
Genome–Wide Identification of the GRAS Family Genes in Melilotus albus and Expression Analysis under Various Tissues and Abiotic Stresses


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Identification and Analysis of GRAS Genes in Melilotus albus
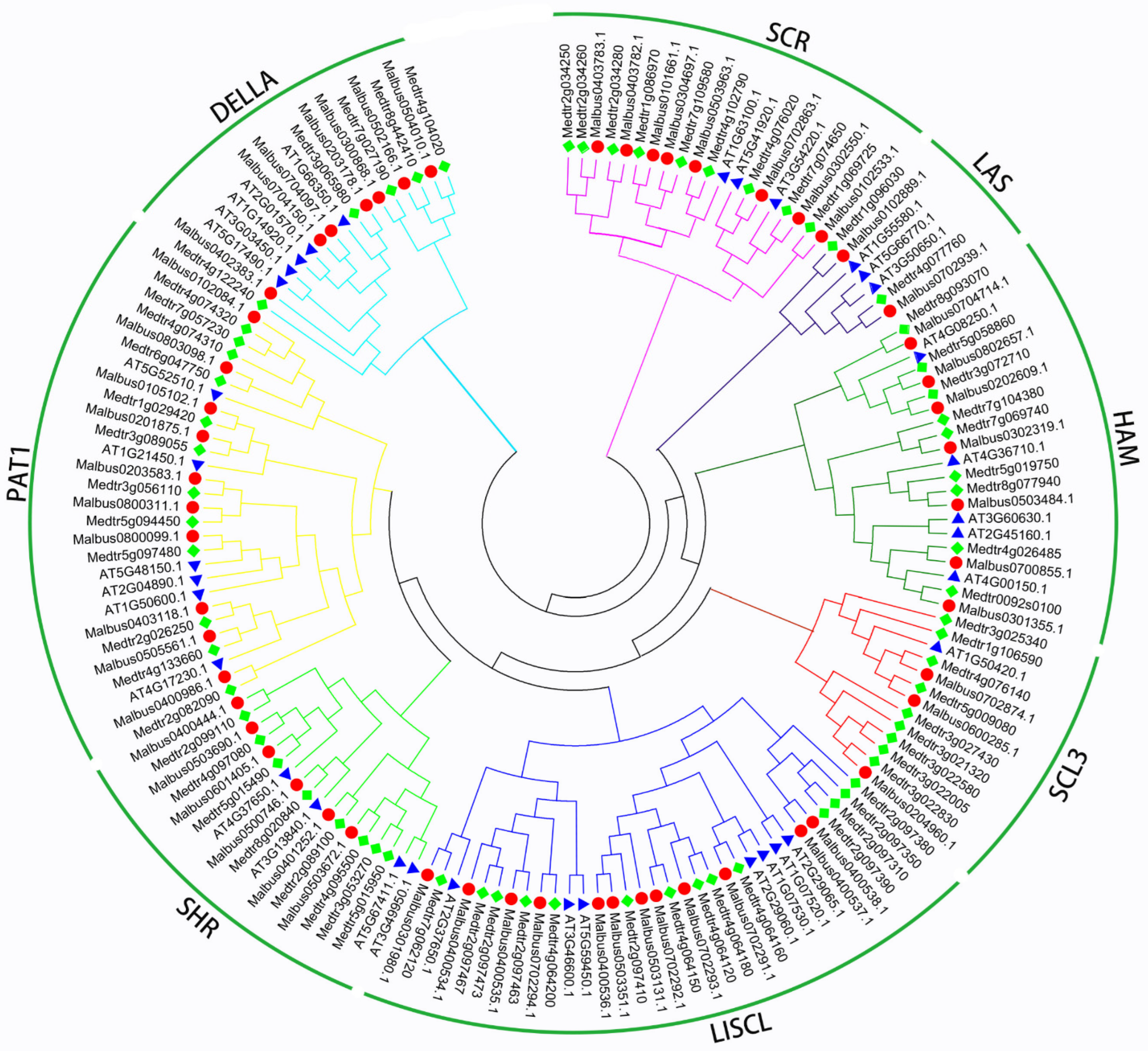
2.2. Phylogenetic Categories Analysis of MaGRAS Genes
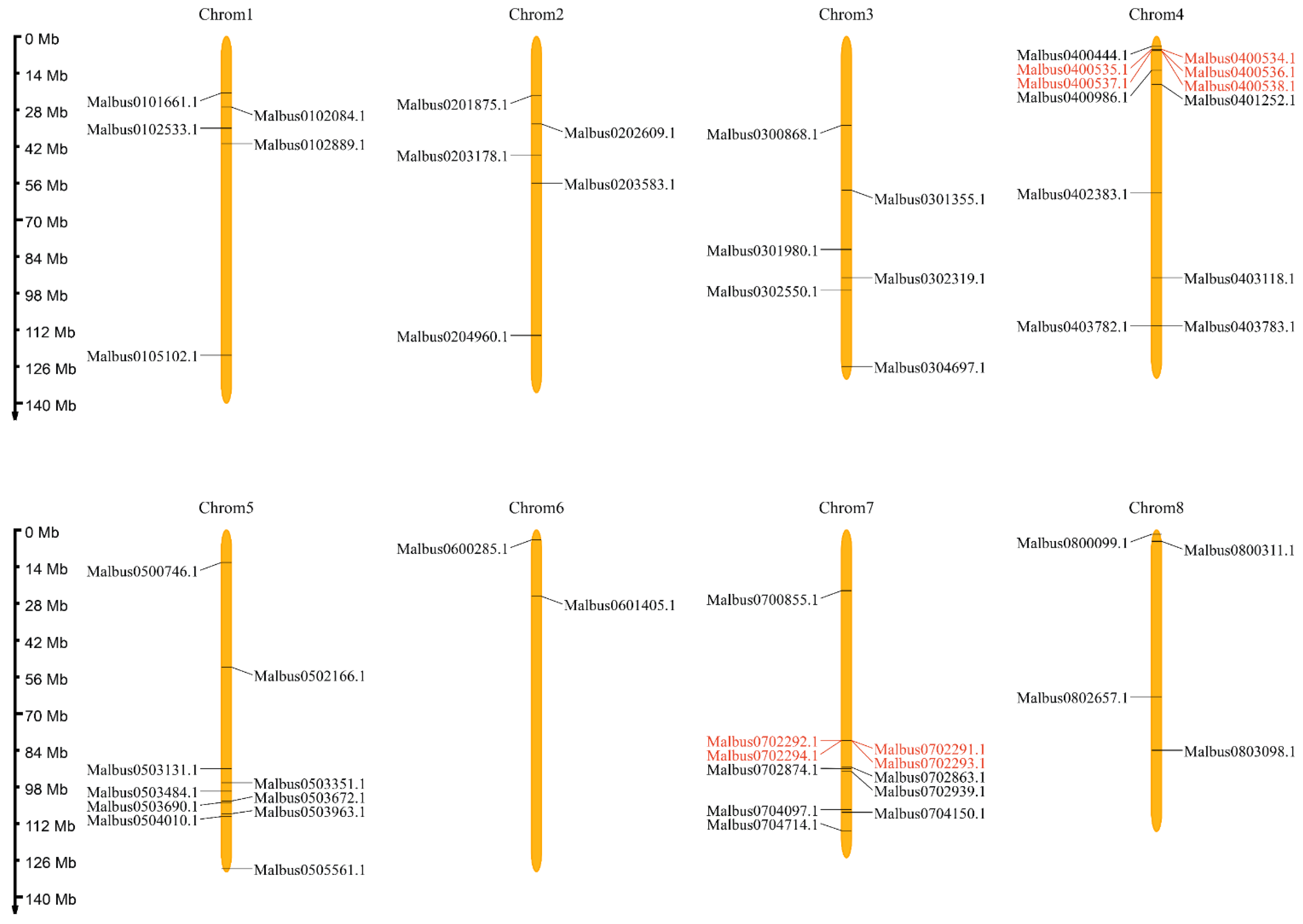
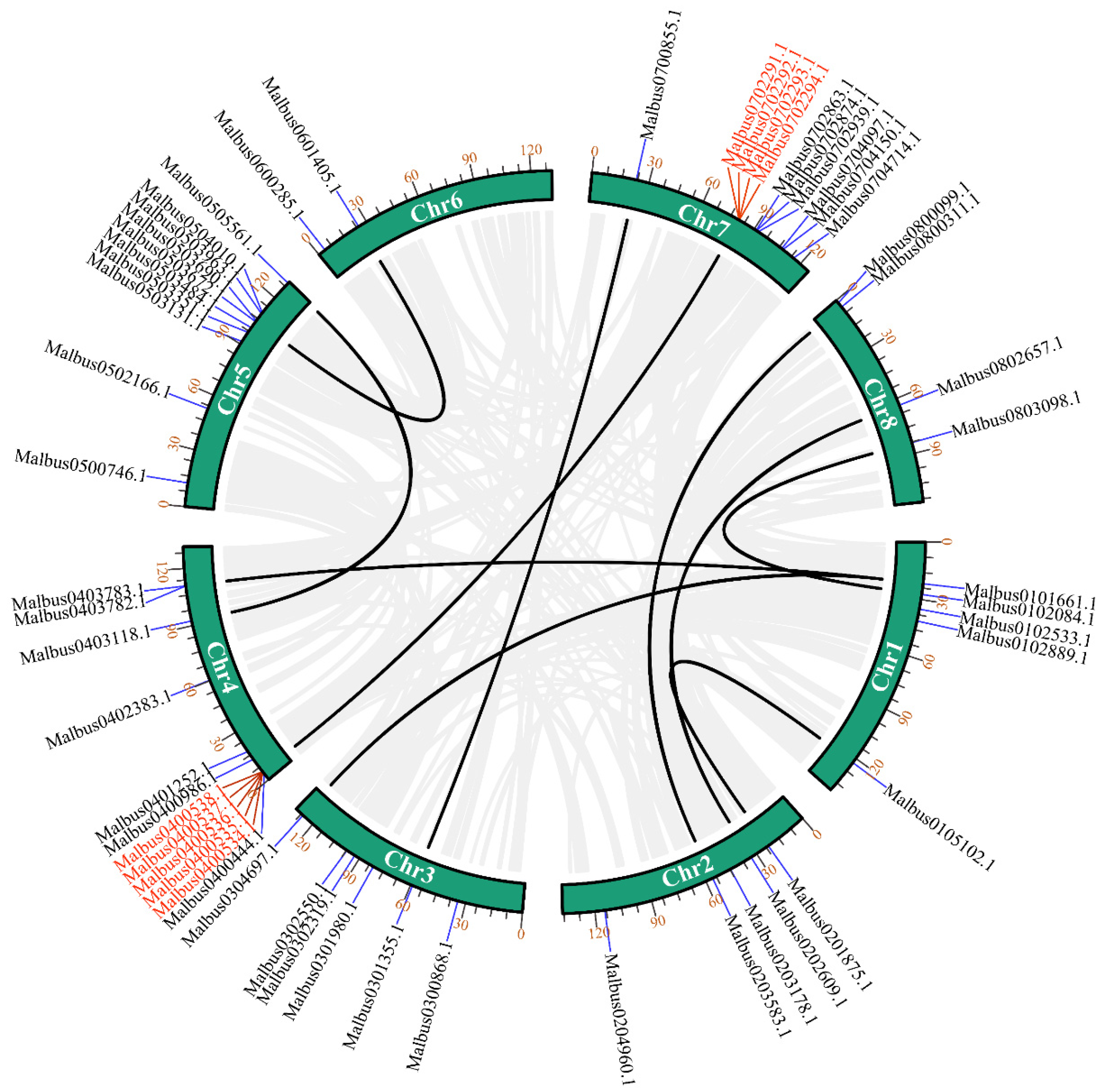
2.3. Chromosomal Locations and Gene Duplication Analysis of MaGRAS Genes
2.4. Structure and Conserved Motifs of MaGRAS Genes
2.5. Identification of Cis–Elements in the MaGRAS Gene Promoters
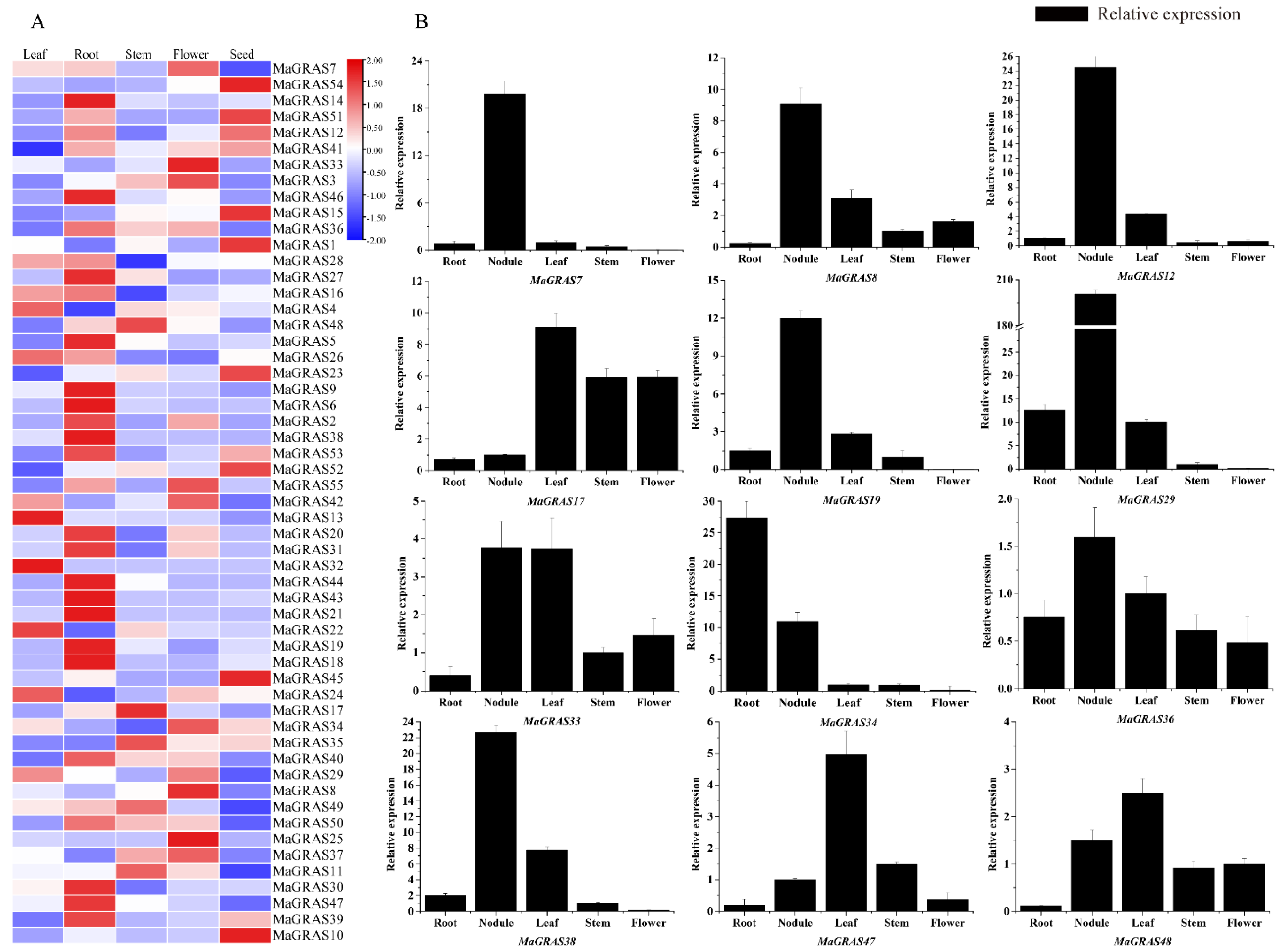
2.6. Expression Pattern Analysis of MaGRAS Genes in M. albus tissues
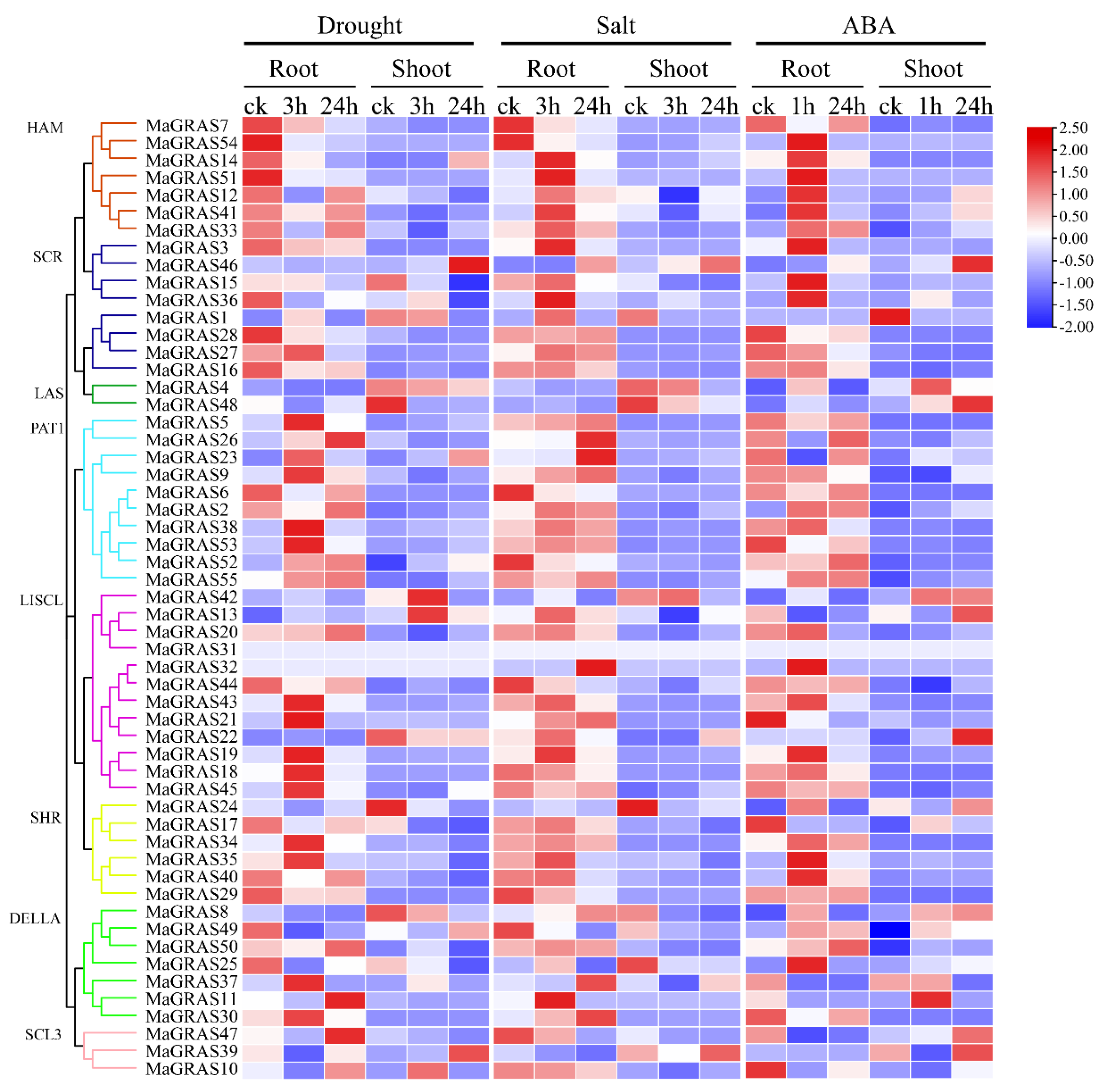
2.7. Expression Analysis of MaGRAS Genes Responding to ABA and Abiotic Stresses
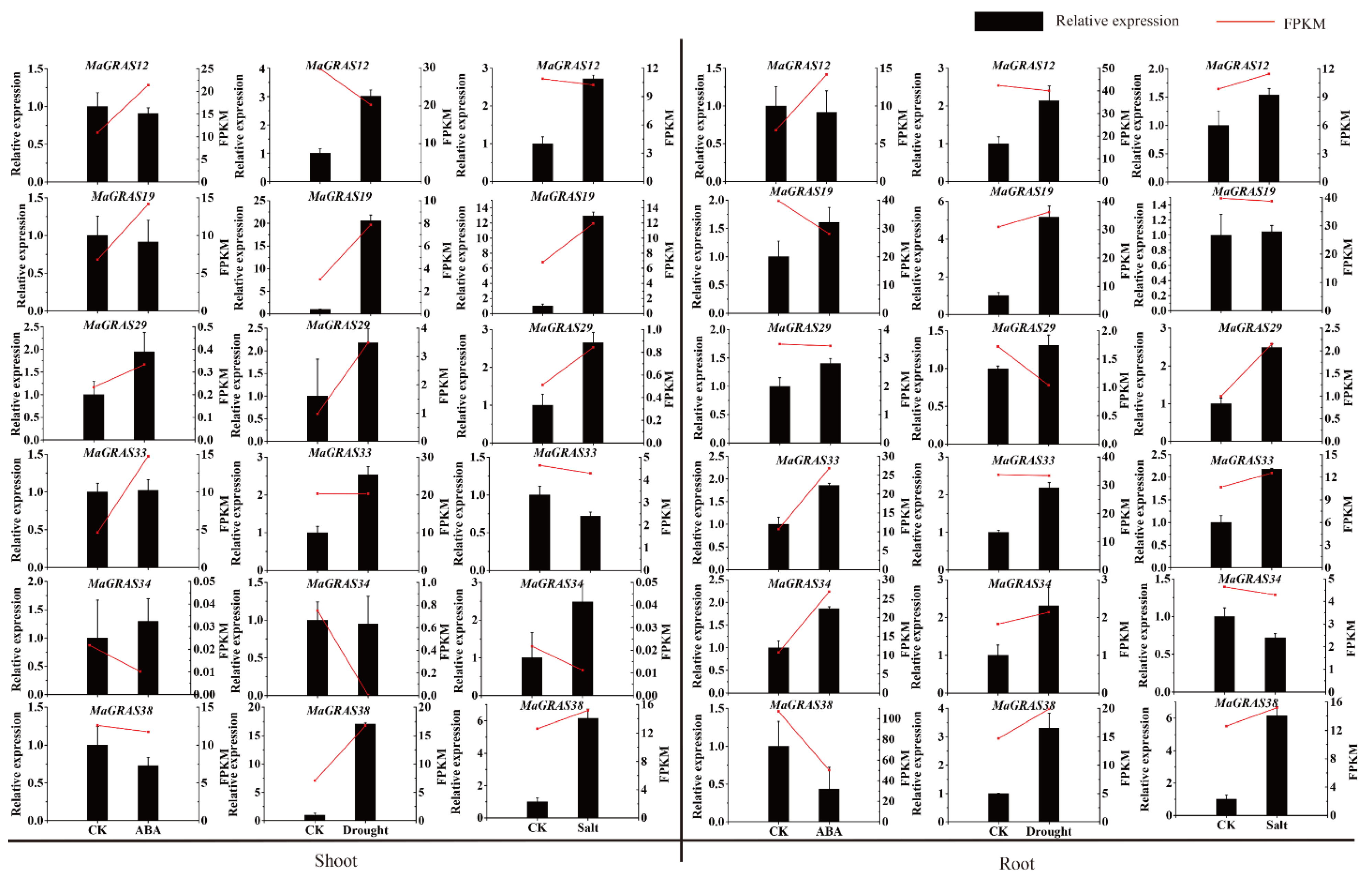
2.8. MaGRAS Genes Improved the Tolerance of Yeast to Abiotic Stresses
3. Discussion
4. Materials and Methods
4.1. Identification of the GRAS Genes in M. albus
4.2. Phylogenetic Analysis and Classification of MaGRASs
4.3. Chromosomal Mapping and Gene Duplication Analysis of MaGRAS Genes
4.4. Analysis of Gene Structures and Motif Composition of MaGRAS Genes
4.5. The Prediction of Protein–Protein for MaGRAS Proteins
4.6. Identification of Putative Cis–Elements in MaGRAS Genes
4.7. Transcriptome Analysis for Tissue–Specific Expression and Stress Treatment
4.8. Quantitative Real–Time PCR Analysis for Tissue–Specific Expression and Stress Treatment
4.9. Functional Verification of Heterologous Expression Genes in Yeast
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Peng, J.R.; Carol, P.; Richards, D.E.; King, K.E.; Cowling, R.J.; Murphy, G.P.; Harberd, N.P. The Arabidopsis GAI gene defines a signaling pathway that negatively regulates gibberellin responses. Genes Dev. 1997, 11, 3194–3205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silverstone, A.L.; Ciampaglio, C.N.; Sun, T.P. The Arabidopsis RGA gene encodes a transcriptional regulator repressing the gibberellin signal transduction pathway. Plant Cell 1998, 10, 155–169. [Google Scholar] [CrossRef] [Green Version]
- DiLaurenzio, L.; WysockaDiller, J.; Malamy, J.E.; Pysh, L.; Helariutta, Y.; Freshour, G.; Hahn, M.G.; Feldmann, K.A.; Benfey, P.N. The SCARECROW gene regulates an asymmetric cell division that is essential for generating the radial organization of the Arabidopsis root. Cell 1996, 86, 423–433. [Google Scholar] [CrossRef] [Green Version]
- Bolle, C. The role of GRAS proteins in plant signal transduction and development. Planta 2004, 218, 683–692. [Google Scholar] [CrossRef]
- Pysh, L.D.; Wysocka–Diller, J.W.; Camilleri, C.; Bouchez, D.; Benfey, P.N. The GRAS gene family in Arabidopsis: Sequence characterization and basic expression analysis of the SCARECROW–LIKE genes. Plant J. 1999, 18, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Xue, B.; Jones, W.T.; Rikkerink, E.; Dunker, A.K.; Uversky, V.N. A functionally required unfoldome from the plant kingdom: Intrinsically disordered N–terminal domains of GRAS proteins are involved in molecular recognition during plant development. Plant Mol. Biol. 2011, 77, 205–223. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Jones, W.T.; Harvey, D.; Edwards, P.J.B.; Pascal, S.M.; Kirk, C.; Considine, T.; Sheerin, D.J.; Rakonjac, J.; Oldfield, C.J.; et al. N–Terminal Domains of DELLA Proteins Are Intrinsically Unstructured in the Absence of Interaction with GID1/Gibberellic Acid Receptors. J. Biol. Chem. 2010, 285, 11557–11571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirsch, S.; Oldroyd, G.E.D. GRAS–domain transcription factors that regulate plant development. Plant Signal. Behav. 2009, 4, 698–700. [Google Scholar] [CrossRef] [Green Version]
- Tian, C.; Wan, P.; Sun, S.; Li, J.; Chen, M.J.P.M.B. Genome–Wide Analysis of the GRAS Gene Family in Rice and Arabidopsis. Plant Mol. Biol. 2004, 54, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Li, H.; Chen, Y.; Zheng, Q.; Li, B.; Li, Z. TaSCL14, a Novel Wheat (Triticum aestivum L.) GRAS Gene, Regulates Plant Growth, Photosynthesis, Tolerance to Photooxidative Stress, and Senescence. J. Genet. Genom. 2015, 42, 21–32. [Google Scholar] [CrossRef]
- Song, L.; Tao, L.; Cui, H.; Ling, L.; Guo, C. Genome–wide identification and expression analysis of the GRAS family proteins in Medicago truncatula. Acta Physiol. Plant. 2017, 39, 93. [Google Scholar] [CrossRef]
- Helariutta, Y.; Fukaki, H.; Wysocka–Diller, J.; Nakajima, K.; Jung, J.; Sena, G.; Hauser, M.T.; Benfey, P.N. The SHORT–ROOT gene controls radial patterning of the Arabidopsis root through radial signaling. Cell 2000, 101, 555–567. [Google Scholar] [CrossRef] [Green Version]
- Cui, H.; Levesque, M.P.; Vernoux, T.; Jung, J.W.; Paquette, A.J.; Gallagher, K.L.; Wang, J.Y.; Blilou, I.; Scheres, B.; Benfey, P.N. An evolutionarily conserved mechanism delimiting SHR movement defines a single layer of endodermis in plants. Science 2007, 316, 421–425. [Google Scholar] [CrossRef] [Green Version]
- Heo, J.-O.; Chang, K.S.; Kim, I.A.; Lee, M.-H.; Lee, S.A.; Song, S.-K.; Lee, M.M.; Lim, J. Funneling of gibberellin signaling by the GRAS transcription regulator SCARECROW–LIKE 3 in the Arabidopsis root. Proc. Natl. Acad. Sci. USA 2011, 108, 2166–2171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho–Plagaro, T.; Molinero–Rosales, N.; Farina Flores, D.; Villena Diaz, M.; Manuel Garcia–Garrido, J. Identification and Expression Analysis of GRAS Transcription Factor Genes Involved in the Control of Arbuscular Mycorrhizal Development in Tomato. Front. Plant Sci. 2019, 10, 268. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Yu, R.; Fan, L.-M.; Wei, N.; Chen, H.; Deng, X.W. DELLA–mediated PIF degradation contributes to coordination of light and gibberellin signalling in Arabidopsis. Nat. Commun. 2016, 7, 11868. [Google Scholar] [CrossRef] [Green Version]
- Navarro, L.; Bari, R.; Achard, P.; Lisón, P.; Nemri, A.; Harberd, N.P.; Jones, J.D.G. DELLAs control plant immune responses by modulating the balance of jasmonic acid and salicylic acid signaling. Curr. Biol. 2008, 18, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Dill, A.; Jung, H.S.; Sun, T.P. The DELLA motif is essential for gibberellin–induced degradation of RGA. Proc. Natl. Acad. Sci. USA 2001, 98, 14162–14167. [Google Scholar] [CrossRef] [Green Version]
- Muntha, S.T.; Zhang, L.; Zhou, Y.; Zhao, X.; Hu, Z.; Yang, J.; Zhang, M. Phytochrome A signal transduction 1 and CONSTANS–LIKE 13 coordinately orchestrate shoot branching and flowering in leafy Brassica juncea. Plant Biotechnol. J. 2019, 17, 1333–1343. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Xian, Z.; Kang, X.; Tang, N.; Li, Z. Genome–wide identification, phylogeny and expression analysis of GRAS gene family in tomato. BMC Plant Biol. 2015, 15, 209. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, S.; Kim, J.; Munoz, A.; Heckmann, A.B.; Downie, J.A.; Oldroyd, G.E.D. GRAS Proteins Form a DNA Binding Complex to Induce Gene Expression during Nodulation Signaling in Medicago truncatula. Plant Cell 2009, 21, 545–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalo, P.; Gleason, C.; Edwards, A.; Marsh, J.; Mitra, R.M.; Hirsch, S.; Jakab, J.; Sims, S.; Long, S.R.; Rogers, J.; et al. Nodulation signaling in legumes requires NSP2, a member of the GRAS family of transcriptional regulators. Science 2005, 308, 1786–1789. [Google Scholar] [CrossRef]
- Fode, B.; Siemsen, T.; Thurow, C.; Weigel, R.; Gatz, C. The Arabidopsis GRAS Protein SCL14 Interacts with Class II TGA Transcription Factors and Is Essential for the Activation of Stress–Inducible Promoters. Plant Cell 2008, 20, 3122–3135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Y.; Fang, L.; Karungo, S.K.; Zhang, L.; Gao, Y.; Li, S.; Xin, H. Overexpression of VaPAT1, a GRAS transcription factor from Vitis amurensis, confers abiotic stress tolerance in Arabidopsis. Plant Cell Rep. 2016, 35, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.-S.; Liang, D.; Shuai, P.; Xia, X.-L.; Yin, W.-L. The salt– and drought–inducible poplar GRAS protein SCL7 confers salt and drought tolerance in Arabidopsis thaliana. J. Exp. Bot. 2010, 61, 4011–4019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, K.; Chen, S.; Li, T.; Ma, X.; Liang, X.; Ding, X.; Liu, H.; Luo, L. OsGRAS23, a rice GRAS transcription factor gene, is involved in drought stress response through regulating expression of stress–responsive genes. BMC Plant Biol. 2015, 15, 141. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Zhang, B.; Su, T.; Li, P.; Xin, X.; Wang, W.; Zhao, X.; Yu, Y.; Zhang, D.; Yu, S.; et al. BrLAS, a GRAS Transcription Factor from Brassica rapa, Is Involved in Drought Stress Tolerance in Transgenic Arabidopsis. Front. Plant Sci. 2018, 9, 1792. [Google Scholar] [CrossRef] [Green Version]
- Awan, M.; Habib, S.; Li, N.; Yang, L.; Li, Z. Overexpression of SlGRAS7 Affects Multiple Behaviors Leading to Confer Abiotic Stresses Tolerance and Impacts Gibberellin and Auxin Signaling in Tomato. Int. J. Genom. 2019, 2019, 4051981. [Google Scholar]
- Liu, Y.; Huang, W.; Xian, Z.; Hu, N.; Lin, D.; Ren, H.; Chen, J.; Su, D.; Li, Z. Overexpression of SIGRAS40 in Tomato Enhances Tolerance to Abiotic Stresses and Influences Auxin and Gibberellin Signaling. Front. Plant Sci. 2017, 8, 1659. [Google Scholar] [CrossRef]
- Khan, Y.; Xiong, Z.; Zhang, H.; Liu, S.; Yaseen, T.; Hui, T. Expression and roles of GRAS gene family in plant growth, signal transduction, biotic and abiotic stress resistance and symbiosis formation—A review. Plant Biol. 2022, 24, 404–416. [Google Scholar] [CrossRef]
- Wang, N.; Wang, K.; Li, S.; Jiang, Y.; Li, L.; Zhao, M.; Jiang, Y.; Zhu, L.; Wang, Y.; Su, Y.; et al. Transcriptome–Wide Identification, Evolutionary Analysis, and GA Stress Response of the GRAS Gene Family in Panax ginseng CA Meyer. Plants 2020, 9, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Wang, X.; Li, C.; Li, H.; Zhang, J.; Ye, Z. Silencing GRAS2 reduces fruit weight in tomato. Chin. Bull. Bot. 2018, 60, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, F.; Luo, K.; Yan, Z.; Zhang, D.; Yan, Q.; Zhang, Y.; Yi, X.; Zhang, J. Analysis of miRNAs and their target genes in five Melilotus albus NILs with different coumarin content. Sci. Rep. 2018, 8, 14138. [Google Scholar] [CrossRef]
- Zhang, J.; Di, H.; Kai, L.; Jahufer, Z.; Wu, F.; Duan, Z.; Stewart, A.; Yan, Z.; Wang, Y. Coumarin Content, Morphological Variation, and Molecular Phylogenetics of Melilotus. Molecules 2018, 23, 810. [Google Scholar] [CrossRef] [Green Version]
- Zabala, J.M.; Marinoni, L.; Giavedoni, J.A.; Schrauf, G.E. Breeding strategies in Melilotus albus Desr., a salt–tolerant forage legume. Euphytica 2018, 214, 22. [Google Scholar] [CrossRef]
- Wu, F.; Duan, Z.; Xu, P.; Yan, Q.; Meng, M.; Cao, M.; Jones, C.S.; Zong, X.; Zhou, P.; Wang, Y.; et al. Genome and systems biology of Melilotus albus provides insights into coumarins biosynthesis. Plant Biotechnol. J. 2022, 20, 592–609. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Shi, H.; Xia, Z.; Tie, W.; Ding, Z.; Yan, Y.; Wang, W.; Hu, W.; Li, K. Genome–Wide Identification and Expression Analysis of the WRKY Gene Family in Cassava. Front. Plant Sci. 2016, 7, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Ding, X.; Gao, Y.; Yang, S. Genome–wide identification and characterization of GRAS genes in soybean (Glycine max). BMC Plant Biol. 2020, 20, 415. [Google Scholar] [CrossRef] [PubMed]
- Hirano, Y.; Nakagawa, M.; Suyama, T.; Murase, K.; Shirakawa, M.; Takayama, S.; Sun, T.-p.; Hakoshima, T. Structure of the SHR–SCR heterodimer bound to the BIRD/IDD transcriptional factor JKD. Nat. Plants 2017, 3, 17010. [Google Scholar] [CrossRef] [Green Version]
- Long, Y.; Goedhart, J.; Schneijderberg, M.; Terpstra, I.; Shimotohno, A.; Bouchet, B.P.; Akhmanova, A.; Gadella, T.W.J., Jr.; Heidstra, R.; Scheres, B.; et al. SCARECROW–LIKE23 and SCARECROW jointly specify endodermal cell fate but distinctly control SHORT–ROOT movement. Plant J. 2015, 84, 773–784. [Google Scholar] [CrossRef]
- To, V.-T.; Shi, Q.; Zhang, Y.; Shi, J.; Shen, C.; Zhang, D.; Cai, W. Genome–Wide Analysis of the GRAS Gene Family in Barley (Hordeum vulgare L.). Genes 2020, 11, 553. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Widmer, A.J.P.M.B.R. Genome–wide Comparative Analysis of the GRAS Gene Family in Populus, Arabidopsis and Rice. Plant Mol. Biol. Rep. 2014, 32, 1129–1145. [Google Scholar] [CrossRef]
- Zhang, H.; Cao, Y.; Shang, C.; Li, J.; Wang, J.; Wu, Z.; Ma, L.; Qi, T.; Fu, C.; Bai, Z.; et al. Genome–wide characterization of GRAS family genes in Medicago truncatula reveals their evolutionary dynamics and functional diversification. PLoS ONE 2017, 12, e0185439. [Google Scholar] [CrossRef]
- Niu, Y.; Zhao, T.; Xu, X.; Li, J. Genome–wide identification and characterization of GRAS transcription factors in tomato (Solanum lycopersicum). PeerJ 2017, 5, e3955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Iyer, L.M.; Aravind, L. Bacterial GRAS domain proteins throw new light on gibberellic acid response mechanisms. Bioinformatics 2012, 28, 2407–2411. [Google Scholar] [CrossRef] [Green Version]
- Jain, M.; Tyagi, A.K.; Khurana, J.P. Genome–wide analysis, evolutionary expansion, and expression of early auxin–responsive SAUR gene family in rice (Oryza sativa). Genomics 2006, 88, 360–371. [Google Scholar] [CrossRef] [Green Version]
- Jain, M.; Nijhawan, A.; Arora, R.; Agarwal, P.; Ray, S.; Sharma, P.; Kapoor, S.; Tyagi, A.K.; Khurana, J.P. F–box proteins in rice. Genome–wide analysis, classification, temporal and spatial gene expression during panicle and seed development, and regulation by light and abiotic stress. Plant Physiol. 2007, 143, 1467–1483. [Google Scholar] [CrossRef] [Green Version]
- Iwamoto, M.; Maekawa, M.; Saito, A.; Higo, H.; Higo, K. Evolutionary relationship of plant catalase genes inferred from exon–intron structures: Isozyme divergence after the separation of monocots and dicots. Theor. Appl. Genet. 1998, 97, 9–19. [Google Scholar] [CrossRef]
- Liu, L.; White, M.J.; MacRae, T.H. Transcription factors and their genes in higher plants. Eur. J. Biochem. 1999, 262, 247–257. [Google Scholar] [CrossRef]
- Liu, M.; Huang, L.; Ma, Z.; Sun, W.; Wu, Q.; Tang, Z.; Bu, T.; Li, C.; Chen, H. Genome–wide identification, expression analysis and functional study of the GRAS gene family in Tartary buckwheat (Fagopyrum tataricum). BMC Plant Biol. 2019, 19, 342. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Liu, J.; Yang, Z.E.; Chen, E.Y.; Zhang, C.J.; Zhang, X.Y.; Li, F.G. Genome–wide analysis of GRAS transcription factor gene family in Gossypium hirsutum L. BMC Genom. 2018, 19, 348. [Google Scholar] [CrossRef] [PubMed]
- Heckmann, A.B.; Lombardo, F.; Miwa, H.; Perry, J.A.; Bunnewell, S.; Parniske, M.; Wang, T.L.; Downie, J.A. Lotus japonicus nodulation requires two GRAS domain regulators, one of which is functionally conserved in a non–legume. Plant Physiol. 2006, 142, 1739–1750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres–Galea, P.; Huang, L.-F.; Chua, N.-H.; Bolle, C. The GRAS protein SCL13 is a positive regulator of phytochrome–dependent red light signaling, but can also modulate phytochrome A responses. Mol. Genet. Genom. 2006, 276, 13–30. [Google Scholar] [CrossRef]
- Bolle, C.; Koncz, C.; Chua, N.H. PAT1, a new member of the GRAS family, is involved in phytochrome A signal transduction. Genes Dev. 2000, 14, 1269–1278. [Google Scholar] [CrossRef]
- Zhang, Z.-L.; Ogawa, M.; Fleet, C.M.; Zentella, R.; Hu, J.; Heo, J.-O.; Lim, J.; Kamiya, Y.; Yamaguchi, S.; Sun, T.-P. SCARECROW–LIKE 3 promotes gibberellin signaling by antagonizing master growth repressor DELLA in Arabidopsis. Proc. Natl. Acad. Sci. USA 2011, 108, 2160–2165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolle, C. Functional Aspects of GRAS Family Proteins. In Plant Transcription Factors; Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Wang, T.-T.; Yu, T.-F.; Fu, J.-D.; Su, H.-G.; Chen, J.; Zhou, Y.-B.; Chen, M.; Guo, J.; Ma, Y.-Z.; Wei, W.-L.; et al. Genome–Wide Analysis of the GRAS Gene Family and Functional Identification of GmGRAS37 in Drought and Salt Tolerance. Front. Plant Sci. 2020, 11, 4690. [Google Scholar] [CrossRef]
- Song, X.-M.; Liu, T.-K.; Duan, W.-K.; Ma, Q.-H.; Ren, J.; Wang, Z.; Li, Y.; Hou, X.-L. Genome–wide analysis of the GRAS gene family in Chinese cabbage (Brassica rapa ssp. pekinensis). Genomics 2014, 103, 135–146. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Sharma, H.; Rajput, R.; Pandey, A.; Upadhyay, S.K. Molecular Characterization Revealed the Role of Thaumatin–Like Proteins of Bread Wheat in Stress Response. Front. Plant Sci. 2022, 12, 807448. [Google Scholar] [CrossRef]
- Shumayla; Tyagi, S.; Sharma, A.; Singh, K.; Upadhyay, S.K. Genomic dissection and transcriptional profiling of Cysteine–rich receptor–like kinases in five cereals and functional characterization of TaCRK68–A. Int. J. Biol. Macromol. 2019, 134, 316–329. [Google Scholar] [CrossRef]
- Zong, X.; Wang, S.; Han, Y.; Zhao, Q.; Xu, P.; Yan, Q.; Wu, F.; Zhang, J. Genome–wide profiling of the potential regulatory network of lncRNA and mRNA in Melilotus albus under salt stress. Environ. Exp. Bot. 2021, 189, 104548. [Google Scholar] [CrossRef]
- Duan, Z.; Yan, Q.; Wu, F.; Wang, Y.; Wang, S.; Zong, X.; Zhou, P.; Zhang, J. Genome–Wide Analysis of the UDP–Glycosyltransferase Family Reveals Its Roles in Coumarin Biosynthesis and Abiotic Stress in Melilotus albus. Int. J. Mol. Sci. 2021, 22, 10826. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Hoogland, C.; Appel, R.D.; Ivanyi, I.; Bairoch, A.; Gattiker, A. ExPASy: The proteomics server for in–depth protein knowledge and analysis. Nucleic Acids Res. 2013, 31, 3784–3788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, K.C.; Shen, H.B. Cell–PLoc: A package of Web servers for predicting subcellular localization of proteins in various organisms. Nat. Protoc. 2007, 3, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Jeanmougin, F.; Thompson, J.D.; Gouy, M.; Higgins, D.G.; Gibson, T.J. Multiple sequence alignment with Clustal x. Trends Biochem. Sci. 1998, 23, 403–405. [Google Scholar] [CrossRef]
- Kumar, S.; Tamura, K.; Nei, M. MEGA: Molecular Evolutionary Genetics Analysis software for microcomputers. Comput. Appl. Biosci. 1994, 10, 189–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Brown, P.; Baxter, L.; Hickman, R.; Beynon, J.; Moore, J.D.; Ott, S. MEME–LaB: Motif analysis in clusters. Bioinformatics 2013, 29, 1696–1697. [Google Scholar] [CrossRef] [Green Version]
- Lescot, M.; Dehais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouze, P.; Rombauts, S. PlantCARE, a database of plant cis–acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Wang, Y.R.; Nan, Z.B. Relative and absolute quantification expression analysis of CsSAMDC gene as a case. China Biotechnol. 2009, 29, 86–91. [Google Scholar]
- Zhang, Z.; Jin, X.; Liu, Z.; Zhang, J.; Liu, W. Genome–wide identification of FAD gene family and functional analysis of MsFAD3.1 involved in the accumulation of α–linolenic acid in alfalfa. Crop Sci. 2020, 61, 566–579. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Duan, Z.; Yan, Q.; Wu, F.; Zhou, P.; Zhang, J. Genome–Wide Identification of the GRAS Family Genes in Melilotus albus and Expression Analysis under Various Tissues and Abiotic Stresses. Int. J. Mol. Sci. 2022, 23, 7403. https://doi.org/10.3390/ijms23137403
Wang S, Duan Z, Yan Q, Wu F, Zhou P, Zhang J. Genome–Wide Identification of the GRAS Family Genes in Melilotus albus and Expression Analysis under Various Tissues and Abiotic Stresses. International Journal of Molecular Sciences. 2022; 23(13):7403. https://doi.org/10.3390/ijms23137403
Chicago/Turabian StyleWang, Shengsheng, Zhen Duan, Qi Yan, Fan Wu, Pei Zhou, and Jiyu Zhang. 2022. "Genome–Wide Identification of the GRAS Family Genes in Melilotus albus and Expression Analysis under Various Tissues and Abiotic Stresses" International Journal of Molecular Sciences 23, no. 13: 7403. https://doi.org/10.3390/ijms23137403
APA StyleWang, S., Duan, Z., Yan, Q., Wu, F., Zhou, P., & Zhang, J. (2022). Genome–Wide Identification of the GRAS Family Genes in Melilotus albus and Expression Analysis under Various Tissues and Abiotic Stresses. International Journal of Molecular Sciences, 23(13), 7403. https://doi.org/10.3390/ijms23137403