
ICOSL Stimulation by ICOS-Fc Accelerates Cutaneous Wound Healing In Vivo


 , , , , , ,
, , , , , ,  ,
, 
 and
and 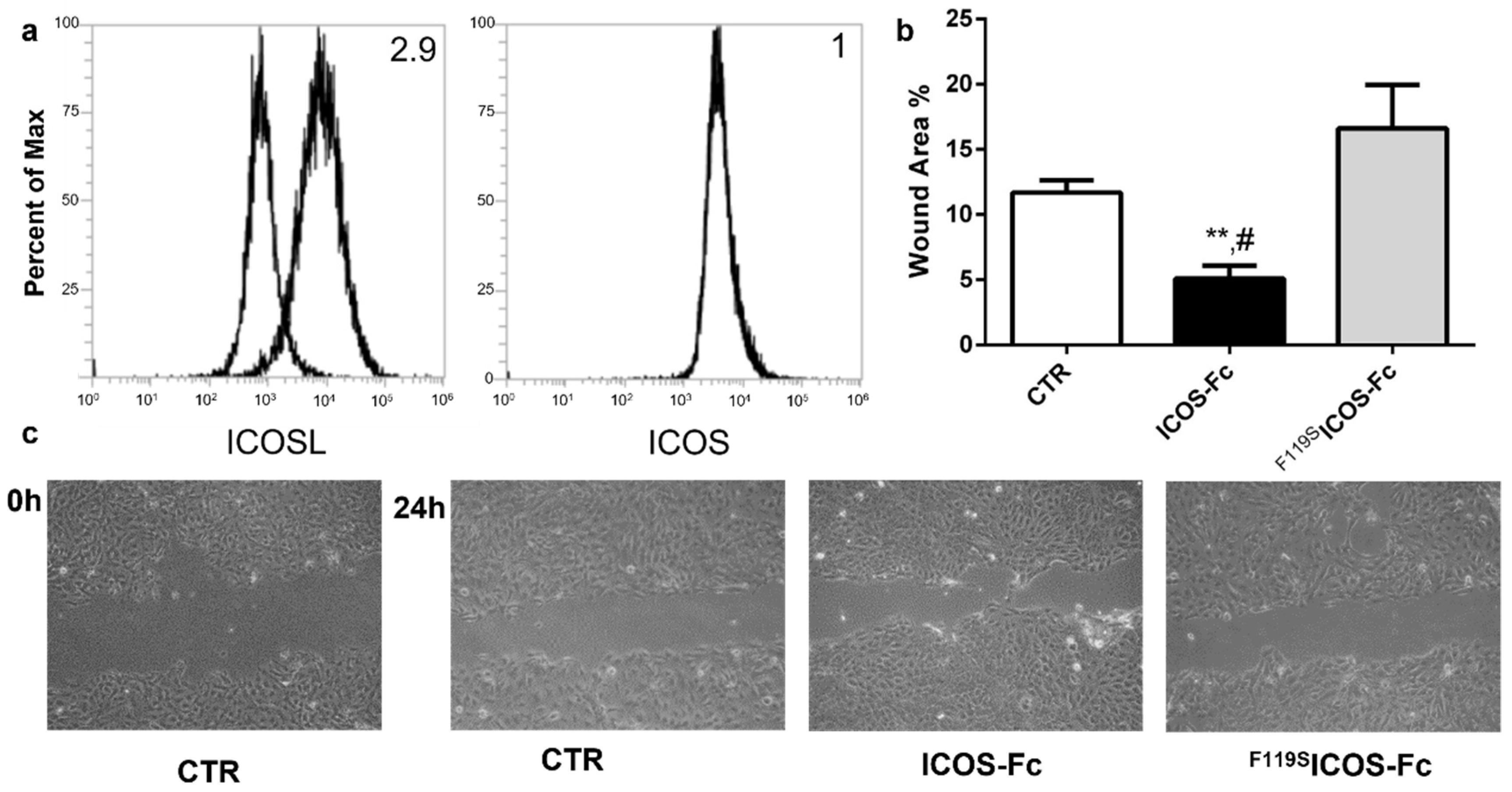{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. ICOSL Activation by ICOS-Fc Increases Keratinocyte Migration In Vitro
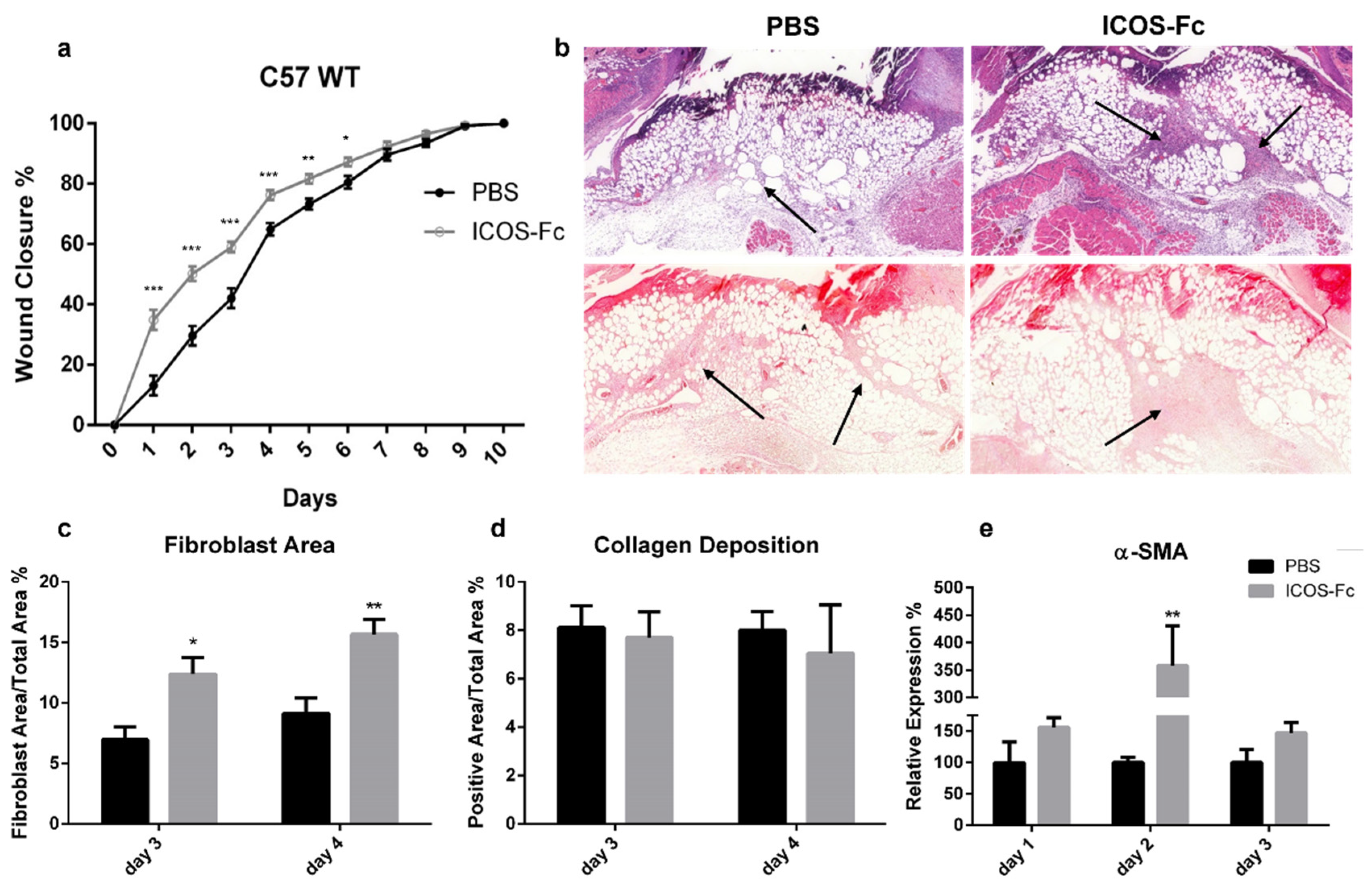
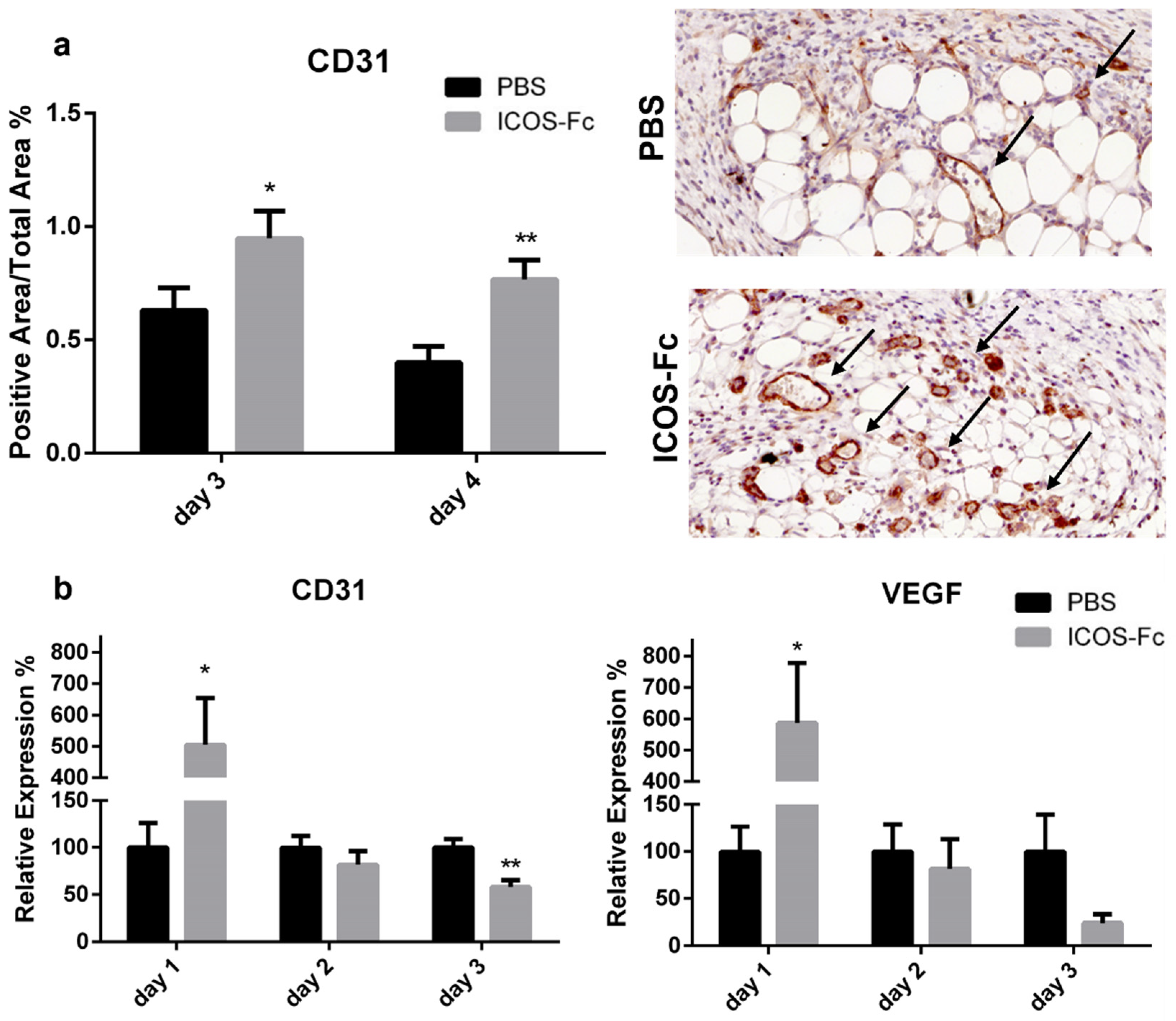
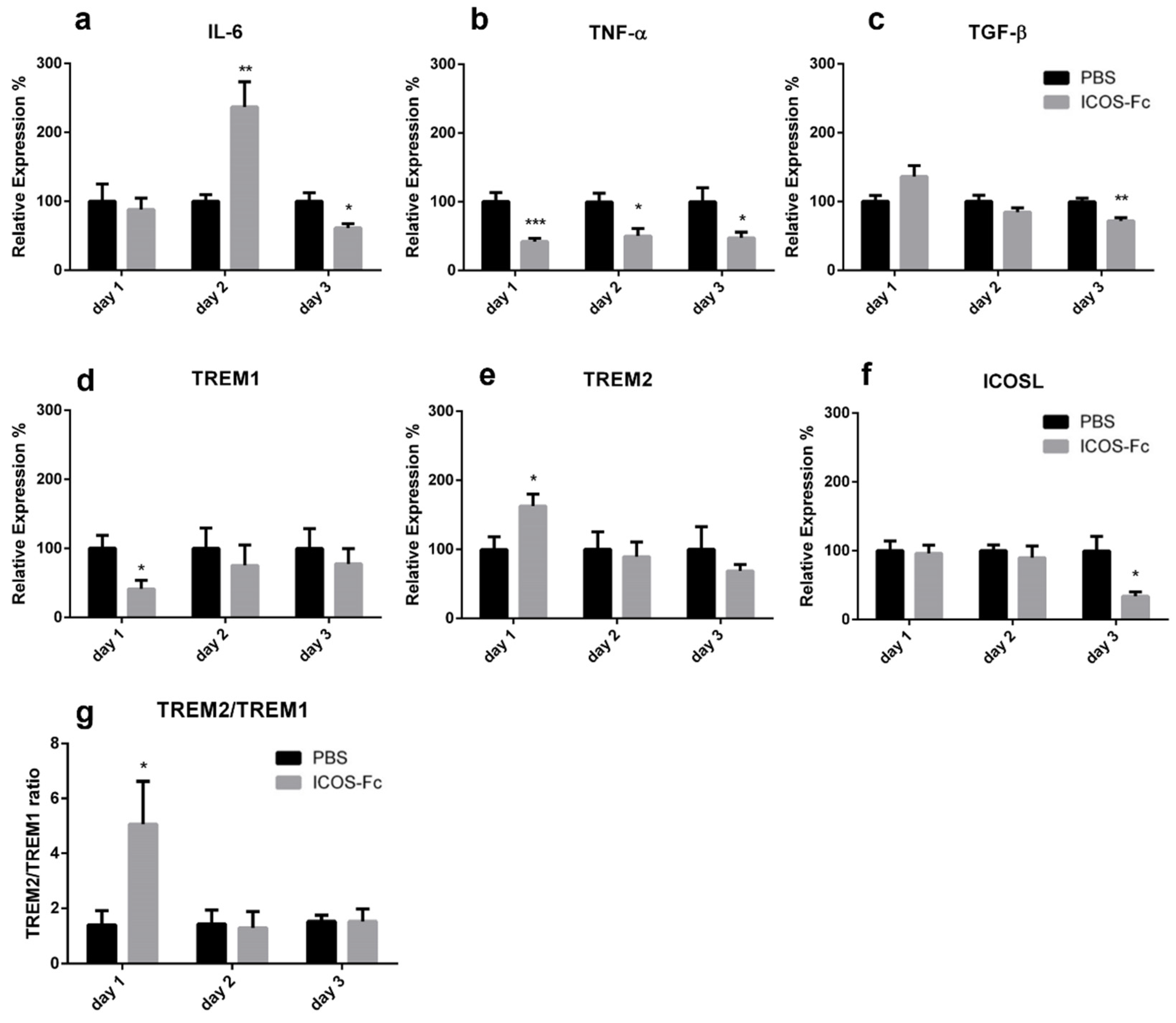
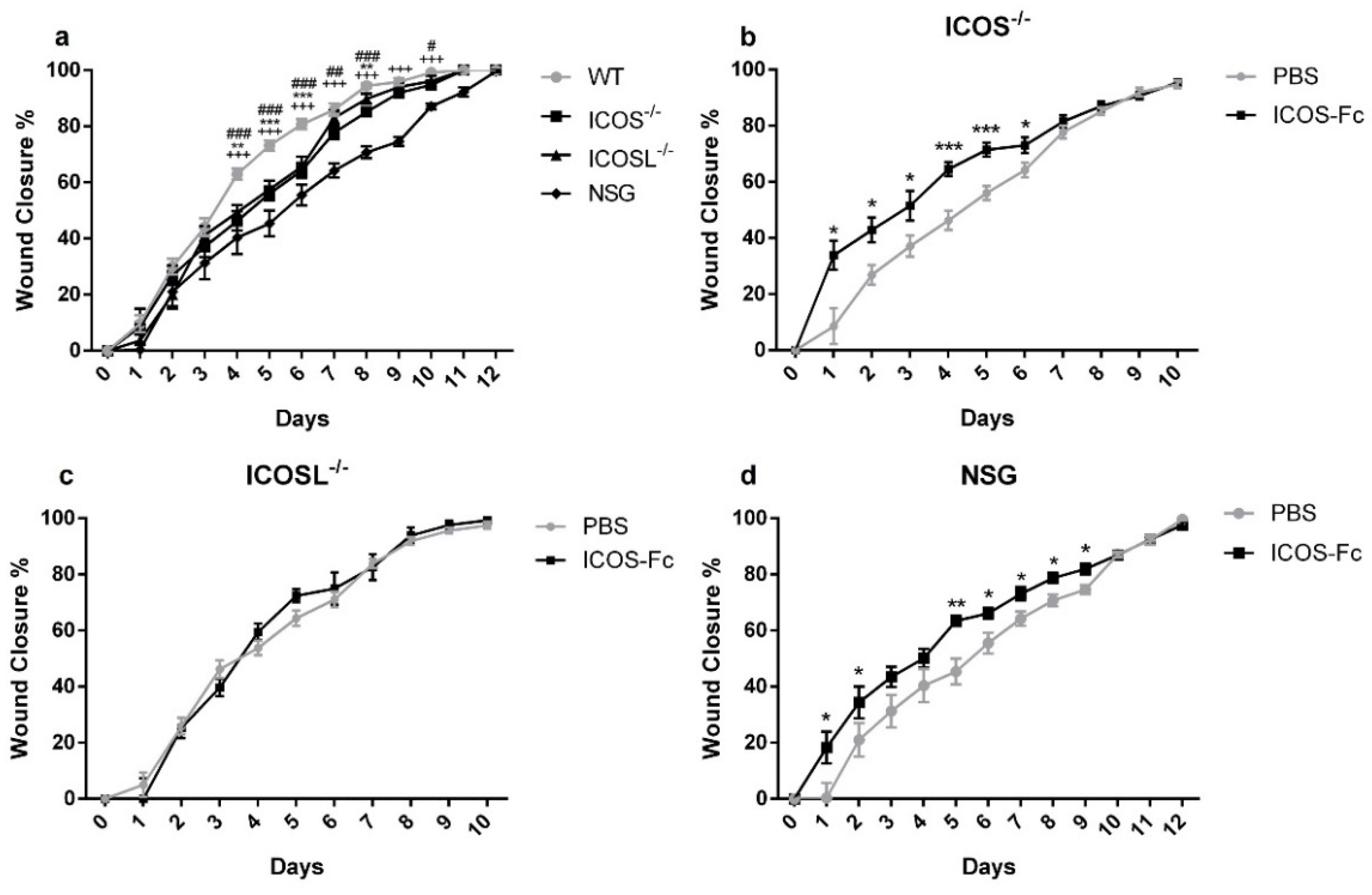
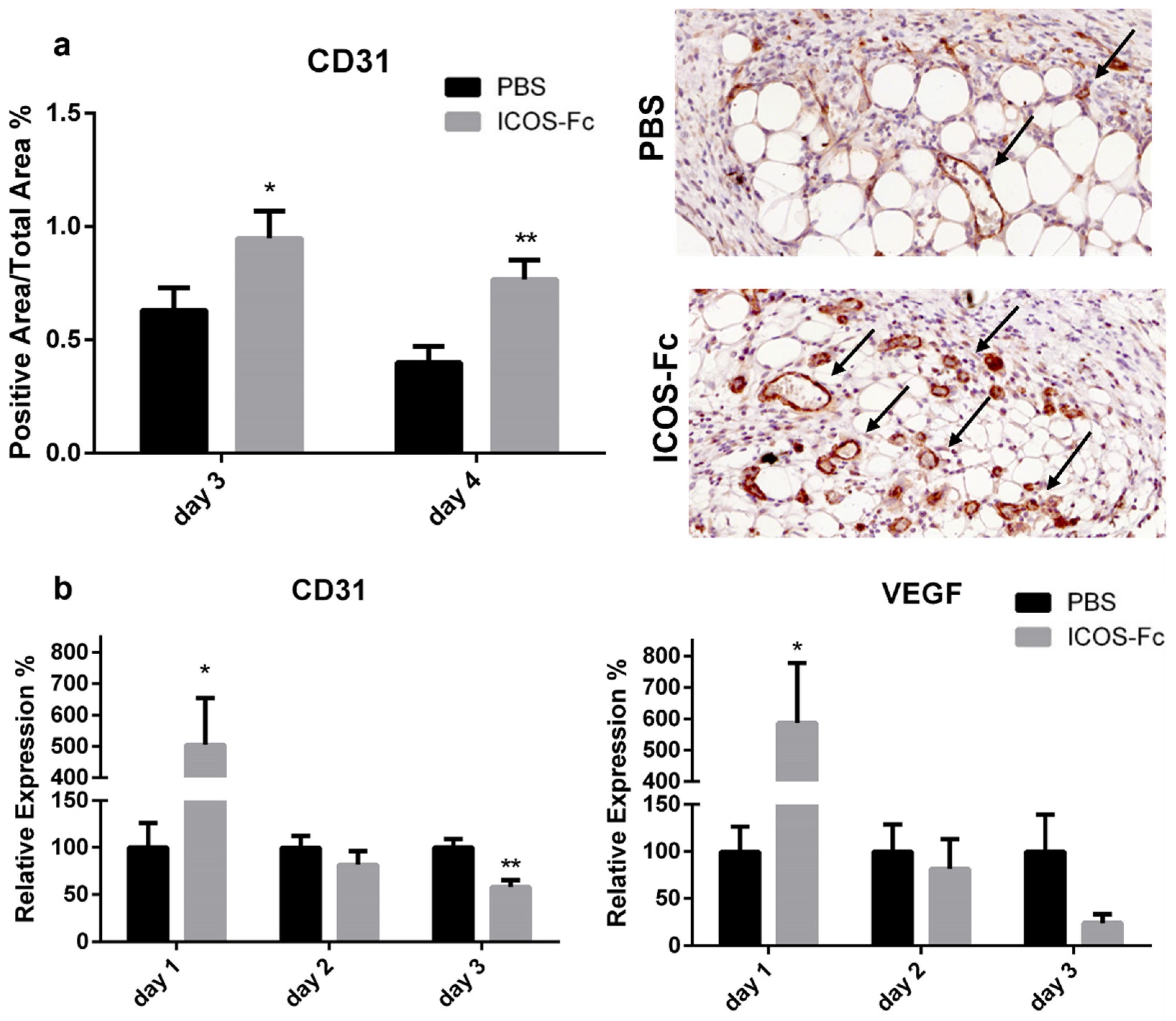
2.2. ICOS-Fc Treatment Accelerates Skin Wound Healing In Vivo
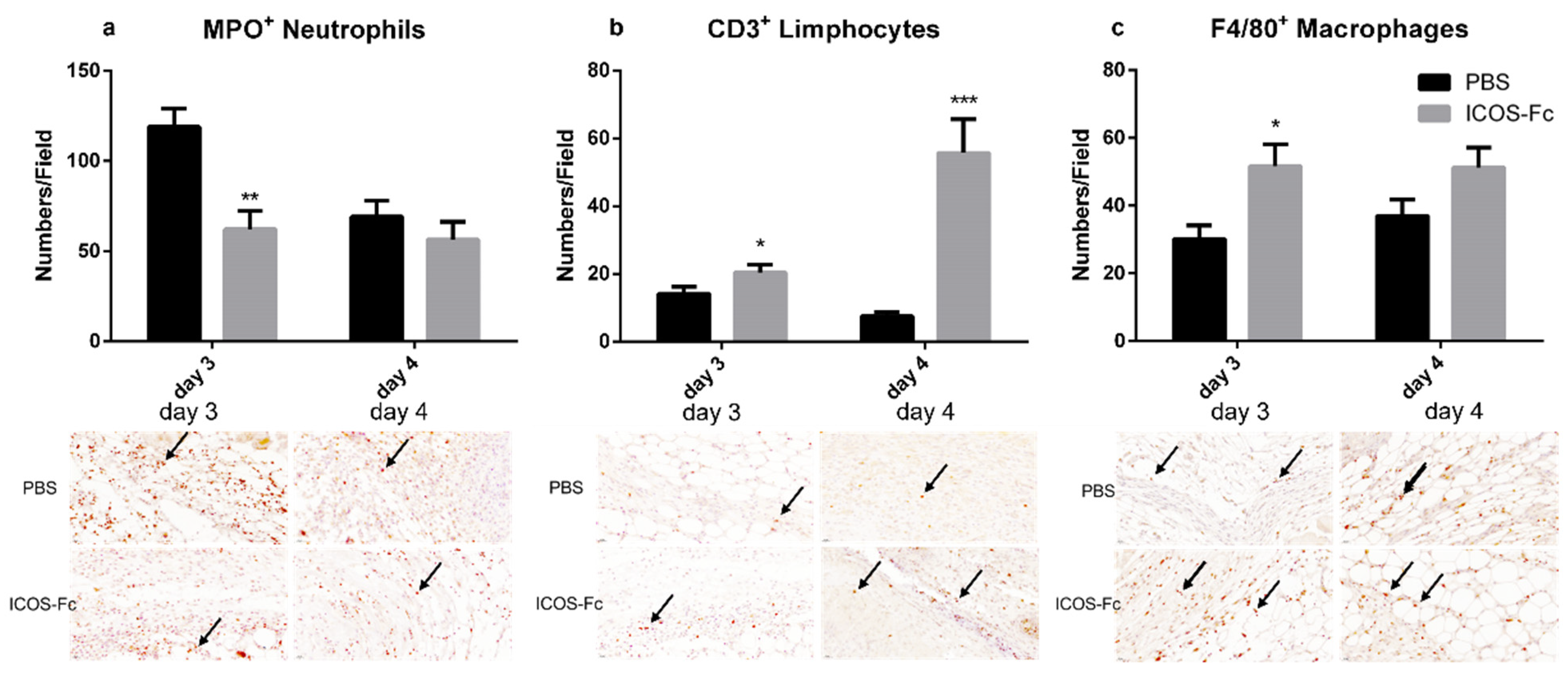
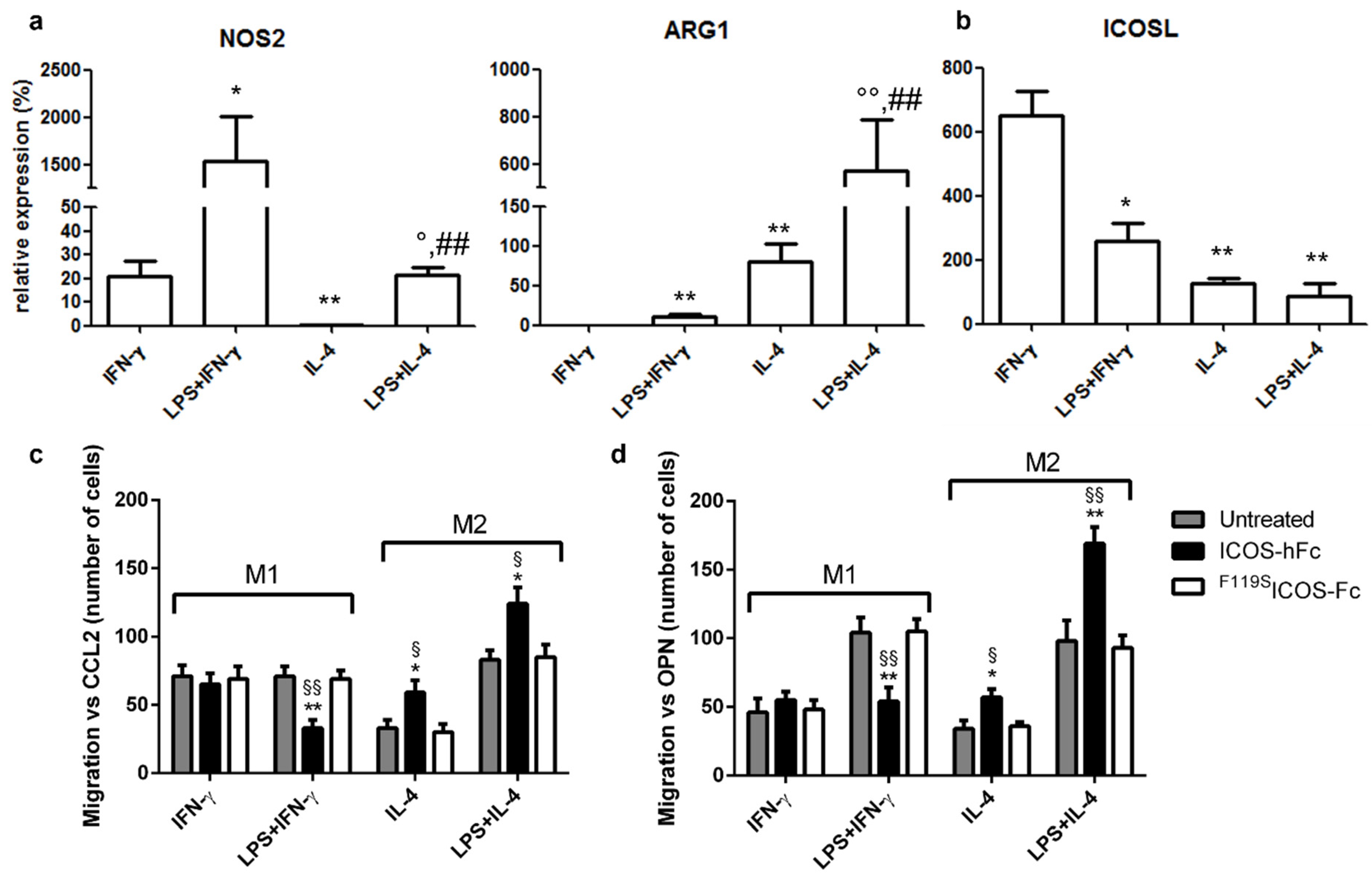
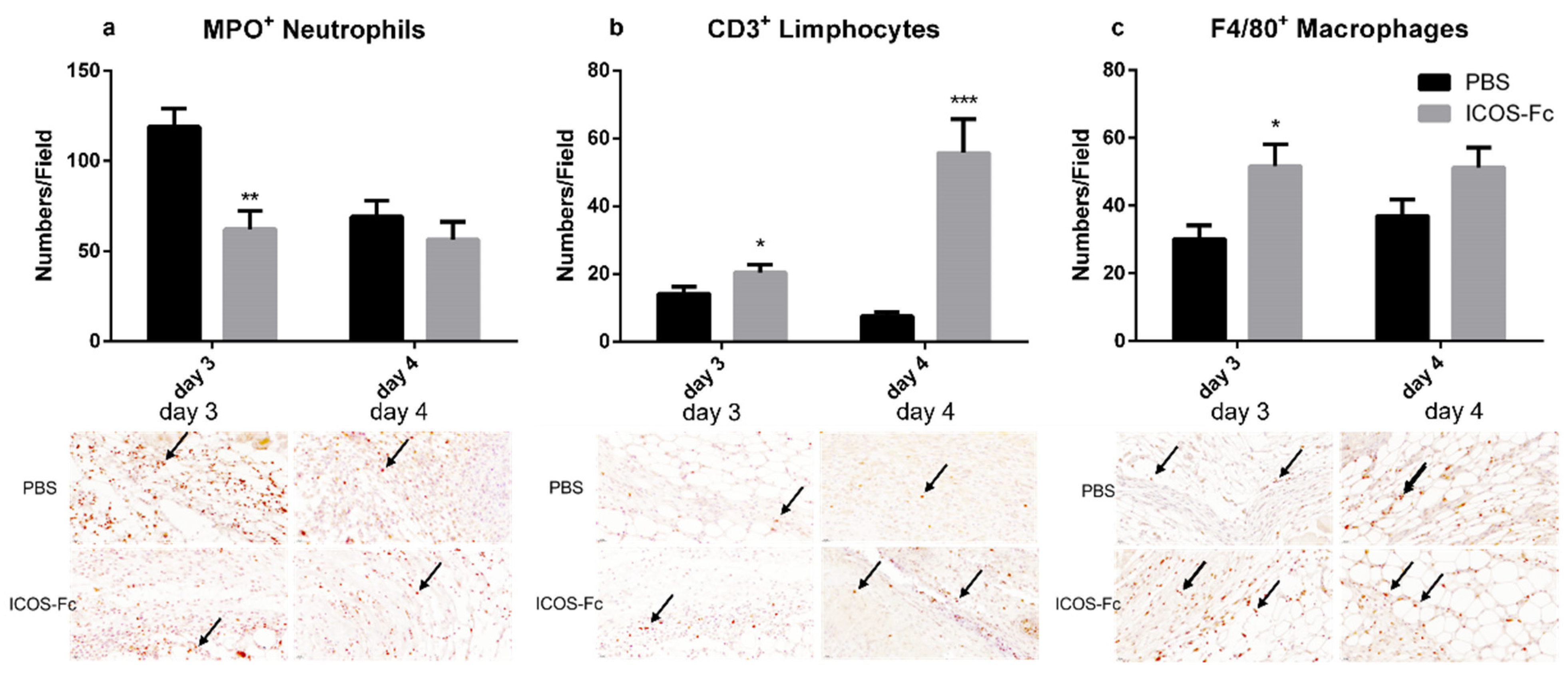
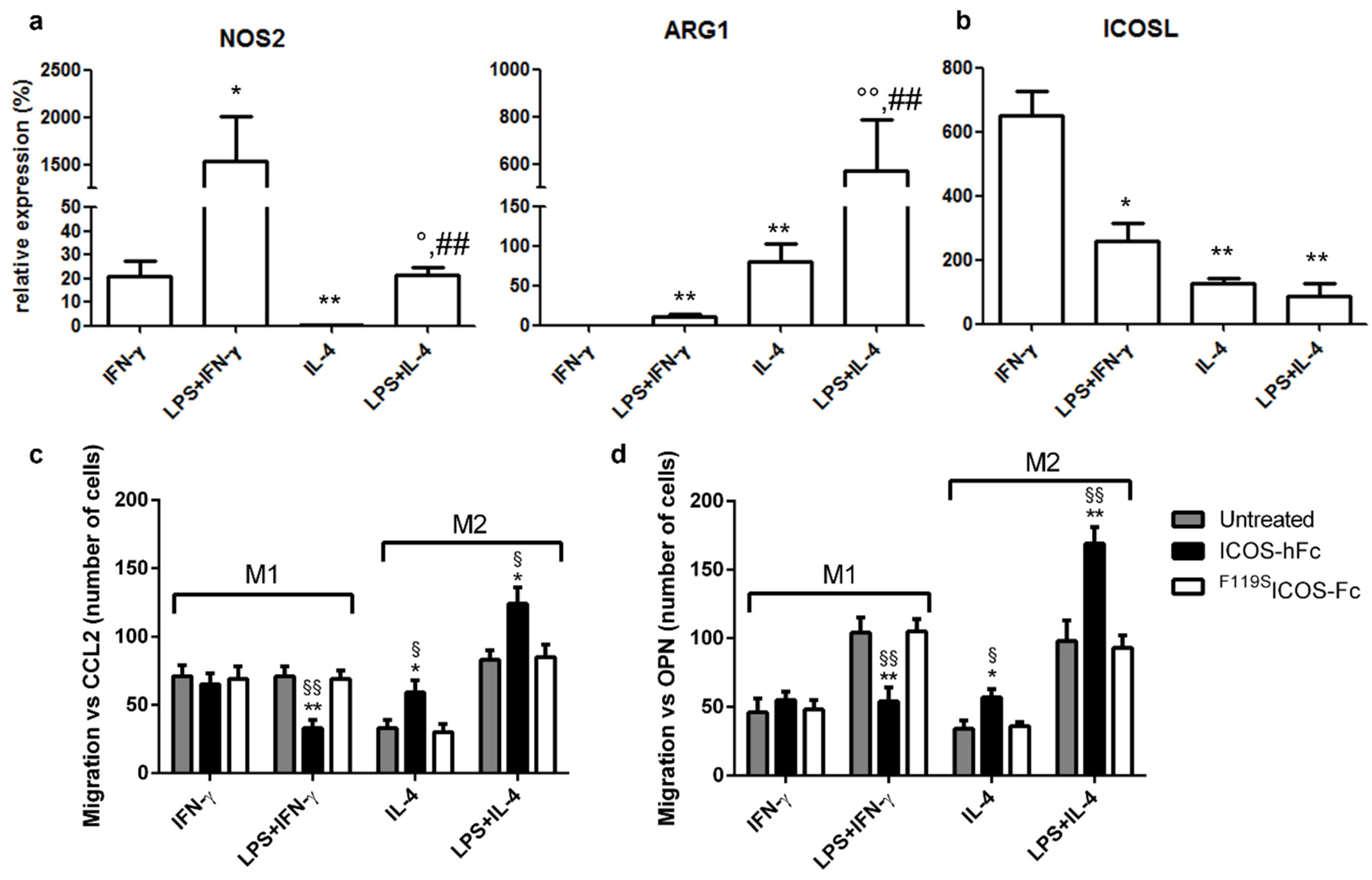
2.3. Effects of ICOSL Triggering on Macrophages
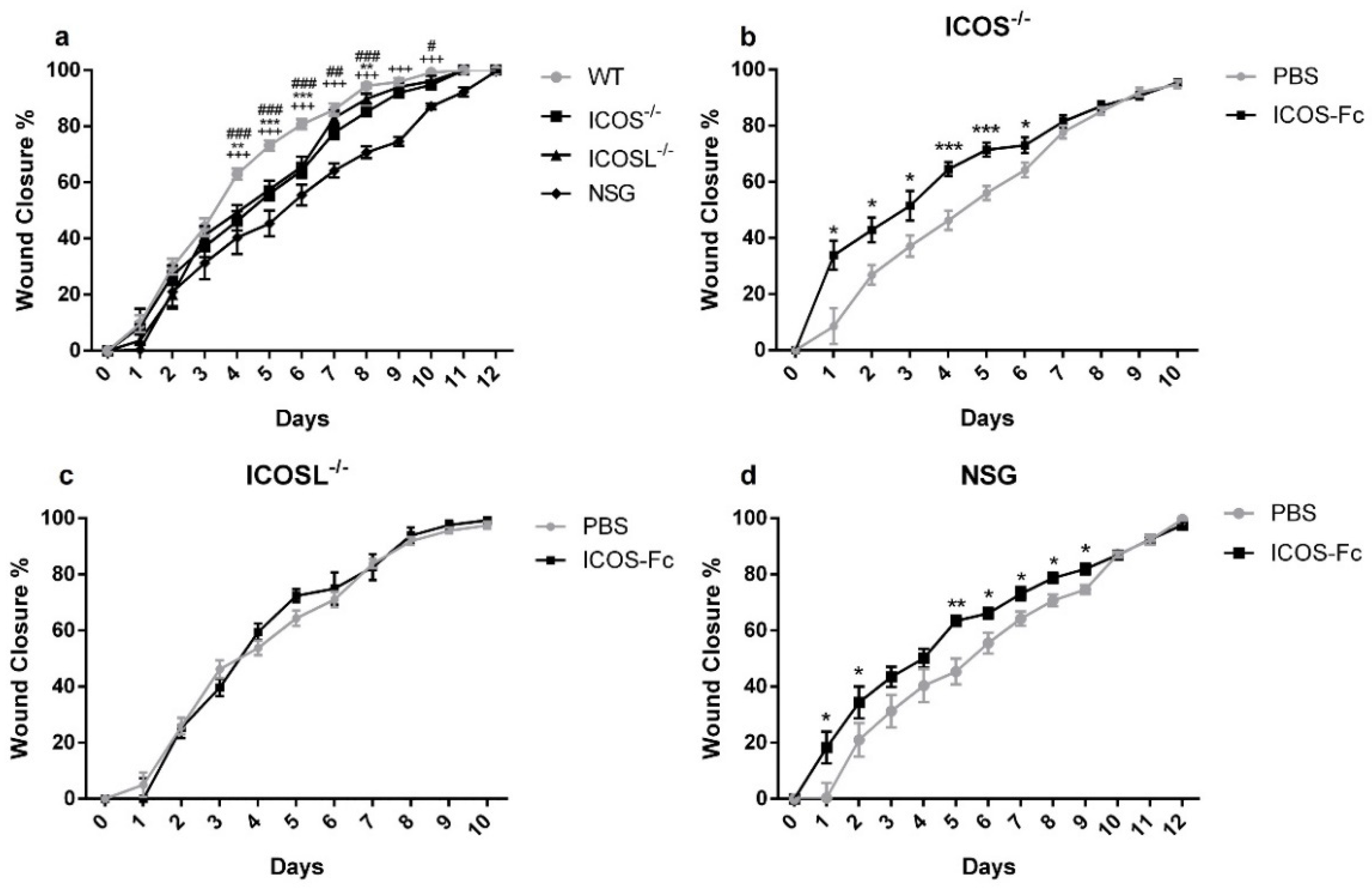
2.4. Wound Healing in KO Mice
3. Discussion
4. Materials and Methods
4.1. Scratch Assay
4.2. Mice
4.3. In Vivo Wounds
4.4. Real-Time PCR Analysis
4.5. Histological Analysis
4.6. Macrophage Migration Assay
4.7. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Singer, A.J.; Clark, R.A. Cutaneous wound healing. N. Engl. J. Med. 1999, 341, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Singampalli, K.L.; Balaji, S.; Wang, X.; Parikh, U.M.; Kaul, A.; Gilley, J.; Birla, R.K.; Bollyky, P.L.; Keswani, S.G. The Role of an IL-10/Hyaluronan Axis in Dermal Wound Healing. Front. Cell Dev. Biol. 2020, 8, 636. [Google Scholar] [CrossRef] [PubMed]
- Aicher, A.; Hayden-Ledbetter, M.; Brady, W.A.; Pezzutto, A.; Richter, G.; Magaletti, D.; Buckwalter, S.; Ledbetter, J.A.; Clark, E.A. Characterization of human inducible costimulator ligand expression and function. J. Immunol. 2000, 164, 4689–4696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Xiong, X. ICOS+ Tregs: A Functional Subset of Tregs in Immune Diseases. Front. Immunol. 2020, 11, 2104, Correction in Front. Immunol. 2021, 12, 701515. [Google Scholar] [CrossRef]
- Yoshinaga, S.K.; Whoriskey, J.S.; Khare, S.D.; Sarmiento, U.; Guo, J.; Horan, T.; Shih, G.; Zhang, M.; Coccia, M.A.; Kohno, T.; et al. T-cell co-stimulation through B7RP-1 and ICOS. Nature 1999, 402, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, A.H.; Freeman, G.J. The B7-CD28 superfamily. Nat. Rev. Immunol. 2002, 2, 116–126. [Google Scholar] [CrossRef]
- Tang, G.; Qin, Q.; Zhang, P.; Wang, G.; Liu, M.; Ding, Q.; Qin, Y.; Shen, Q. Reverse signaling using an inducible costimulator to enhance immunogenic function of dendritic cells. Cell. Mol. Life Sci. 2009, 66, 3067–3080. [Google Scholar] [CrossRef]
- Dianzani, C.; Minelli, R.; Mesturini, R.; Chiocchetti, A.; Barrera, G.; Boscolo, S.; Sarasso, C.; Gigliotti, C.L.; Sblattero, D.; Yagi, J.; et al. B7h triggering inhibits umbilical vascular endothelial cell adhesiveness to tumor cell lines and polymorphonuclear cells. J. Immunol. 2010, 185, 3970–3979. [Google Scholar] [CrossRef]
- Dianzani, C.; Minelli, R.; Gigliotti, C.L.; Occhipinti, S.; Giovarelli, M.; Conti, L.; Boggio, E.; Shivakumar, Y.; Baldanzi, G.; Malacarne, V.; et al. B7h triggering inhibits the migration of tumor cell lines. J. Immunol. 2014, 192, 4921–4931. [Google Scholar] [CrossRef] [Green Version]
- Occhipinti, S.; Dianzani, C.; Chiocchetti, A.; Boggio, E.; Clemente, N.; Gigliotti, C.L.; Soluri, M.F.; Minelli, R.; Fantozzi, R.; Yagi, J.; et al. Triggering of B7h by the ICOS modulates maturation and migration of monocyte-derived dendritic cells. J. Immunol. 2013, 190, 1125–1134. [Google Scholar] [CrossRef] [Green Version]
- Gigliotti, C.L.; Boggio, E.; Clemente, N.; Shivakumar, Y.; Toth, E.; Sblattero, D.; D’Amelio, P.; Isaia, G.C.; Dianzani, C.; Yagi, J.; et al. ICOS-Ligand Triggering Impairs Osteoclast Differentiation and Function In Vitro and In Vivo. J. Immunol. 2016, 197, 3905–3916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raineri, D.; Dianzani, C.; Cappellano, G.; Maione, F.; Baldanzi, G.; Iacobucci, I.; Clemente, N.; Baldone, G.; Boggio, E.; Gigliotti, C.L.; et al. Osteopontin binds ICOSL promoting tumor metastasis. Commun. Biol. 2020, 3, 615. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.E.; Li, N.Y.; Wai, P.Y.; Kuo, P.C. Epithelial-mesenchymal transition, TGF-β, and osteopontin in wound healing and tissue remodeling after injury. J. Burn Care Res. 2012, 33, 311–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, R.; Shaw, T.J.; Martin, P. Molecular mechanisms linking wound inflammation and fibrosis: Knockdown of osteopontin leads to rapid repair and reduced scarring. J. Exp. Med. 2008, 205, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Maeda, S.; Fujimoto, M.; Matsushita, T.; Hamaguchi, Y.; Takehara, K.; Hasegawa, M. Inducible costimulator (ICOS) and ICOS ligand signaling has pivotal roles in skin wound healing via cytokine production. Am. J. Pathol. 2011, 179, 2360–2369. [Google Scholar] [CrossRef]
- Johnson, B.Z.; Stevenson, A.W.; Prêle, C.M.; Fear, M.W.; Wood, F.M. The Role of IL-6 in Skin Fibrosis and Cutaneous Wound Healing. Biomedicines 2020, 8, 101. [Google Scholar] [CrossRef]
- Ramavath, N.N.; Gadipudi, L.L.; Provera, A.; Gigliotti, L.C.; Boggio, E.; Bozzola, C.; Albano, E.; Dianzani, U.; Sutti, S. Inducible T-Cell Costimulator Mediates Lymphocyte/Macrophage Interactions During Liver Repair. Front. Immunol. 2021, 12, 786680. [Google Scholar] [CrossRef]
- Clemente, N.; Boggio, E.; Gigliotti, L.C.; Raineri, D.; Ferrara, B.; Miglio, G.; Argenziano, M.; Chiocchetti, A.; Cappellano, G.; Trotta, F.; et al. Immunotherapy of experimental melanoma with ICOS-Fc loaded in biocompatible and biodegradable nanoparticles. J. Control. Release 2020, 320, 112–124. [Google Scholar] [CrossRef]
- Darby, I.A.; Laverdet, B.; Bonté, F.; Desmoulière, A. Fibroblasts and myofibroblasts in wound healing. Clin. Cosmet. Investig. Dermatol. 2014, 7, 301–311. [Google Scholar]
- Raineri, D.; Cappellano, G.; Vilardo, B.; Maione, F.; Clemente, N.; Canciani, E.; Boggio, E.; Gigliotti, C.L.; Monge, C.; Dianzani, C.; et al. Inducible T-Cell Costimulator Ligand Plays a Dual Role in Melanoma Metastasis upon Binding to Osteopontin or Inducible T-Cell Costimulator. Biomedicines 2021, 10, 51. [Google Scholar] [CrossRef]
- Boggio, E.; Gigliotti, C.L.; Moia, R.; Scotta, A.; Crespi, I.; Boggione, P.; De Paoli, L.; Deambrogi, C.; Garzaro, M.; Vidali, M.; et al. Inducible T-cell co-stimulator (ICOS) and ICOS ligand are novel players in the multiple-myeloma microenvironment. Br. J. Haematol. 2022, 196, 1369–1380. [Google Scholar] [CrossRef] [PubMed]
- Cui, K.; Ardell, C.L.; Podolnikova, N.P.; Yakubenko, V.P. Distinct Migratory Properties of M1, M2, and Resident Macrophages Are Regulated by αDβ2 and αMβ2 Integrin-Mediated Adhesion. Front. Immunol. 2018, 9, 2650. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Chen, S.; Liu, H.B.; Parent, C.A.; Coulombe, P.A. Keratin 6 regulates collective keratinocyte migration by altering cell-cell and cell-matrix adhesion. J. Cell Biol. 2018, 217, 4314–4330. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stoppa, I.; Gigliotti, C.L.; Clemente, N.; Pantham, D.; Dianzani, C.; Monge, C.; Puricelli, C.; Rolla, R.; Sutti, S.; Renò, F.; et al. ICOSL Stimulation by ICOS-Fc Accelerates Cutaneous Wound Healing In Vivo. Int. J. Mol. Sci. 2022, 23, 7363. https://doi.org/10.3390/ijms23137363
Stoppa I, Gigliotti CL, Clemente N, Pantham D, Dianzani C, Monge C, Puricelli C, Rolla R, Sutti S, Renò F, et al. ICOSL Stimulation by ICOS-Fc Accelerates Cutaneous Wound Healing In Vivo. International Journal of Molecular Sciences. 2022; 23(13):7363. https://doi.org/10.3390/ijms23137363
Chicago/Turabian StyleStoppa, Ian, Casimiro Luca Gigliotti, Nausicaa Clemente, Deepika Pantham, Chiara Dianzani, Chiara Monge, Chiara Puricelli, Roberta Rolla, Salvatore Sutti, Filippo Renò, and et al. 2022. "ICOSL Stimulation by ICOS-Fc Accelerates Cutaneous Wound Healing In Vivo" International Journal of Molecular Sciences 23, no. 13: 7363. https://doi.org/10.3390/ijms23137363
APA StyleStoppa, I., Gigliotti, C. L., Clemente, N., Pantham, D., Dianzani, C., Monge, C., Puricelli, C., Rolla, R., Sutti, S., Renò, F., Boldorini, R., Boggio, E., & Dianzani, U. (2022). ICOSL Stimulation by ICOS-Fc Accelerates Cutaneous Wound Healing In Vivo. International Journal of Molecular Sciences, 23(13), 7363. https://doi.org/10.3390/ijms23137363