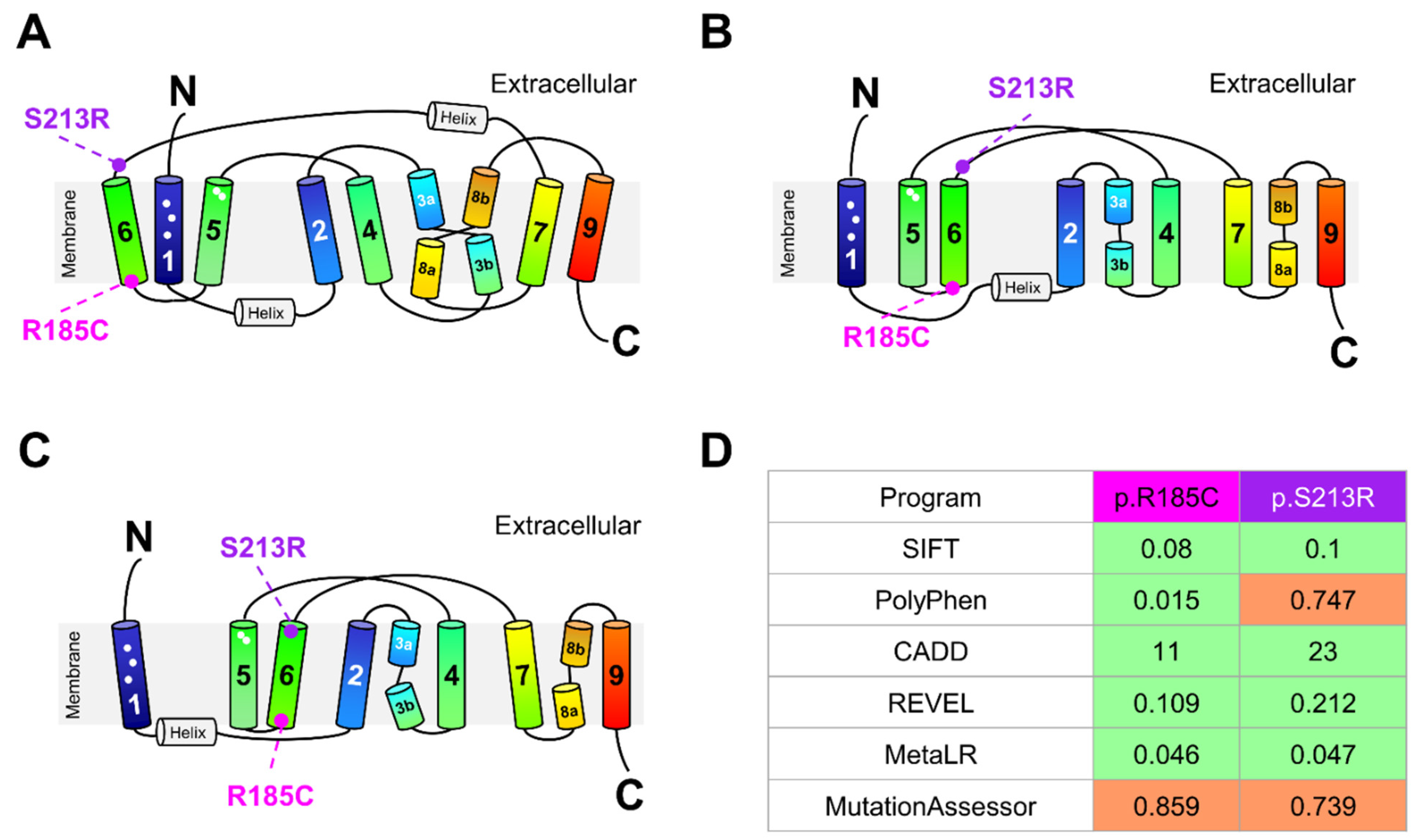
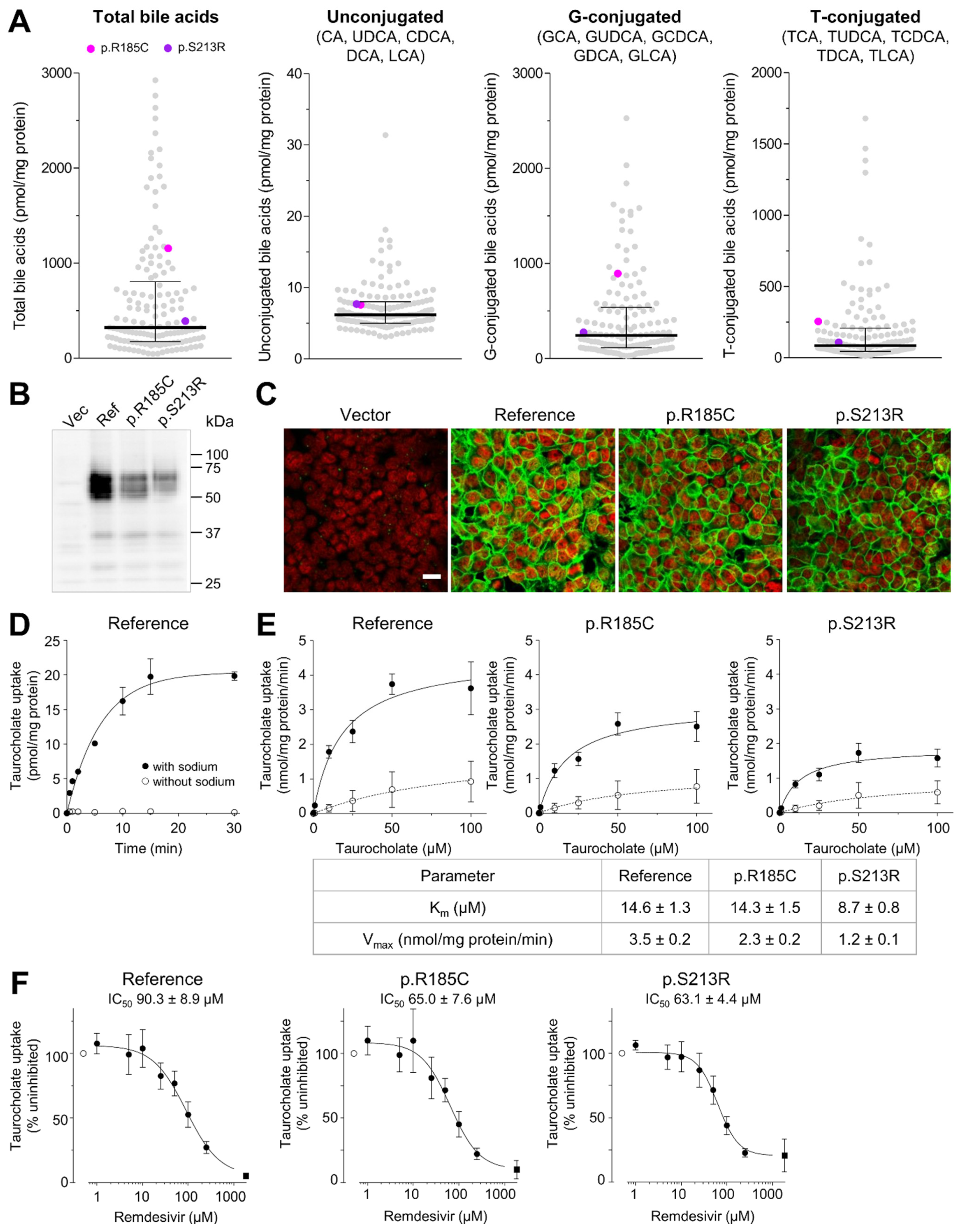
2.5. Impact of SLC10A1 Missense Variants on SLC10A1/NTCP Function
2.6. Genome-Wide Association Study (GWAS)
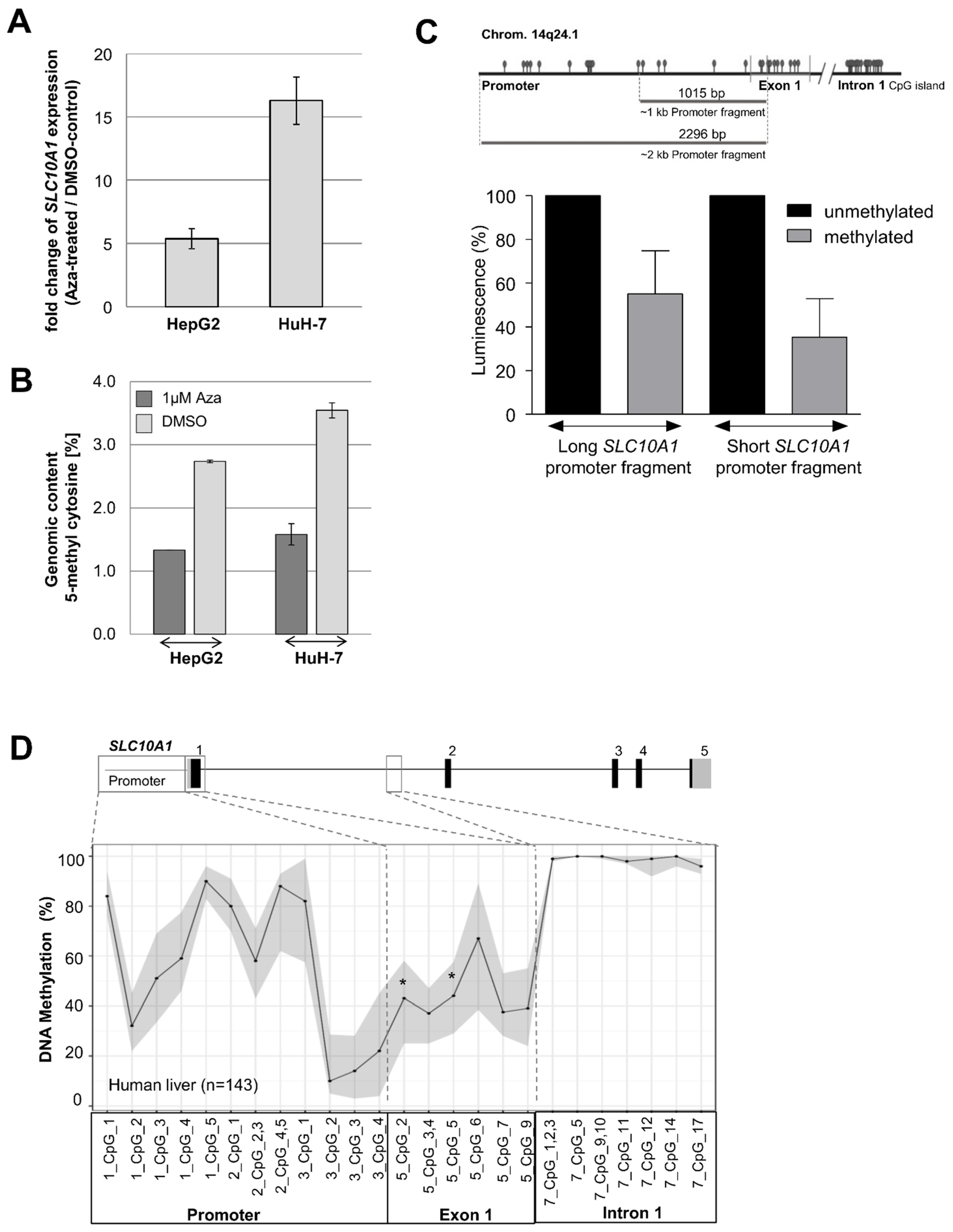
2.7. Epigenetic Regulation of SLC10A1/NTCP
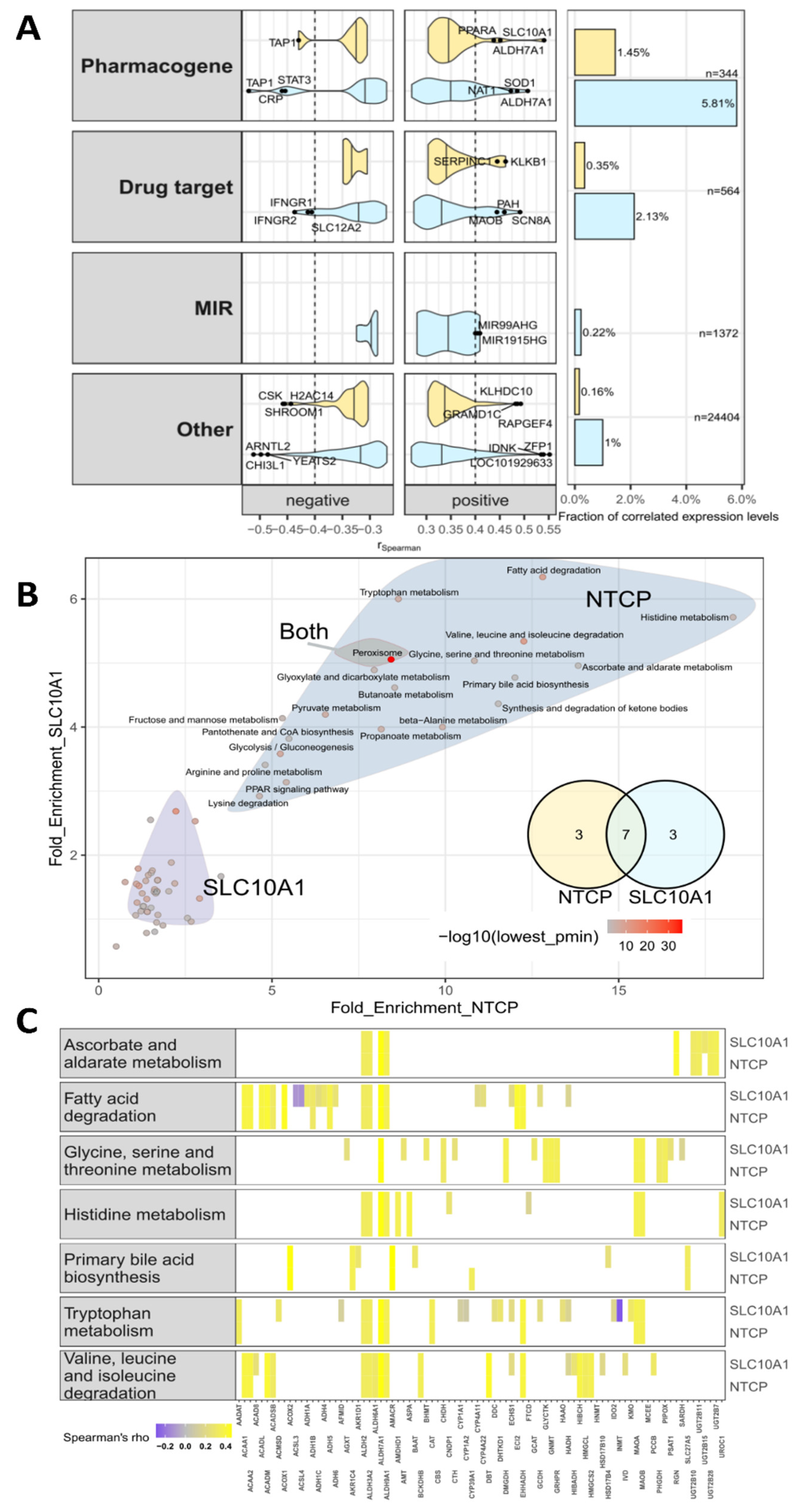
2.8. Genome-Wide Expression Correlation Analysis and Gene Set Enrichment Analysis
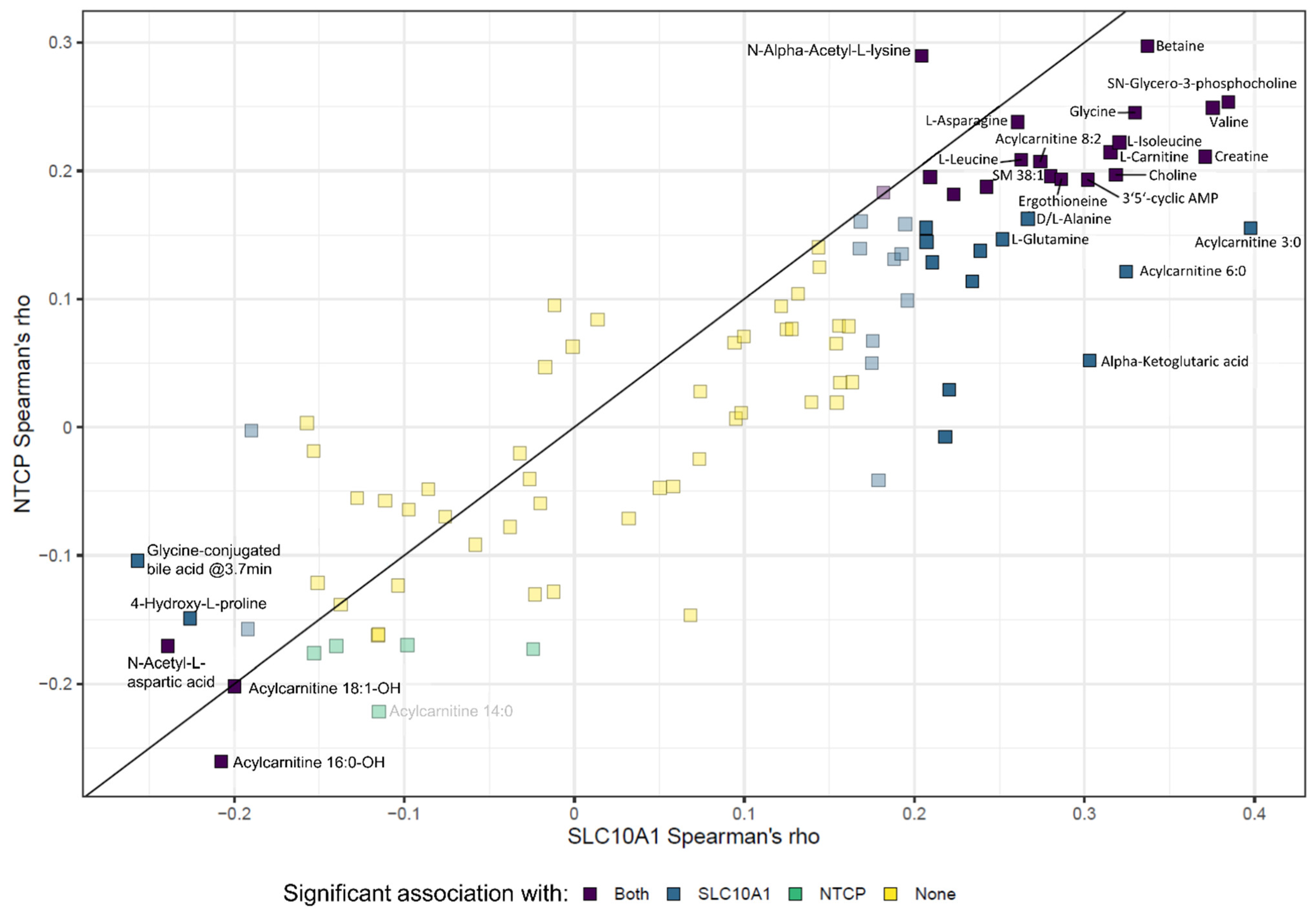
2.9. SLC10A1/NTCP Expression and Hepatic Metabolism
3. Discussion
4. Materials and Methods
4.1. Liver Tissue Samples
4.2. Analysis of SLC10A1 Expression and Genetic Variability in the TCGA LIHC Cohort
4.3. RNA Isolation and Quantification
4.4. LC–MS/MS-Based Targeted Proteomics
4.4.1. Sample Preparation and Digestion
4.4.2. LC-MS/MS and Data Analysis
4.5. SLC10A1 Sequencing and Genotyping
4.5.1. Next Generation Sequencing (NGS)
4.5.2. MALDI-TOF MS-Based Genotyping
4.5.3. Sanger Sequencing
4.5.4. Genome-Wide SNP Microarray
4.6. Location of NTCP Variants in the Human NTCP Structure and In Silico Prediction of Their Functional Effects
4.7. Generation and Characterization of Cell Lines Overexpressing NTCP and NTCP Variants
4.8. Uptake Studies
4.9. DNA Methylation Analyses Using MALDI-TOF Mass Spectrometry
4.10. Cell Culture Experiments and Treatment with 5-Aza-2′-deoxycytidine
4.11. SLC10A1 Promoter-Reporter-Gene Constructs
4.12. Quantification of Bile Acids
4.13. Untargeted Metabolomics Analyses
4.14. Transcriptome and Gene Set Enrichment Analysis
4.15. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Anwer, M.S.; Stieger, B. Sodium-dependent bile salt transporters of the SLC10A transporter family: More than solute transporters. Pflug. Arch. 2014, 466, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Döring, B.; Lütteke, T.; Geyer, J.; Petzinger, E. The SLC10 carrier family: Transport functions and molecular structure. Curr. Top. Membr. 2012, 70, 105–168. [Google Scholar] [CrossRef]
- Stieger, B. The role of the sodium-taurocholate cotransporting polypeptide (NTCP) and of the bile salt export pump (BSEP) in physiology and pathophysiology of bile formation. Handb. Exp. Pharmacol. 2011, 201, 205–259. [Google Scholar] [CrossRef]
- Claro da Silva, T.; Polli, J.E.; Swaan, P.W. The solute carrier family 10 (SLC10): Beyond bile acid transport. Mol. Asp. Med. 2013, 34, 252–269. [Google Scholar] [CrossRef] [PubMed]
- Ho, R.H.; Tirona, R.G.; Leake, B.F.; Glaeser, H.; Lee, W.; Lemke, C.J.; Wang, Y.; Kim, R.B. Drug and bile acid transporters in rosuvastatin hepatic uptake: Function, expression, and pharmacogenetics. Gastroenterology 2006, 130, 1793–1806. [Google Scholar] [CrossRef] [PubMed]
- Toth, P.P.; Dayspring, T.D. Drug safety evaluation of rosuvastatin. Expert Opin. Drug Saf. 2011, 10, 969–986. [Google Scholar] [CrossRef]
- Cadranel, J.F.; Erlinger, S.; Desruenne, M.; Luciani, J.; Lunel, F.; Grippon, P.; Cabrol, A.; Opolon, P. Chronic administration of cyclosporin A induces a decrease in hepatic excretory function in man. Digest. Dis. Sci. 1992, 37, 1473–1476. [Google Scholar] [CrossRef]
- Blank, A.; Eidam, A.; Haag, M.; Hohmann, N.; Burhenne, J.; Schwab, M.; van de Graaf, S.; Meyer, M.R.; Maurer, H.H.; Meier, K.; et al. The NTCP-inhibitor Myrcludex B: Effects on bile acid disposition and tenofovir pharmacokinetics. Clin. Pharmacol. Ther. 2018, 103, 341–348. [Google Scholar] [CrossRef]
- McRae, M.; Rezk, N.L.; Bridges, A.S.; Corbett, A.H.; Tien, H.-C.; Brouwer, K.L.R.; Kashuba, A.D.M. Plasma bile acid concentrations in patients with human immunodeficiency virus infection receiving protease inhibitor therapy: Possible implications for hepatotoxicity. Pharmacotherapy 2010, 30, 17–24. [Google Scholar] [CrossRef]
- Stauber, R.E.; Fauler, G.; Rainer, F.; Leber, B.; Posch, A.; Streit, A.; Spindelboeck, W.; Stadlbauer, V.; Kessler, H.H.; Mangge, H. Anti-HCV treatment with ombitasvir/paritaprevir/ritonavir ± dasabuvir is associated with increased bile acid levels and pruritus. Wien. Klin. Wochenschr. 2017, 129, 848–851. [Google Scholar] [CrossRef]
- Trauner, M.; Fuchs, C.D.; Halilbasic, E.; Paumgartner, G. New therapeutic concepts in bile acid transport and signaling for management of cholestasis. Hepatology 2017, 65, 1393–1404. [Google Scholar] [CrossRef] [PubMed]
- Welling, P.G. Pharmacokinetics of the thiazide diuretics. Biopharm. Drug Dispos. 1986, 7, 501–535. [Google Scholar] [CrossRef] [PubMed]
- Ingle, B.L.; Veber, B.C.; Nichols, J.W.; Tornero-Velez, R. Informing the human plasma protein binding of environmental chemicals by machine learning in the pharmaceutical space: Applicability domain and limits of predictability. J. Chem. Inf. Model. 2016, 56, 2243–2252. [Google Scholar] [CrossRef] [PubMed]
- Dingemanse, J.; van Giersbergen, P.L.M. Clinical pharmacology of bosentan, a dual endothelin receptor antagonist. Clin. Pharmacokinet. 2004, 43, 1089–1115. [Google Scholar] [CrossRef] [PubMed]
- Venitz, J.; Zack, J.; Gillies, H.; Allard, M.; Regnault, J.; Dufton, C. Clinical pharmacokinetics and drug-drug interactions of endothelin receptor antagonists in pulmonary arterial hypertension. J. Clin. Pharmacol. 2012, 52, 1784–1805. [Google Scholar] [CrossRef]
- Leslie, E.M.; Watkins, P.B.; Kim, R.B.; Brouwer, K.L.R. Differential inhibition of rat and human Na+-dependent taurocholate cotransporting polypeptide (NTCP/SLC10A1) by bosentan: A mechanism for species differences in hepatotoxicity. J. Pharmacol. Exp. Ther. 2007, 321, 1170–1178. [Google Scholar] [CrossRef]
- Kaiser, H.; Aaronson, D.; Dockhorn, R.; Edsbäcker, S.; Korenblat, P.; Källén, A. Dose-proportional pharmacokinetics of budesonide inhaled via Turbuhaler. Br. J. Clin. Pharmacol. 1999, 48, 309–316. [Google Scholar] [CrossRef]
- Blank, A.; Markert, C.; Hohmann, N.; Carls, A.; Mikus, G.; Lehr, T.; Alexandrov, A.; Haag, M.; Schwab, M.; Urban, S.; et al. First-in-human application of the novel hepatitis B and hepatitis D virus entry inhibitor myrcludex B. J. Hepatol. 2016, 65, 483–489. [Google Scholar] [CrossRef]
- Nkongolo, S.; Ni, Y.; Lempp, F.A.; Kaufman, C.; Lindner, T.; Esser-Nobis, K.; Lohmann, V.; Mier, W.; Mehrle, S.; Urban, S. Cyclosporin A inhibits hepatitis B and hepatitis D virus entry by cyclophilin-independent interference with the NTCP receptor. J. Hepatol. 2014, 60, 723–731. [Google Scholar] [CrossRef]
- Kim, J.-R.; Kim, S.; Huh, W.; Ko, J.-W. No pharmacokinetic interactions between candesartan and amlodipine following multiple oral administrations in healthy subjects. Drug Des. Devel. Ther. 2018, 12, 2475–2483. [Google Scholar] [CrossRef]
- Ito, K.; Chiba, K.; Horikawa, M.; Ishigami, M.; Mizuno, N.; Aoki, J.; Gotoh, Y.; Iwatsubo, T.; Kanamitsu, S.; Kato, M.; et al. Which concentration of the inhibitor should be used to predict in vivo drug interactions from in vitro data? AAPS PharmSci. 2002, 4, E25. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.B.; Leake, B.; Cvetkovic, M.; Roden, M.M.; Nadeau, J.; Walubo, A.; Wilkinson, G.R. Modulation by drugs of human hepatic sodium-dependent bile acid transporter (sodium taurocholate cotransporting polypeptide) activity. J. Pharmacol. Exp. Ther. 1999, 291, 1204–1209. [Google Scholar] [PubMed]
- Vaidyanathan, J.; Yoshida, K.; Arya, V.; Zhang, L. Comparing various in vitro prediction criteria to assess the potential of a new molecular entity to inhibit organic anion transporting polypeptide 1B1. J. Clin. Pharmacol. 2016, 56 (Suppl. S7), S59–S72. [Google Scholar] [CrossRef] [PubMed]
- Unger, M.S.; Mudunuru, J.; Schwab, M.; Hopf, C.; Drewes, G.; Nies, A.T.; Zamek-Gliszczynski, M.J.; Reinhard, F. Clinically-relevant OATP2B1 inhibitors in marketed drug space. Mol. Pharm. 2020, 17, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Rekić, D.; Röshammar, D.; Mukonzo, J.; Ashton, M. In silico prediction of efavirenz and rifampicin drug-drug interaction considering weight and CYP2B6 phenotype. Br. J. Clin. Pharmacol. 2011, 71, 536–543. [Google Scholar] [CrossRef] [PubMed]
- McRae, M.P.; Lowe, C.M.; Tian, X.; Bourdet, D.L.; Ho, R.H.; Leake, B.F.; Kim, R.B.; Brouwer, K.L.R.; Kashuba, A.D.M. Ritonavir, saquinavir, and efavirenz, but not nevirapine, inhibit bile acid transport in human and rat hepatocytes. J. Pharmacol. Exp. Ther. 2006, 318, 1068–1075. [Google Scholar] [CrossRef] [PubMed]
- Kelly, M.R.; Cutler, R.E.; Forrey, A.W.; Kimpel, B.M. Pharmacokinetics of orally administered furosemide. Clin. Pharmacol. Ther. 1974, 15, 178–186. [Google Scholar]
- Manvelian, G.; Daniels, S.; Altman, R. A phase I study evaluating the pharmacokinetic profile of a novel, proprietary, nano-formulated, lower-dose oral indomethacin. Postgrad. Med. 2012, 124, 197–205. [Google Scholar] [CrossRef]
- Ishizaki, T.; Sasaki, T.; Suganuma, T.; Horai, Y.; Chiba, K.; Watanabe, M.; Asuke, W.; Hoshi, H. Pharmacokinetics of ketoprofen following single oral, intramuscular and rectal doses and after repeated oral administration. Eur. J. Clin. Pharmacol. 1980, 18, 407–414. [Google Scholar] [CrossRef]
- Niemi, M.; Backman, J.T.; Neuvonen, M.; Neuvonen, P.J. Effect of rifampicin on the pharmacokinetics and pharmacodynamics of nateglinide in healthy subjects. Br. J. Clin. Pharmacol. 2003, 56, 427–432. [Google Scholar] [CrossRef]
- Barbhaiya, R.H.; Shukla, U.A.; Chaikin, P.; Greene, D.S.; Marathe, P.H. Nefazodone pharmacokinetics: Assessment of nonlinearity, intra-subject variability and time to attain steady-state plasma concentrations after dose escalation and de-escalation. Eur. J. Clin. Pharmacol. 1996, 50, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Blardi, P.; Urso, R.; Lalla, A.; de Volpi, L.; Di Perri, T.; Auteri, A. Nimodipine: Drug pharmacokinetics and plasma adenosine levels in patients affected by cerebral ischemia. Clin. Pharmacol. Ther. 2002, 72, 556–561. [Google Scholar] [CrossRef]
- Kirch, W.; Hutt, H.J.; Heidemann, H.; Rämsch, K.; Janisch, H.D.; Ohnhaus, E.E. Drug interactions with nitrendipine. J. Cardiovasc. Pharmacol. 1984, 6 (Suppl. S7), S982–S985. [Google Scholar] [CrossRef] [PubMed]
- Selen, A.; Amidon, G.L.; Welling, P.G. Pharmacokinetics of probenecid following oral doses to human volunteers. J. Pharm. Sci. 1982, 71, 1238–1242. [Google Scholar] [CrossRef] [PubMed]
- Donkers, J.M.; Zehnder, B.; van Westen, G.J.P.; Kwakkenbos, M.J.; IJzerman, A.P.; Oude Elferink, R.P.J.; Beuers, U.; Urban, S.; van de Graaf, S.F.J. Reduced hepatitis B and D viral entry using clinically applied drugs as novel inhibitors of the bile acid transporter NTCP. Sci. Rep. 2017, 7, 15307. [Google Scholar] [CrossRef]
- Martin, P.D.; Warwick, M.J.; Dane, A.L.; Hill, S.J.; Giles, P.B.; Phillips, P.J.; Lenz, E. Metabolism, excretion, and pharmacokinetics of rosuvastatin in healthy adult male volunteers. Clin. Ther. 2003, 25, 2822–2835. [Google Scholar] [CrossRef]
- Ryu, S.; Novak, J.J.; Patel, R.; Yates, P.; Di, L. The impact of low temperature on fraction unbound for plasma and tissue. Biopharm. Drug Dispos. 2018, 39, 437–442. [Google Scholar] [CrossRef]
- FDA Guidance. In Vitro Drug Interaction Studies—Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions Guidance for Industry. 2020. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/vitro-drug-interaction-studies-cytochrome-p450-enzyme-and-transporter-mediated-drug-interactions (accessed on 10 February 2022).
- Brouwer, K.L.; Keppler, D.; Hoffmaster, K.A.; Bow, D.A.; Cheng, Y.; Lai, Y.; Palm, J.E.; Stieger, B.; Evers, R. In vitro methods to support transporter evaluation in drug discovery and development. Clin. Pharmacol. Ther. 2013, 94, 95–112. [Google Scholar] [CrossRef]
- Dong, Z.; Ekins, S.; Polli, J.E. Quantitative NTCP pharmacophore and lack of association between DILI and NTCP Inhibition. Eur. J. Pharm. Sci. 2015, 66, 1–9. [Google Scholar] [CrossRef]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 2012, 1, e00049. [Google Scholar] [CrossRef]
- Watashi, K.; Urban, S.; Li, W.; Wakita, T. NTCP and beyond: Opening the door to unveil hepatitis B virus entry. Int. J. Mol. Sci. 2014, 15, 2892–2905. [Google Scholar] [CrossRef]
- Ni, Y.; Lempp, F.A.; Mehrle, S.; Nkongolo, S.; Kaufman, C.; Fälth, M.; Stindt, J.; Königer, C.; Nassal, M.; Kubitz, R.; et al. Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes. Gastroenterology 2014, 146, 1070–1083. [Google Scholar] [CrossRef] [PubMed]
- Hu, N.-J.; Iwata, S.; Cameron, A.D.; Drew, D. Crystal structure of a bacterial homologue of the bile acid sodium symporter ASBT. Nature 2011, 478, 408–411. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Levin, E.J.; Pan, Y.; McCoy, J.G.; Sharma, R.; Kloss, B.; Bruni, R.; Quick, M.; Zhou, M. Structural basis of the alternating-access mechanism in a bile acid transporter. Nature 2014, 505, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Asami, J.; Kimura, K.T.; Fujita-Fujiharu, Y.; Ishida, H.; Zhang, Z.; Nomura, Y.; Liu, K.; Uemura, T.; Sato, Y.; Ono, M.; et al. Structure of bile acid transporter NTCP crucial for hepatitis B virus entry. Nature 2022, 606, 1021–1026. [Google Scholar] [CrossRef]
- Goutam, K.; Ielasi, F.S.; Pardon, E.; Steyaert, J.; Reyes, N. Structural basis of sodium-dependent bile salt uptake into the liver. Nature 2022, 606, 1015–1020. [Google Scholar] [CrossRef]
- Park, J.-H.; Iwamoto, M.; Yun, J.-H.; Uchikubo-Kamo, T.; Son, D.; Jin, Z.; Yoshida, H.; Ohki, M.; Ishimoto, N.; Mizutani, K.; et al. Structural insights into the HBV receptor and bile acid transporter NTCP. Nature 2022, 606, 1027–1031. [Google Scholar] [CrossRef]
- Kubitz, R.; Dröge, C.; Kluge, S.; Stindt, J.; Häussinger, D. Genetic variations of bile salt transporters. Drug Discov. Today Technol. 2014, 12, e55–e67. [Google Scholar] [CrossRef]
- Russell, L.E.; Zhou, Y.; Lauschke, V.M.; Kim, R.B. In vitro functional characterization and in silico prediction of rare genetic variation in the bile acid and drug transporter, Na+-taurocholate cotransporting polypeptide (NTCP, SLC10A1). Mol. Pharm. 2020, 17, 1170–1181. [Google Scholar] [CrossRef]
- Vaz, F.M.; Paulusma, C.C.; Huidekoper, H.; Ru, M.; de Lim, C.; Koster, J.; Ho-Mok, K.; Bootsma, A.H.; Groen, A.K.; Schaap, F.G.; et al. Sodium taurocholate cotransporting polypeptide (SLC10A1) deficiency: Conjugated hypercholanemia without a clear clinical phenotype. Hepatology 2015, 61, 260–267. [Google Scholar] [CrossRef]
- Deng, M.; Mao, M.; Guo, L.; Chen, F.-P.; Wen, W.-R.; Song, Y.-Z. Clinical and molecular study of a pediatric patient with sodium taurocholate cotransporting polypeptide deficiency. Exp. Ther. Med. 2016, 12, 3294–3300. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.-J.; Deng, M.; Qiu, J.-W.; Wu, J.-F.; Song, Y.-Z. Monozygotic twins suffering from sodium taurocholate cotransporting polypeptide deficiency: A case report. Front. Pediatr. 2018, 6, 354. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Peng, B.; Liu, Y.; Xu, G.; He, W.; Ren, B.; Jing, Z.; Sui, J.; Li, W. Viral entry of hepatitis B and D viruses and bile salts transportation share common molecular determinants on sodium taurocholate cotransporting polypeptide. J. Virol. 2014, 88, 3273–3284. [Google Scholar] [CrossRef]
- Binh, M.T.; Hoan, N.X.; van Tong, H.; Sy, B.T.; Trung, N.T.; Bock, C.-T.; Toan, N.L.; Le Song, H.; Bang, M.H.; Meyer, C.G.; et al. NTCP S267F variant associates with decreased susceptibility to HBV and HDV infection and decelerated progression of related liver diseases. Int. J. Infect. Dis. 2019, 80, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Nies, A.T.; Niemi, M.; Burk, O.; Winter, S.; Zanger, U.M.; Stieger, B.; Schwab, M.; Schaeffeler, E. Genetics is a major determinant of expression of the human hepatic uptake transporter OATP1B1, but not of OATP1B3 and OATP2B1. Genome Med. 2013, 5, 65. [Google Scholar] [CrossRef] [PubMed]
- Schaeffeler, E.; Hellerbrand, C.; Nies, A.T.; Winter, S.; Kruck, S.; Hofmann, U.; van der Kuip, H.; Zanger, U.M.; Koepsell, H.; Schwab, M. DNA methylation is associated with downregulation of the organic cation transporter OCT1 (SLC22A1) in human hepatocellular carcinoma. Genome Med. 2011, 3, 82. [Google Scholar] [CrossRef] [PubMed]
- Kojima, H.; Nies, A.T.; König, J.; Hagmann, W.; Spring, H.; Uemura, M.; Fukui, H.; Keppler, D. Changes in the expression and localization of hepatocellular transporters and radixin in primary biliary cirrhosis. J. Hepatol. 2003, 39, 693–702. [Google Scholar] [CrossRef]
- Eastman, R.T.; Roth, J.S.; Brimacombe, K.R.; Simeonov, A.; Shen, M.; Patnaik, S.; Hall, M.D. Remdesivir: A review of its discovery and development leading to emergency use authorization for treatment of COVID-19. ACS Cent. Sci. 2020, 6, 672–683. [Google Scholar] [CrossRef]
- EMA. Summary on Compassionate Use; Procedure No. EMEA/H/K/5622/CU; European Medicines Agency: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Ahlin, G.; Chen, L.; Lazorova, L.; Chen, Y.; Ianculescu, A.G.; Davis, R.L.; Giacomini, K.M.; Artursson, P. Genotype-dependent effects of inhibitors of the organic cation transporter, OCT1: Predictions of metformin interactions. Pharm. J. 2011, 11, 400–411. [Google Scholar] [CrossRef]
- Winter, S.; Fisel, P.; Büttner, F.; Rausch, S.; D’Amico, D.; Hennenlotter, J.; Kruck, S.; Nies, A.T.; Stenzl, A.; Junker, K.; et al. Methylomes of renal cell lines and tumors or metastases differ significantly with impact on pharmacogenes. Sci. Rep. 2016, 6, 29930. [Google Scholar] [CrossRef]
- Klein, K.; Tremmel, R.; Winter, S.; Fehr, S.; Battke, F.; Scheurenbrand, T.; Schaeffeler, E.; Biskup, S.; Schwab, M.; Zanger, U.M. A new panel-based next-generation sequencing method for ADME genes Reveals novel associations of common and rare variants with expression in a human liver cohort. Front. Genet. 2019, 10, 7. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Arribas, G.H.; Turku, A.; Jürgenson, T.; Mkrtchian, S.; Krebs, K.; Wang, Y.; Svobodova, B.; Milani, L.; Schulte, G.; et al. Rare genetic variability in human drug target genes modulates drug response and can guide precision medicine. Sci. Adv. 2021, 7, eabi6856. [Google Scholar] [CrossRef]
- Gaude, E.; Frezza, C. Tissue-specific and convergent metabolic transformation of cancer correlates with metastatic potential and patient survival. Nat. Commun. 2016, 7, 13041. [Google Scholar] [CrossRef] [PubMed]
- Leuthold, P.; Schaeffeler, E.; Winter, S.; Büttner, F.; Hofmann, U.; Mürdter, T.E.; Rausch, S.; Sonntag, D.; Wahrheit, J.; Fend, F.; et al. Comprehensive metabolomic and lipidomic profiling of human kidney tissue: A platform comparison. J. Proteome Res. 2017, 16, 933–944. [Google Scholar] [CrossRef] [PubMed]
- Nies, A.T.; Koepsell, H.; Winter, S.; Burk, O.; Klein, K.; Kerb, R.; Zanger, U.M.; Keppler, D.; Schwab, M.; Schaeffeler, E. Expression of organic cation transporters OCT1 (SLC22A1) and OCT3 (SLC22A3) is affected by genetic factors and cholestasis in human liver. Hepatology 2009, 50, 1227–1240. [Google Scholar] [CrossRef]
- Kurzawski, M.; Szeląg-Pieniek, S.; Łapczuk-Romańska, J.; Wrzesiński, M.; Sieńko, J.; Oswald, S.; Droździk, M. The reference liver—ABC and SLC drug transporters in healthy donor and metastatic livers. Pharmacol. Rep. 2019, 71, 738–745. [Google Scholar] [CrossRef] [PubMed]
- Drozdzik, M.; Szelag-Pieniek, S.; Post, M.; Zeair, S.; Wrzesinski, M.; Kurzawski, M.; Prieto, J.; Oswald, S. Protein abundance of hepatic drug transporters in patients with different forms of liver damage. Clin. Pharmacol. Ther. 2019, 107, 1138–1148. [Google Scholar] [CrossRef] [PubMed]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Fickert, P.; Wagner, M. Biliary bile acids in hepatobiliary injury—What is the link? J. Hepatol. 2017, 67, 619–631. [Google Scholar] [CrossRef]
- Han, C.; Li, H.; Ma, Z.; Dong, G.; Wang, Q.; Wang, S.; Fang, P.; Li, X.; Chen, H.; Liu, T.; et al. MIR99AHG is a noncoding tumor suppressor gene in lung adenocarcinoma. Cell Death Dis. 2021, 12, 424. [Google Scholar] [CrossRef]
- Meng, Q.; Wang, X.; Xue, T.; Zhao, Q.; Wang, W.; Zhao, K. Long noncoding RNA MIR99AHG promotes gastric cancer progression by inducing EMT and inhibiting apoptosis via miR577/FOXP1 axis. Cancer Cell Int. 2020, 20, 414. [Google Scholar] [CrossRef] [PubMed]
- Emmrich, S.; Streltsov, A.; Schmidt, F.; Thangapandi, V.R.; Reinhardt, D.; Klusmann, J.-H. LincRNAs MONC and MIR100HG act as oncogenes in acute megakaryoblastic leukemia. Mol. Cancer 2014, 13, 171. [Google Scholar] [CrossRef] [PubMed]
- Rieger, J.K.; Klein, K.; Winter, S.; Zanger, U.M. Expression variability of absorption, distribution, metabolism, excretion-related microRNAs in human liver: Influence of nongenetic factors and association with gene expression. Drug Metab. Dispos. 2013, 41, 1752–1762. [Google Scholar] [CrossRef] [PubMed]
- Geier, A.; Dietrich, C.G.; Voigt, S.; Ananthanarayanan, M.; Lammert, F.; Schmitz, A.; Trauner, M.; Wasmuth, H.E.; Boraschi, D.; Balasubramaniyan, N.; et al. Cytokine-dependent regulation of hepatic organic anion transporter gene transactivators in mouse liver. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G831–G841. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, L.; Zhang, X.; Jiang, X.; Wang, L. Tanshinone IIA prevents rifampicin-induced liver injury by regulating BSEP/NTCP expression via epigenetic activation of NRF2. Liver Int. 2020, 40, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Takahashi, S.; Brocker, C.N.; He, S.; Chen, L.; Xie, G.; Jang, K.; Gao, X.; Krausz, K.W.; Qu, A.; et al. Hepatocyte peroxisome proliferator-activated receptor α regulates bile acid synthesis and transport. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 1396–1411. [Google Scholar] [CrossRef]
- Zhang, Y.; Lickteig, A.J.; Csanaky, I.L.; Klaassen, C.D. Editor’s highlight: Clofibrate decreases bile acids in livers of male mice by increasing biliary bile acid excretion in a PPARα-dependent manner. Toxicol. Sci. 2017, 160, 351–360. [Google Scholar] [CrossRef]
- Lei, W.; Liu, D.; Sun, M.; Lu, C.; Yang, W.; Wang, C.; Cheng, Y.; Zhang, M.; Shen, M.; Yang, Z.; et al. Targeting STAT3: A crucial modulator of sepsis. J. Cell. Physiol. 2021, 236, 7814–7831. [Google Scholar] [CrossRef]
- Abualsunun, W.A.; Sahin, C.; Cummins, C.L.; Piquette-Miller, M. Essential role of STAT-3 dependent NF-κB activation on IL-6-mediated downregulation of hepatic transporters. Eur. J. Pharm. Sci. 2020, 143, 105151. [Google Scholar] [CrossRef]
- Keane, M.H.; Overmars, H.; Wikander, T.M.; Ferdinandusse, S.; Duran, M.; Wanders, R.J.A.; Faust, P.L. Bile acid treatment alters hepatic disease and bile acid transport in peroxisome-deficient PEX2 Zellweger mice. Hepatology 2007, 45, 982–997. [Google Scholar] [CrossRef]
- Zhang, Q.; He, Z.; Liu, Z.; Gong, L. Integrated plasma and liver gas chromatography mass spectrometry and liquid chromatography mass spectrometry metabolomics to reveal physiological functions of sodium taurocholate cotransporting polypeptide (NTCP) with an Ntcp knockout mouse model. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2021, 1165, 122531. [Google Scholar] [CrossRef] [PubMed]
- Lever, M.; Slow, S. The clinical significance of betaine, an osmolyte with a key role in methyl group metabolism. Clin. Biochem. 2010, 43, 732–744. [Google Scholar] [CrossRef] [PubMed]
- Brocker, C.; Lassen, N.; Estey, T.; Pappa, A.; Cantore, M.; Orlova, V.V.; Chavakis, T.; Kavanagh, K.L.; Oppermann, U.; Vasiliou, V. Aldehyde dehydrogenase 7A1 (ALDH7A1) is a novel enzyme involved in cellular defense against hyperosmotic stress. J. Biol. Chem. 2010, 285, 18452–18463. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhayay, S.; Ananthanarayanan, M.; Stieger, B.; Meier, P.J.; Suchy, F.J.; Anwer, M.S. cAMP increases liver Na+-taurocholate cotransport by translocating transporter to plasma membranes. Am. J. Physiol. 1997, 273, G842–G848. [Google Scholar] [CrossRef] [PubMed]
- Schonhoff, C.M.; Thankey, K.; Webster, C.R.L.; Wakabayashi, Y.; Wolkoff, A.W.; Anwer, M.S. Rab4 facilitates cyclic adenosine monophosphate-stimulated bile acid uptake and Na+-taurocholate cotransporting polypeptide translocation. Hepatology 2008, 48, 1665–1670. [Google Scholar] [CrossRef]
- Wegler, C.; Gaugaz, F.Z.; Andersson, T.B.; Wiśniewski, J.R.; Busch, D.; Gröer, C.; Oswald, S.; Norén, A.; Weiss, F.; Hammer, H.S.; et al. Variability in mass spectrometry-based quantification of clinically relevant drug transporters and drug metabolizing enzymes. Mol. Pharm. 2017, 14, 3142–3151. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Gaugaz, F.Z. Fast and sensitive total protein and peptide assays for proteomic analysis. Anal. Chem. 2015, 87, 4110–4116. [Google Scholar] [CrossRef]
- Wiśniewski, J.R. Quantitative evaluation of filter aided sample preparation (FASP) and multienzyme digestion FASP protocols. Anal. Chem. 2016, 88, 5438–5443. [Google Scholar] [CrossRef]
- Emami Riedmaier, A.; Burk, O.; van Eijck, B.A.C.; Schaeffeler, E.; Klein, K.; Fehr, S.; Biskup, S.; Müller, S.; Winter, S.; Zanger, U.M.; et al. Variability in hepatic expression of organic anion transporter 7/SLC22A9, a novel pravastatin uptake transporter: Impact of genetic and regulatory factors. Pharm. J. 2016, 16, 341–351. [Google Scholar] [CrossRef]
- Schröder, A.; Klein, K.; Winter, S.; Schwab, M.; Bonin, M.; Zell, A.; Zanger, U.M. Genomics of ADME gene expression: Mapping expression quantitative trait loci relevant for absorption, distribution, metabolism and excretion of drugs in human liver. Pharm. J. 2013, 13, 12–20. [Google Scholar] [CrossRef]
- Dickens, D.; Rädisch, S.; Chiduza, G.N.; Giannoudis, A.; Cross, M.J.; Malik, H.; Schaeffeler, E.; Sison-Young, R.L.; Wilkinson, E.L.; Goldring, C.E.; et al. Cellular uptake of the atypical antipsychotic clozapine is a carrier-mediated process. Mol. Pharm. 2018, 15, 3557–3572. [Google Scholar] [CrossRef] [PubMed]
- Nies, A.T.; König, J.; Hofmann, U.; Kölz, C.; Fromm, M.F.; Schwab, M. Interaction of remdesivir with clinically relevant hepatic drug uptake transporters. Pharmaceutics 2021, 13, 369. [Google Scholar] [CrossRef] [PubMed]
- Fisel, P.; Kruck, S.; Winter, S.; Bedke, J.; Hennenlotter, J.; Nies, A.T.; Scharpf, M.; Fend, F.; Stenzl, A.; Schwab, M.; et al. DNA methylation of the SLC16A3 promoter regulates expression of the human lactate transporter MCT4 in renal cancer with consequences for clinical outcome. Clin. Cancer Res. 2013, 19, 5170–5181. [Google Scholar] [CrossRef]
- Haag, M.; Hofmann, U.; Mürdter, T.E.; Heinkele, G.; Leuthold, P.; Blank, A.; Haefeli, W.E.; Alexandrov, A.; Urban, S.; Schwab, M. Quantitative bile acid profiling by liquid chromatography quadrupole time-of-flight mass spectrometry: Monitoring hepatitis B therapy by a novel Na(+)-taurocholate cotransporting polypeptide inhibitor. Anal. Bioanal. Chem. 2015, 407, 6815–6825. [Google Scholar] [CrossRef]
- Tamm, R.; Mägi, R.; Tremmel, R.; Winter, S.; Mihailov, E.; Smid, A.; Möricke, A.; Klein, K.; Schrappe, M.; Stanulla, M.; et al. Polymorphic variation in TPMT is the principal determinant of TPMT phenotype: A meta-analysis of three genome-wide association studies. Clin. Pharmacol. Ther. 2017, 101, 684–695. [Google Scholar] [CrossRef] [PubMed]
- Keizer, R.J.; Jansen, R.S.; Rosing, H.; Thijssen, B.; Beijnen, J.H.; Schellens, J.H.M.; Huitema, A.D.R. Incorporation of concentration data below the limit of quantification in population pharmacokinetic analyses. Pharmacol. Res. Perspect. 2015, 3, e00131. [Google Scholar] [CrossRef] [PubMed]
- Dunn, W.B.; Broadhurst, D.; Begley, P.; Zelena, E.; Francis-McIntyre, S.; Anderson, N.; Brown, M.; Knowles, J.D.; Halsall, A.; Haselden, J.N.; et al. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat. Protoc. 2011, 6, 1060–1083. [Google Scholar] [CrossRef]
- Büttner, F.; Winter, S.; Rausch, S.; Hennenlotter, J.; Kruck, S.; Stenzl, A.; Scharpf, M.; Fend, F.; Agaimy, A.; Hartmann, A.; et al. Clinical utility of the S3-score for molecular prediction of outcome in non-metastatic and metastatic clear cell renal cell carcinoma. BMC Med. 2018, 16, 108. [Google Scholar] [CrossRef]
- Schaeffeler, E.; Büttner, F.; Reustle, A.; Klumpp, V.; Winter, S.; Rausch, S.; Fisel, P.; Hennenlotter, J.; Kruck, S.; Stenzl, A.; et al. Metabolic and lipidomic reprogramming in renal cell carcinoma subtypes reflects regions of tumor origin. Eur. Urol. Focus 2019, 5, 608–618. [Google Scholar] [CrossRef]
- Ulgen, E.; Ozisik, O.; Sezerman, O.U. pathfindR: An R package for comprehensive identification of enriched pathways in omics data through active subnetworks. Front. Genet. 2019, 10, 858. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing. In R Foundation for Statistical Computing; R Core Team: Vienna, Austria, 2016. [Google Scholar]
- Eklund, A. Beeswarm: The Bee Swarm Plot, an Alternative to Stripchart, R Package Version 0.2.3. 2016. Available online: https://CRAN.R-project.org/package=beeswarm (accessed on 10 February 2022).
- Wickham, H. Tidyverse: Easily Install and Load the ‘Tidyverse’, R Package Version 1.2.1. 2017. Available online: https://CRAN.R-project.org/package=tidyverse (accessed on 10 February 2022).
- Turner, S. Qqman: Q-Q and Manhattan Plots for GWAS Data, R Package Version 0.1.4. 2017. Available online: https://CRAN.R-project.org/package=qqman (accessed on 10 February 2022).
- González, J.R.; Armengol, L.; Guinó, E.; Solé, X.; Moreno, V. SNPassoc: SNPs-Based Whole Genome Association Studies, R Package Version 1.9.2. Available online: https://CRAN.R-project.org/package=SNPassoc (accessed on 10 February 2022).
- Fox, J.; Weisberg, S. An R Companion to Applied Regression, 2nd ed.; SAGE Publications: Thousand Oaks, CA, USA, 2011. [Google Scholar]
- Das, S.; Forer, L.; Schönherr, S.; Sidore, C.; Locke, A.E.; Kwong, A.; Vrieze, S.I.; Chew, E.Y.; Levy, S.; McGue, M.; et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016, 48, 1284–1287. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug 1 | Dose (mg) | Imax (µM) 2 | Iin,max (µM) 2 | fU 2 | Iin,max,u (µM) 2 | IC50 (µM) 3 | R 4 |
|---|---|---|---|---|---|---|---|
| Bendroflumethiazide 5 | 10 [12] | 0.2 [12] | 1.68 | 0.06 [13] | 0.101 | 77 | 1.00 |
| Bosentan | 125 [14] | 4.2 [14] | 18.2 | 0.02 [15] | 0.365 | 24 [16] | 1.02 |
| Budesonide 5 | 0.8 [17] | 0.0026 [17] | 0.12 | 0.13 [13] | 0.015 | 320 | 1.00 |
| Bulevirtide (Myrcludex B) | 2 [8] | 0.04 [8] | 0.07 | 0.85 [18] | 0.056 | 0.053 [19] | 2.06 |
| Candesartan 5 | 32 [20] | 0.75 [20] | 5.26 | 0.01 [13] | 0.053 | 339 | 1.00 |
| Cyclosporin A | 700 [21] | 0.45 [21] | 36.6 | 0.12 [21] | 4.39 | 1 [22] | 5.39 |
| Cyclosporin A | 200 [23] | 1.8 [23] | 12.2 | 0.1 [23] | 1.22 | 1 [22] | 2.22 |
| Diltiazem 5 | 180 [21] | 0.29 [21] | 27.3 | 0.3 [21] | 8.17 | 871 | 1.01 |
| Diltiazem 5 | 120 [23] | 0.36 [23] | 18.3 | 0.22 [23] | 4.04 | 871 | 1.00 |
| Doxazosin 5 | 8 [24] | 0.06 [24] | 1.16 | 0.02 [24] | 0.023 | 51 | 1.00 |
| Efavirenz | 600 [25] | 11.6 [25] | 129.7 | 0.01 [13] | 1.3 | 43 [26] | 1.03 |
| Ezetimibe 5 | 10 [24] | 0.01 [24] | 1.53 | 0.1 [24] | 0.153 | 36 | 1.00 |
| Fenofibrate 5 | 300 [23] | 23.8 [23] | 75.5 | 0.01 [13] | 0.755 | 188 | 1.00 |
| Flutamide 5 | 250 [24] | 0.4 [24] | 56.6 | 0.06 [24] | 3.4 | 164 | 1.02 |
| Furosemide | 80 [27] | 6.7 [27] | 21.7 | 0.01 [13] | 0.217 | 15 [22] | 1.01 |
| Gemfibrozil 5 | 600 [23] | 60.9 [23] | 209.8 | 0.0065 [23] | 1.36 | 23 | 1.06 |
| Glyburide 5 | 5 [24] | 0.2 [24] | 0.83 | 0.02 [24] | 0.017 | 11 | 1.00 |
| Indomethacin 5 | 50 [28] | 7.7 [28] | 16.4 | 0.03 [13] | 0.492 | 251 | 1.00 |
| Irbesartan 5 | 300 [24] | 7.7 [24] | 51.2 | 0.1 [24] | 5.12 | 17 | 1.30 |
| Ketokonazole | 200 [24] | 8.5 [24] | 31.9 | 0.01 [24] | 0.319 | 264 [22] | 1.00 |
| Ketokonazole | 200 [21] | 6.6 [21] | 30.0 | 0.01 [21] | 0.3 | 264 [22] | 1.00 |
| Ketokonazole | 200 [23] | 3.2 [23] | 26.6 | 0.01 [23] | 0.266 | 264 [22] | 1.00 |
| Ketoprofen 5 | 100 [29] | 39.7 [29] | 64.2 | 0.03 [13] | 1.92 | 467 | 1.00 |
| Lapatinib 5 | 1250 [23] | 4.2 [23] | 137.8 | 0.01 [23] | 1.38 | 415 | 1.00 |
| Losartan 5 | 50 [24] | 0.5 [24] | 7.84 | 0.01 [24] | 0.078 | 105 | 1.00 |
| Methylprednisolone 5 | 1000 [21] | 26.5 [21] | 192.4 | 0.22 [21] | 42.3 | 346 | 1.12 |
| Nateglinide 5 | 60 [30] | 15.5 [30] | 27.3 | 0.02 [13] | 0.546 | 290 | 1.00 |
| Nefazodone 5 | 200 [31] | 4.4 [31] | 30.8 | 0.01 [13] | 0.308 | 183 | 1.00 |
| Nifedipine 5 | 10 [21] | 0.23 [21] | 2.02 | 0.0045 [21] | 0.009 | 91 | 1.00 |
| Nimodipine 5 | 30 [32] | 0.11 [32] | 4.56 | 0.02 [13] | 0.091 | 276 | 1.00 |
| Nitrendipine 5 | 20 [33] | 0.12 [33] | 3.56 | 0.02 [13] | 0.071 | 161 | 1.00 |
| Olmesartan 5 | 160 [24] | 3.8 [24] | 26.1 | 0.01 [24] | 0.261 | 339 | 1.00 |
| Pioglitazone 5 | 30 [23] | 4.2 [23] | 9.44 | 0.009 [23] | 0.084 | 5.8 | 1.01 |
| Probenecid 5 | 2000 [34] | 520.8 [34] | 956.1 | 0.12 [13] | 114.7 | 791 | 1.14 |
| Propranolol | 105 [21] | 0.52 [21] | 25.7 | 0.1 [21] | 2.57 | 6 [22] | 1.43 |
| Propranolol | 80 [23] | 0.19 [23] | 19.4 | 0.13 [23] | 2.51 | 6 [22] | 1.42 |
| Raloxifene 5 | 60 [24] | 0.003 [24] | 7.87 | 0.05 [24] | 0.394 | 438 | 1.00 |
| Rifampicin 5 | 600 [23] | 7.9 [23] | 53.2 | 0.3 [23] | 16.0 | 605 | 1.00 |
| Ritonavir | 600 [21] | 15.5 [21] | 67.2 | 0.006 [21] | 0.403 | 2 [26] | 1.20 |
| Ritonavir | 600 [23] | 15.3 [23] | 66.9 | 0.015 [23] | 1.0 | 2 [26]] | 1.50 |
| Rosiglitazone | 8 [35] | 1.7 [35] | 3.1 | 0.01 [13] | 0.031 | 5.1 [35] | 1.01 |
| Rosuvastatin 5 | 20 [36] | 0.01 [36] | 2.6 | 0.17 [37] | 0.441 | 186 | 1.00 |
| Saquinavir | 400 [23] | 7.6 [23] | 44.6 | 0.02 [23] | 0.892 | 7 [26] | 1.13 |
| Simvastatin 5 | 80 [24] | 0.10 [24] | 12.0 | 0.06 [24] | 0.718 | 70 | 1.01 |
| Sulfasalazine | 1000 [35] | 15.0 [35] | 170.9 | 0.05 [24] | 8.5 | 9.6 [35] | 1.89 |
| Telmisartan 5 | 80 [23] | 1.2 [23] | 10.9 | 0.005 [23] | 0.054 | 87 | 1.00 |
| Zafirlukast | 20 [35] | 0.6 [35] | 2.8 | 0.01 [24] | 0.028 | 6.5 [35] | 1.00 |
| SLC10A1 mRNA | NTCP Protein | |||
|---|---|---|---|---|
| CpG_site | Correlation Coefficient | Unadjusted P | Correlation Coefficient | Unadjusted P |
| 1_CpG_1 | 0.09 | 0.28 | 0.13 | 0.14 |
| 1_CpG_2 | −0.01 | 0.87 | −0.08 | 0.37 |
| 1_CpG_3 | −0.04 | 0.61 | −0.09 | 0.28 |
| 1_CpG_4 | −0.02 | 0.82 | −0.07 | 0.44 |
| 1_CpG_5 | 0.16 | 0.06 | 0.05 | 0.55 |
| 2_CpG_1 | 0.01 | 0.91 | 0.05 | 0.54 |
| 2_CpG_2.3 | −0.01 | 0.89 | −0.03 | 0.72 |
| 2_CpG_4.5 | −0.17 | 0.05 | −0.15 | 0.08 |
| 3_CpG_1 | 0.18 | 0.04 | −0.05 | 0.57 |
| 3_CpG_2 | −0.15 | 0.09 | −0.14 | 0.12 |
| 3_CpG_3 | −0.14 | 0.11 | −0.23 | 0.010 |
| 3_CpG_4 | −0.10 | 0.27 | −0.07 | 0.42 |
| 5_CpG_2 | −0.08 | 0.39 | −0.29 | 0.002 # |
| 5_CpG_3.4 | −0.09 | 0.29 | −0.17 | 0.044 |
| 5_CpG_5 | −0.15 | 0.07 | −0.26 | 0.002 # |
| 5_CpG_6 | −0.04 | 0.65 | −0.02 | 0.79 |
| 5_CpG_7 | −0.10 | 0.25 | −0.14 | 0.09 |
| 5_CpG_9 | −0.06 | 0.51 | −0.15 | 0.08 |
| 7_CpG_1.2.3 | 0.02 | 0.82 | 0.14 | 0.11 |
| 7_CpG_5 | −0.02 | 0.78 | −0.05 | 0.53 |
| 7_CpG_9,10 | −0.04 | 0.66 | −0.13 | 0.15 |
| 7_CpG_11 | 0.00 | 0.96 | −0.02 | 0.85 |
| 7_CpG_12 | −0.01 | 0.95 | −0.01 | 0.91 |
| 7_CpG_14 | −0.05 | 0.66 | 0.01 | 0.91 |
| 7_CpG_17 | −0.10 | 0.24 | 0.00 | 0.98 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tremmel, R.; Nies, A.T.; van Eijck, B.A.C.; Handin, N.; Haag, M.; Winter, S.; Büttner, F.A.; Kölz, C.; Klein, F.; Mazzola, P.; et al. Hepatic Expression of the Na+-Taurocholate Cotransporting Polypeptide Is Independent from Genetic Variation. Int. J. Mol. Sci. 2022, 23, 7468. https://doi.org/10.3390/ijms23137468
Tremmel R, Nies AT, van Eijck BAC, Handin N, Haag M, Winter S, Büttner FA, Kölz C, Klein F, Mazzola P, et al. Hepatic Expression of the Na+-Taurocholate Cotransporting Polypeptide Is Independent from Genetic Variation. International Journal of Molecular Sciences. 2022; 23(13):7468. https://doi.org/10.3390/ijms23137468
Chicago/Turabian StyleTremmel, Roman, Anne T. Nies, Barbara A. C. van Eijck, Niklas Handin, Mathias Haag, Stefan Winter, Florian A. Büttner, Charlotte Kölz, Franziska Klein, Pascale Mazzola, and et al. 2022. "Hepatic Expression of the Na+-Taurocholate Cotransporting Polypeptide Is Independent from Genetic Variation" International Journal of Molecular Sciences 23, no. 13: 7468. https://doi.org/10.3390/ijms23137468
APA StyleTremmel, R., Nies, A. T., van Eijck, B. A. C., Handin, N., Haag, M., Winter, S., Büttner, F. A., Kölz, C., Klein, F., Mazzola, P., Hofmann, U., Klein, K., Hoffmann, P., Nöthen, M. M., Gaugaz, F. Z., Artursson, P., Schwab, M., & Schaeffeler, E. (2022). Hepatic Expression of the Na+-Taurocholate Cotransporting Polypeptide Is Independent from Genetic Variation. International Journal of Molecular Sciences, 23(13), 7468. https://doi.org/10.3390/ijms23137468